

Supplemental Digital Content is available in the text.
Keywords: adherens junctions, angiogenesis, blood vessels, cell adhesion molecules, growth and development
Abstract
Rationale:
Angiogenesis and vessel integrity depend on the adhesion of endothelial cells (ECs) to the extracellular matrix and to adjacent ECs. The focal adhesion protein α-parvin (α-pv) is essential for vascular development. However, the role of α-pv in ECs in vivo is not known.
Objective:
To determine the function of α-pv in ECs during vascular development in vivo and the underlying mechanisms.
Methods and Results:
We deleted the α-pv gene specifically in ECs of mice to study its role in angiogenesis and vascular development. Here, we show that endothelial-specific deletion of α-pv in mice results in late embryonic lethality associated with hemorrhages and reduced vascular density. Postnatal-induced EC-specific deletion of α-pv leads to retinal hypovascularization because of reduced vessel sprouting and excessive vessel regression. In the absence of α-pv, blood vessels display impaired VE-cadherin junction morphology. In vitro, α-pv–deficient ECs show reduced stable adherens junctions, decreased monolayer formation, and impaired motility, associated with reduced formation of integrin-mediated cell–extracellular matrix adhesion structures and an altered actin cytoskeleton.
Conclusions:
Endothelial α-pv is essential for vessel sprouting and for vessel stability.
The expansion of the blood vessel network, known as angiogenesis, is a critical process that occurs in response to an insufficient supply of oxygen and nutrients during development, tissue growth, and tissue regeneration.1 Sprouting angiogenesis involves coordinated endothelial cell (EC) specification, adhesion, migration, and proliferation, and its regulation involves physical interactions of ECs to extracellular matrix (ECM), as well as homotypic adhesions between adjacent ECs. Sprouting angiogenesis is initiated in response to the local production of tissue-derived proangiogenic factors, such as vascular endothelial growth factor, which bind to cognate receptors on the endothelium and induce the specification and migration of leading tip cells, stimulate the proliferation of the neighboring stalk cells to elongate the sprouts and form the lumen.1 Subsequent to sprouting angiogenesis, the initial vascular plexus is remodeled through extensive pruning and selective branch regression, ultimately establishing an efficient and mature hierarchical vascular network.1
The dynamic behavior of ECs during angiogenesis largely depends on the organization and dynamic rearrangement of the actin cytoskeleton.2 For instance, tip cells dynamically extend actin-driven filopodia and lamellipodia to explore the environment for chemotactic guidance cues to provide directionality for the developing vessel network and to migrate.3,4 These cellular projections also facilitate the formation of initial cell–cell contacts between tip cells from different sprouts, allowing anastomosis of the sprouts from different vessel segments.1 These initial cell–cell contacts involve the adherens junctions (AJs), in which adhesion is mediated by the homophilic engagement of free-floating vascular endothelial (VE)-cadherin molecules in the plasma membrane of neighboring cells.5 AJs are also needed for stabilization of newly formed vessels.5 VE-cadherin interacts, via its cytoplasmic domains, with proteins associated with the actin cytoskeleton, thereby anchoring the junctions to the cellular scaffold and allowing the maturation of the junction.6–8 Angiogenesis requires fast and local remodeling of endothelial cell–cell junctions.9 This rapid remodeling of endothelial AJs is, in part, achieved by local spatiotemporal rearrangements of the actin cytoskeleton.10 Recently, it has been shown that actin-driven lamellipodia that develop at VE-cadherin–free spaces within stable cell–cell junctions regulate the local dynamics and patterning of VE-cadherin, thereby controlling AJ integrity and monolayer formation.11 These new lamellipodia projections are named junction-associated intermittent lamellipodia (JAIL).
In addition to its contact to neighboring cells by AJs, the actin cytoskeleton is physically connected to the ECM by integrin-mediated adhesions, which are necessary for cell spreading and migration.12 Integrins are heterodimeric transmembrane receptors composed of an α and a β subunit.13 On ECM binding, integrins recruit adaptor and signaling proteins to their cytoplasmic domains and form focal adhesions (FAs), through which they relay signals into cells. The assembly of FAs is initiated by the formation of nascent adhesions and focal complexes (FXs) at the edge of the lamellipodium. Then, a subset of FXs grows and matures into FAs, which become the primary anchorage sites of stress fibers.12 As such, integrin-mediated adhesion and signaling are essential for angiogenesis.13 β1 integrins regulate EC adhesion, migration, and survival,14 and their importance is illustrated in studies in which endothelial-specific deletion of β1 integrins causes early embryonic lethality of mice because of vascular defects.15,16 Integrin-mediated cell adhesion to the ECM also regulates critical cell–cell communication processes required for coordinated collective behavior of ECs. For instance, integrin-ECM binding regulates the specification of tip and stalk cells through modulation of Dll4/Notch signaling, thus controlling vessel sprouting.17 Integrins also regulate assembly and integrity of cell–cell junctions by controlling local rearrangement of the actin cytoskeleton at the cell–cell contacts.18,19
Parvins are a family of adaptor proteins that localize to FAs and facilitate the interaction of integrins with the actin cytoskeleton and consist of 3 members: α-parvin (α-pv), β-parvin (β-pv), and γ-parvin (γ-pv).20 Parvins contain an actin-binding domain comprising 2 tandem calponin homology domains which enable parvins to recruit actin filaments (F-actin) to FAs and associate to stress fibers. Parvins also interact with actin binding and regulatory proteins including, paxillin, integrin-linked kinase, and regulators of the Rho GTPases, thereby playing a critical role in actin-dependent processes, including cell shape regulation and cell migration.20 Specifically, α-pv is a critical regulator of vascular development.21 Its ubiquitous gene deletion in mice leads to embryonic lethality associated with severe cardiovascular defects, including aberrant organization of vessel networks, impaired coverage of vessels by mural cells, and abnormal heart development.21 The functions of α-pv in ECs are, however, not known.
Here, we show that deletion of α-pv in ECs in mice results in multiple vascular defects characterized by decreased vascular density because of reduced vessel sprouting and compromised vessel stability. Our results show that α-pv is essential for the coordinated changes in cell shape of ECs required for maintenance of cell–cell junctions.
Methods
Mutant Mice and Inducible Genetic Experiments
To delete α-pv in ECs, Tie2-Cre transgenic mice22 were bred into a background of α-pvfloxed/floxed (α-pvfl/fl) mice, and embryos were analyzed for the expression of α-pv at different stages. α-pvfl/fl mice were generously provided by Reinhard Fässler. For postnatal EC-specific loss-of-function experiments, Cdh5(PAC)-CreERT223;α-pvfl/fl mice (males) were mated with α-pvfl/fl mice (females). Gene inactivation in pups was triggered by intraperitoneal injection of 50 μL of tamoxifen solution (Sigma; 1 mg/mL; generated by diluting a 10 mg/mL tamoxifen stock solution in 1:4 ethanol:peanut oil with additional peanut oil) once daily at postnatal days (P) 1, P2, and P3.
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Promocell (C-12203) or alternatively isolated from embryos as described elsewhere24 and cultured with EC medium (Promocell). For the isolation of ECs, embryos were harvested at E13.5 and washed in PBS. For each embryo, the tail was removed and used for genotyping. Embryos were treated with digestion buffer (collagenase A [Sigma; 1 mg/mL] and dispase-II [Boehringer; 1 mg/mL] in PBS) at 37°C for 60 minutes. Samples were filtered through cell strainers and incubated with VE-cadherin–coated Dynabeads for 30 minutes at room temperature. VE-cadherin–positive cells coupled to Dynabeads were purified with a magnet, centrifuged, and resuspended in fresh EC growth medium (Promocell) for culturing.
Small Interfering RNA Transfection
HUVECs were transfected with a small interfering RNA (siRNA) duplex against α-pv (Sigma, SASI_Hs01_00165014 and SASI_Hs01_00165015) and scrambled control (Sigma, SIC001) using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s protocol. Experiments were performed 48 hours after transfection. All experiments were performed with 2 independent siRNA against α-pv.
Expression of Lifeact-Enhanced Green Fluorescent Protein in HUVECs by Lentiviral Gene Transfer and Spinning Disc Live-Cell Imaging
Expression of Lifeact-enhanced green fluorescent protein (EGFP) was performed by lentiviral gene transfer as described in detail recently.11 Briefly, passage 1 HUVECs were seeded on cross-linked gelatin at a cell density of about 3×104 and subsequently infected with replication-deficient lentivirus carrying Lifeact-EGFP. After 2 to 3 days, cells Lifeact-EGFP displayed a sufficient signal usable to perform fluorescent live-cell imaging by confocal spinning disc microscopy (Carl Zeiss) at 37°C and 5% CO2.
Quantification of Reticular Junctional Index, Intercellular Space Index, Gap Index, and Gap Size Index
Reticular junctional index (reticular junctional area/cell number) was calculated using the formula (total reticular junctional area/total cell area)/cell number. Intercellular space index (number of intercellular spaces per cell number) was calculated using the formula ([intercellular space number/total cell area]×1000)/cell number. Gap index (number of intercellular gaps/cell number) was calculated using the formula ([intercellular gap number/total cell area]×1000)/cell number. Gap size index (intercellular gap area/cell number) was determined using the formula ([intercellular gap area/total cell area]×1000)/cell number. Reticular junctional area, intercellular gap area, and total cell area were defined manually using ImageJ. In each case, a minimum of 5 fields were quantified (≈40 cells per field) per experiment, and data shown represent the mean of at least 3 independent experiments.
Quantification of JAIL Frequency
To quantify the frequency of occurrence of JAIL, Lifeact-EGFP was expressed in HUVEC cultures (4 HUVEC primary isolates) and then treated with either siRNA against α-pv or scrambled control, respectively, as indicated. Sixteen different movies were acquired in total for each α-pv and scrambled siRNA-treated cultures. The leading edges of JAIL were marked in 2 fields of view (200×200 pixel) for each movie >50 frames. The number of the marked leading edges was counted, and the values were normalized to the scrambled siRNA control experiments.
Statistical Analysis
Statistical analysis was performed using Student t test. At least 3 independent experiments were performed.
Results
Deletion of α-pv From ECs Leads to Vascular Defects, Hemorrhages, and Lethality at Late Embryogenesis
To gain insight into the functions of α-pv in ECs, we intercrossed mice carrying a loxP-flanked α-pv gene (α-pvfl/fl) with mice expressing the Cre recombinase under the control of the Tie2 promoter (Tie2-Cre).22 Intercrosses between α-pvfl/+;Tie2-Cre males and α-pvfl/+ females failed to yield viable newborn α-pvfl/fl;Tie2-Cre (referred to herein as α-pvΔEC) mice, indicating that Tie2-mediated deletion of α-pv gene is embryonically lethal (Online Table I). Western blot analysis of lung and EC lysates from α-pvΔEC embryos at embryonic day (E) 13.5 showed downregulation of α-pv expression when compared with lysates from controls littermates (Online Figure IA). Timed mating intercrosses between α-pvfl/+;Tie2-Cre males and α-pvfl/fl females showed that α-pvΔEC embryos were present at expected Mendelian ratio up to E15.5, and that lethality of α-pvΔEC embryos commenced at around E14.5 (Online Table II). By E13.5, α-pvΔEC embryos were slightly smaller than control littermates and showed subcutaneous hemorrhages primarily in the head and trunk regions (Figure 1A). Serial histological cross-sections of E15.5 embryos confirmed the presence of hemorrhages in α-pvΔEC embryos (Online Figure IB). CD31 whole-mount immunostaining of E15.5 control and α-pvΔEC embryos and yolk sacs revealed the presence of tortuous vascular plexuses and reduced vascular density in α-pvΔEC embryos (Figure 1B; Online Figure IC). Together, these results indicate that α-pv is required for embryonic blood vessel development.
Figure 1.
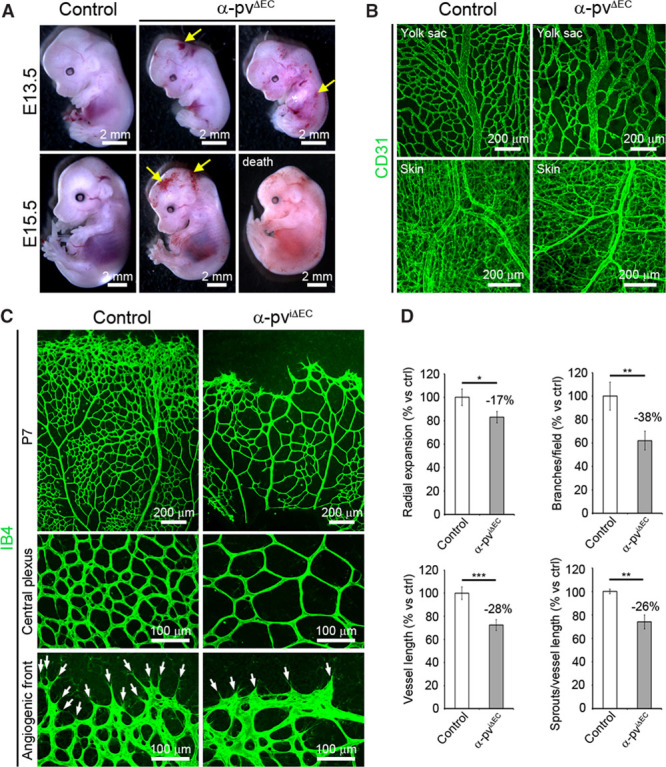
Loss of endothelial α-parvin (α-pv) leads to vascular defects and embryonic lethality in mice. A, Freshly dissected E13.5 and E15.5 control and α-pvΔEC embryos. Arrows point to subcutaneous hemorrhages. B, CD31 whole-mount immunostaining of E15.5 yolk sac and dermal vasculature. C, Visualization of the vasculature by isolectin-B4 (IB4) immunofluorescence in retinas from control and α-pviΔEC mice at P7. Arrows point to vessel sprouts. D, Quantification of vascular parameters in the control and α-pviΔEC retinas as indicated. Values represent percentages of mean vs respective controls±SEM. P values are 0.024, 0.002, 0.001, and 0.004, respectively. EC indicates endothelial cell. ns P>0.05, *P≤0.05, **P≤0.01, ***P≤0.001.
Postnatal EC-Specific α-pv Deletion Results in Reduced Vessel Sprouting and Decreased Vessel Density
Next, we investigated the functions of endothelial α-pv in the retinal vasculature. From postnatal day (P) 1 until P8, a primary vascular plexus grows progressively within the ganglion layer of the mouse retina from the optic stalk toward the periphery.1 We crossed α-pvfl/fl mice with Cadh5(PAC)-CreERT2 mice,23 induced α-pv gene deletion in ECs by administering 3 consecutive intraperitoneal injections of tamoxifen in newborns starting at P1, and analyzed retinal vascularization over time.25 Western blot analysis of lung lysates from P6 α-pvfl/fl;Cadh5(PAC)-CreERT2 (referred to herein as α-pviΔEC) mice showed downregulation of α-pv expression when compared with lysates from Cre-negative control littermates (Online Figure IIA). Isolectin-B4 (IB4) labeling of control and α-pviΔEC retinas showed a significant reduction in radial expansion of the vasculature from the center to the periphery in α-pviΔEC retinas compared with control retinas (Figure 1C and 1D; Online Figure IIB). Vessel density (quantified by the number of branch points) and vessel sprouting (quantified by the number of sprouts per vessel length) at the angiogenic front were also significantly reduced in α-pviΔEC retinas (Figure 1C and 1D; Online Figure IIB). Number of filopodia was not altered in the absence of α-pv (Online Figure IIC). These results indicate that endothelial α-pv is also essential for postnatal angiogenesis.
Loss of Endothelial α-pv Alters Vessel Morphology and Compromises EC Proliferation
A closer morphological analysis showed that vessels from α-pviΔEC retinas displayed irregular shapes and appeared unstable compared with the regular shape of vessels from control retinas (Online Figure IIIA). Similar morphological defects were also observed in vessels from α-pvΔEC embryos (Online Figure IIIB). The analysis also revealed a higher occurrence of small caliber vessel segments, IB4-labeled connections between 2 branch points, in α-pviΔEC retinas (Figure 2A). These segments were not lumenized because they were negative for intercellular adhesion molecule 2, a marker of the apical/luminal side of the vessels (Figure 2A and 2B).
Figure 2.
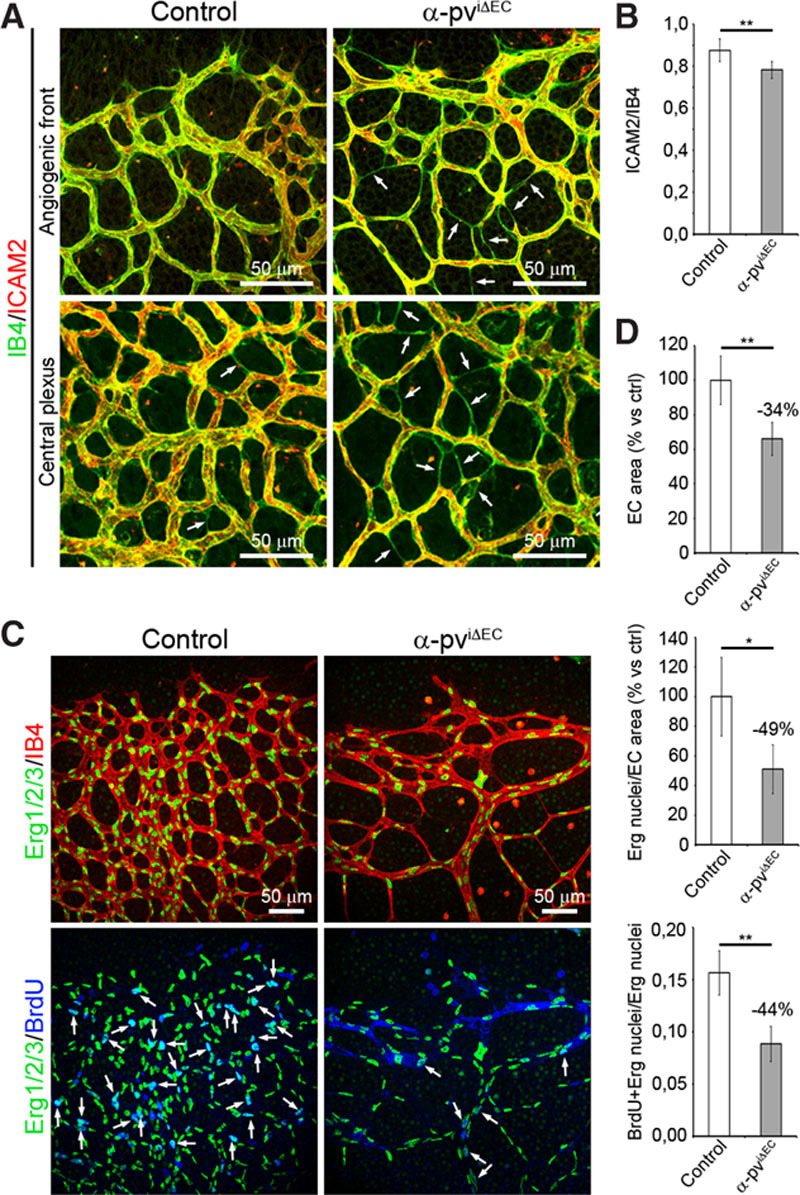
Depletion of α-parvin (α-pv) from endothelial cells (ECs) alters vessel morphology and compromises EC proliferation. A, P6 control and α-pviΔEC retinas labeled for isolectin-B4 (IB4) and intercellular adhesion molecule 2 (ICAM2). B, Ratio of ICAM2-positive vessel segments to IB4-positive vessel segments. Values represent percentages of mean±SEM. P value is 0.01. C, P6 control and α-pviΔEC retinas labeled for Erg1/2/3, IB4, and bromodeoxyuridine (BrdU). D, Quantification of EC area (IB4-positive area), EC nuclei (Erg1/2/3-positive nuclei) per EC area, and ratio of EC/BrdU-positive nuclei to total EC nuclei. Values represent percentages of mean±SEM. P values are 0.007, 0.014, and 0.002, respectively. ns P>0.05, *P≤0.05, **P≤0.01.
Angiogenic growth of blood vessels requires proliferation of ECs.1 Bromodeoxyuridine incorporation assay in control and α-pviΔEC mice followed by a colabeling of bromodeoxyuridine, the EC-specific transcription factor Erg1/2/3 and IB4 showed a reduced number of proliferating ECs in α-pviΔEC retinas compared with control retinas, indicating that endothelial α-pv positively controls proliferation of ECs (Figure 2C and 2D).
Loss of Endothelial α-pv Results in Ectopic Vessel Regression
Because vessel instability results in reduced vessel density1 and regressing ECs leave empty basal membrane sleeves rich in collagen IV26, we determined whether depletion of α-pv from ECs impairs vessel stability by performing whole-mount immunostaining of control and α-pviΔEC retinas using an antibody against collagen IV and IB4. The analysis showed a significant increase in collagen IV segments lacking IB4 in α-pviΔEC retinas compared with control retinas, indicating that endothelial loss of α-pv indeed compromises vessel stability (Figure 3A and 3B). Furthermore, whole-mount immunostaining for cleaved caspase-3 and IB4 showed a significant increase in apoptotic vessel segments in α-pviΔEC retinas compared with control retinas (Figure 3C and 3D).
Figure 3.
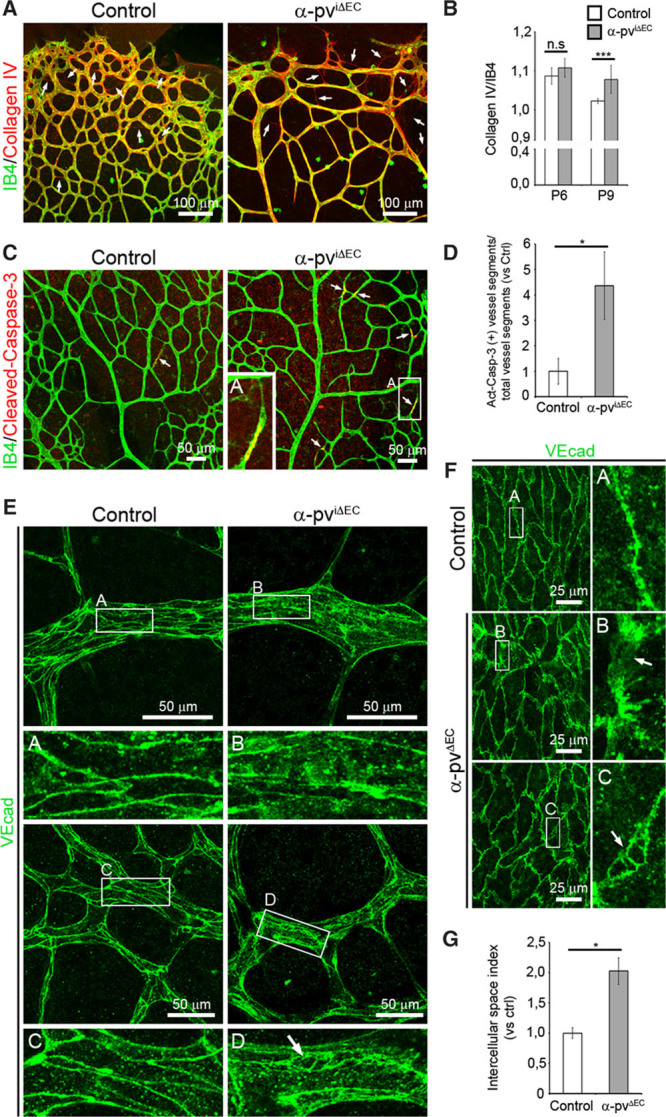
Loss of endothelial α-parvin (α-pv) results in increased vessel regression. A, P8 control and α-pviΔEC retinas labeled for isolectin-B4 (IB4) and collagen IV. Arrows point to empty collagen IV sleeves. B, Ratio of collagen IV–positive vessel segments to IB4-positive vessel segments. Values represent mean±SEM. P values are 0.18 and 0.0004, respectively. C, P7.5 control and α-pviΔEC retinas labeled for IB4 and cleaved (active) caspase-3. Arrows point to cleaved caspase-3–positive vessel segments. D, Relative ratio of cleaved (Act) caspase-3–positive vessel segments to total vessel segments. Values represent mean vs control±SEM. P value is 0.05. E, P7 control and α-pviΔEC retinas labeled for VE-cadherin. Arrow highlights intercellular space between endothelial cells (ECs). F, VE-cadherin whole-mount immunostaining of E15.5 yolk sacs from control and α-pvΔEC mice. Arrows highlight intercellular space between ECs. G, Quantification of intercellular space index in the YS vasculature. Values represent mean vs control±SEM. P value is 0.02. ns P>0.05, *P≤0.05, ***P≤0.001.
Blood vessel stability and the maintenance of vascular integrity greatly depend on cell–cell junctions between ECs.5 We therefore performed whole-mount immunostaining for the junctional marker VE-cadherin in retinas of both α-pviΔEC and control littermates. Indeed, we observed a diffuse and discontinuous stain around cell boundaries in vessels of α-pviΔEC retinas compared with the sharp and continuous stain observed in vessels of control retinas (Figure 3E). In α-pviΔEC retinas, we also observed several cases of vessel segments with a cytoplasmic dotted VE-cadherin stain and fragments of vessels partially disconnected from the vascular bed (Online Figure IV). In addition, mutant vessels displayed a higher incidence of intercellular spaces between ECs compared with control vessels (Figure 3E–3G). Endothelial junctions were also similarly disorganized in vessels of α-pvΔEC embryos (Figure 3F and 3G).
Mural cells, namely vascular smooth muscle cells and pericytes, are recruited to newly formed vessels to add vessel stability.27 Because α-pv is crucial for the recruitment of mural cells to the vessel wall in embryos,21 we analyzed the coverage of vessels by mural cells in α-pvΔEC embryos and α-pviΔEC mice. Whole-mount immunostaining of control and α-pvΔEC embryos and control and α-pviΔEC retinas using antibodies against α-smooth muscle actin and antineuron glial 2 showed that the mural cell coverage of the embryonic and retinal vessels lacking α-pv was comparable with control vessels (Online Figure V), suggesting that endothelial depletion of α-pv did not have a significant effect on the recruitment of the mural cells to the vessel wall. Collectively, these results indicate that depletion of endothelial α-pv impairs vessel stability and that endothelial α-pv is required for a proper junctional formation.
Depletion of α-pv in ECs Impairs VE-Cadherin–Mediated Cell–Cell Junctions and Delays Monolayer Formation
Two types of AJs can be distinguished in ECs: continuous, stable AJs, in which VE-cadherin is localized linearly along cell–cell borders, and discontinuous AJs, in which VE-cadherin is distributed in many short linear structures perpendicular to cell–cell borders.10,28 In addition, in in vitro cultures of primary ECs, VE-cadherin shows a particularly broad reticular pattern at sites of cellular overlap (reticular junctions).29,30
To investigate the role of α-pv in the regulation of EC junctions, we depleted α-pv in primary HUVECs by siRNA and performed double immunostaining for VE-cadherin and β-catenin. All experiments were performed with 2 independent siRNA against α-pv (Online Figure VIA). After 24 hours in culture, α-pv–deficient cells displayed irregular shapes with VE-cadherin and β-catenin distributed in filopodia-like structures perpendicular to the cell borders, whereas control cells (HUVECs transfected with scrambled siRNA) had a round cobblestone-like morphology (Figure 4A; Online Figure VIB). Quantitative analysis indicated that depletion of α-pv significantly decreased the levels of stable AJs (Figure 4B). Next, we determined the reticular junctional area per cell (reticular junctional index; see Methods), and found that α-pv–deficient cells also showed a significant reduction of reticular junctions (Figure 4B). In addition, quantification also showed a higher incidence of intercellular gaps per cell (gap index; see Methods) in α-pv–deficient cells compared with control cells (Online Figure VIC).
Figure 4.
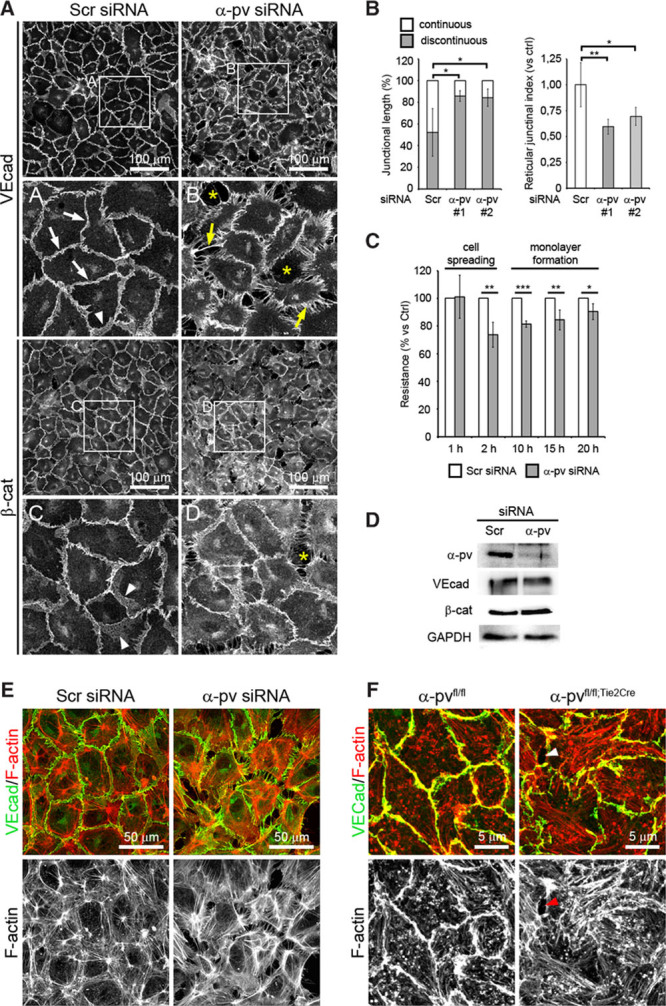
Depletion of α-parvin (α-pv) in endothelial cells (ECs) impairs adherens junction (AJ) stability. A, VE-cadherin and β-catenin immunostaining of human umbilical vein endothelial cells (HUVECs) transfected either with control (Scramble) or α-pv small interfering RNA (siRNA) and cultured on gelatin-coated slides for 24 hours. White arrows point to stable AJs, arrowheads point to overlapping junctions, and yellow arrows point to radial VE-cadherin bundles. Asterisks highlight intercellular gaps. B, Quantification of percentages of continuous and discontinuous AJs, and the reticular junctional index in control and α-pv–depleted HUVECs. Values mean±SEM. P values are 0.03, 0.02, 0.005, and 0.03, respectively. C, Transendothelial resistance measurements over time in cultures of control and α-pv–depleted HUVECs. Values represent percentages of mean vs controls±SEM. P values are 0.007, 0.0001, 0.02, and 0.05, respectively. D, Western blot of α-pv, VE-cadherin, and β-catenin protein levels in control and α-pv–depleted HUVECs cultured on gelatin-coated plates for 24 hours. GAPDH was used as a loading control. Double-fluorescence labeling for VE-cadherin and F-actin of (E) control and α-pv–depleted HUVECs, and (F) primary ECs isolated from α-pvfl/fl and α-pvfl/fl;Tie2Cre embryos cultured on gelatin-coated slides for 48 hours. Arrowheads point to intercellular gap. ns P>0.05, *P≤0.05, **P≤0.01, ***P≤0.001.
To confirm the relevance of α-pv for endothelial monolayer integrity, control and α-pv–depleted HUVECs were seeded on gold electrodes, and transendothelial resistance was recorded by electric cell-substrate impedance sensing. In this assay, cells are seeded in sufficient numbers to cover the electrode during cell spreading, and transendothelial resistance is recorded continuously in a noninvasive fashion. The increase in transendothelial resistance in the first hours after seeding reflects cell spreading and at later time points represents junctional integrity.31 Depletion of α-pv did not affect initial cell spreading, but transendothelial resistance in the second phase of the experiment was significantly lower in α-pv–depleted cells compared with control cells, indicating that loss of α-pv leads to reduced monolayer integrity (Figure 4C). VE-cadherin or β-catenin protein levels were not affected in α-pv deficiency, indicating that cell junction defects and impaired monolayer formation were not caused by low VE-cadherin or β-catenin expression (Figure 4D).
Stable AJs are associated with cortical actin and are aligned by thin parallel actin bundles, whereas discontinuous AJs are attached to radial stress fibers.10,28 F-actin staining of control and α-pv–deficient cells showed that loss of α-pv also induced changes in the actin cytoskeleton, which were characterized by an increase in staining of short radial actin bundles (Figure 4E; Online Figure VIB).
To corroborate these findings, we isolated ECs from control and α-pvΔEC embryos (Online Figure IA), plated them on gelatin for 48 hours and performed double-fluorescence labeling for VE-cadherin and F-actin. Similar to α-pv–deficient HUVECs, ECs from α-pvΔEC embryos showed discontinuous VE-cadherin staining, reduced levels of VE-cadherin–associated cortical actin, increased levels of radial actin bundles, and a higher incidence of intercellular gaps (Figure 4F; Online Figure VID and VIE). Collectively, these results indicate that α-pv is required for cell–cell junction integrity and actin cytoskeleton organization in ECs.
α-pv Localizes to JAIL at VE-Cadherin Junctions and Is Required for Its Formation
To elucidate the mechanism by which α-pv regulates cell–cell junctions, we first investigated the subcellular localization of α-pv in HUVECs. Under sparse culture conditions, α-pv localized at FXs close to the edge of the lamellipodium (Online Figure VIIA; arrowheads) and at FAs at the tip of stress fibers (Online Figure VIIA; arrows). At sites where 2 adjacent cells overlap, α-pv also localized in small, punctate clusters that resemble FXs along the edge of the overlapping membranes (Figure 5A). Immunostaining revealed that initial VE-cadherin clusters localize between and in close proximity to α-pv clusters along the edge of these overlapping areas (Figure 5A). Similarly, α-pv was also distributed along the edge of JAIL at overlapping plasma membranes (Figure 5A; Online Figure VIIB; Online Movie I). Triple-fluorescence labeling for α-pv, VE-cadherin, and F-actin showed that α-pv dot-like structures at the cell–cell junctions are associated with the F-actin and occasionally connected via actin filaments to α-pv–positive FA-like structures (Figure 5B).
Figure 5.
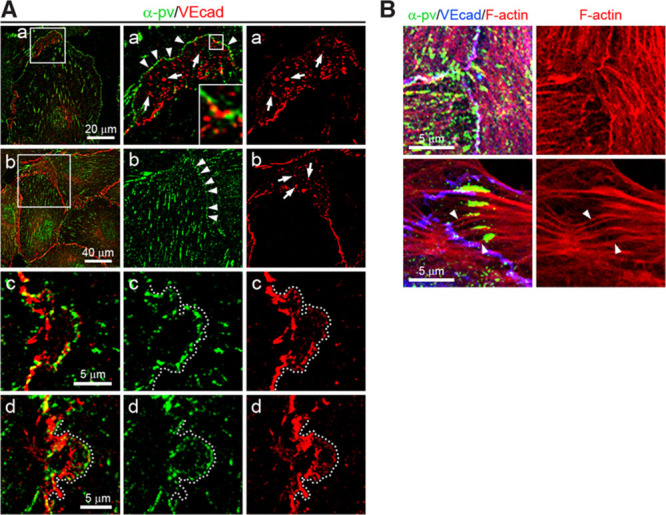
Subcellular localization of α-parvin (α-pv) in endothelial cells (ECs). A, Double immunostaining of α-pv and VE-cadherin of human umbilical vein endothelial cells (HUVECs) cultured under sparse (a) and subconfluent (b–d) conditions on gelatin for 24 hours. Arrows point to VE-cadherin clusters and arrowheads indicate α-pv clusters. Dotted lines highlight the edge of small junction-associated intermittent lamellipodia. B, Triple-fluorescent labeling for α-pv, F-actin, and VE-cadherin of HUVECs. Notice that focal adhesions are linked to adherens junctions by F-actin cables (arrowheads).
Next, we investigated whether α-pv is needed for the formation of JAIL. Because JAIL are more prominent when ECs are cultured at low confluence,11 we depleted α-pv in HUVECs expressing Lifeact-EGFP32 and performed spinning disc fluorescence live-cell imaging under subconfluent conditions. In control cells, JAIL were characterized by round edges, broad lamella, and a growing period of about 5 minutes before retraction (Figure 6A; Online Movie II). In contrast, α-pv–deficient cells extended irregular and discontinuous lamellipodial projections, which were frequently interrupted by edge withdrawal (Figure 6B; Online Video II). These abnormal projections usually displayed multiple filopodia-like structures along the membrane cortex (Figure 6B; Online Video II). Moreover, quantification showed reduced number of JAIL in α-pv–deficient cells compared with control cells (Figure 6C). The analysis also showed that depletion of α-pv led to increased formation of intercellular gaps (Figure 6B, arrowheads). Collectively, these results indicate that α-pv is required for proper JAIL formation.
Figure 6.
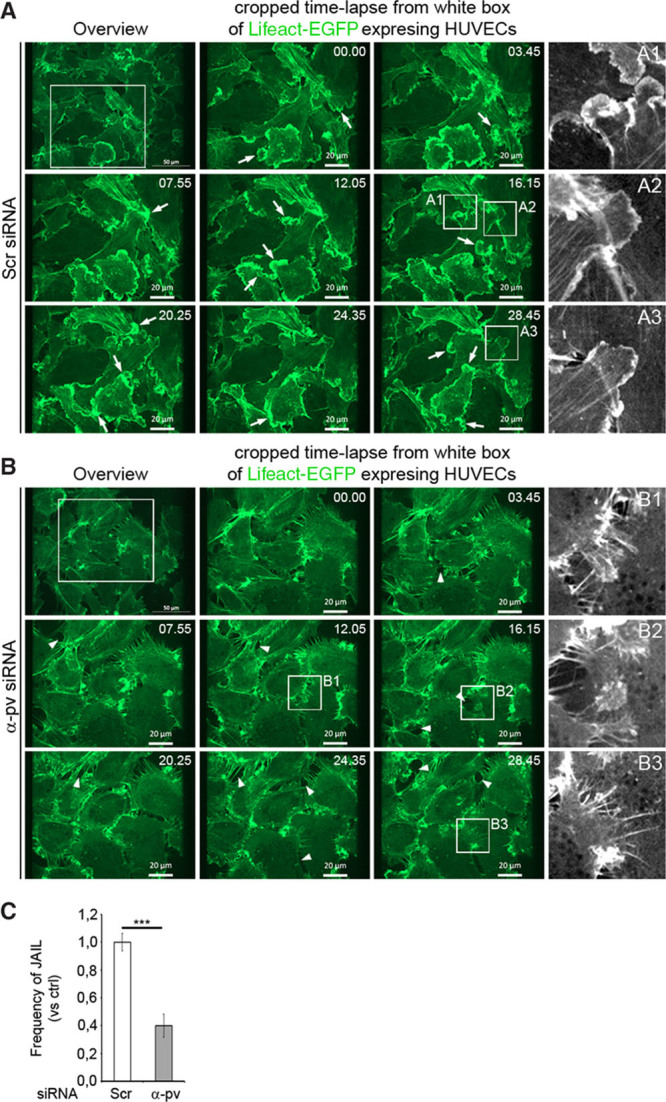
Depletion of α-parvin (α-pv) in endothelial cells (ECs) reduces junction-associated intermittent lamellipodia (JAIL) and augments intercellular gap formation. Still images (upper left images) and enlarged views (white boxes) from time-lapse recordings of control and α-pv–depleted human umbilical vein endothelial cells (HUVECs) expressing Lifeact-enhanced green fluorescent protein (EGFP) and cultured under subconfluent conditions on cross-linked gelatin-coated plates. A, Multiple correctly formed Lifeact-EGFP–positive JAIL (arrows) develop in the control cell. Black and white images on the right highlight normal development of JAILs in control cells. B, Dysfunctional JAIL in α-pv–depleted cells. Black and white images on the right highlight JAIL disruption (B1 and B2) and abnormal JAIL with multiple filopodia-like structures (B3). Arrowheads point to intercellular gaps. C, Quantification of JAIL number in control and α-pv–depleted HUVECs. Values represent mean vs control±SEM. P value is ≤0.001. siRNA indicates small interfering RNA. ns P>0.05, ***P≤0.001.
Depletion of α-pv in ECs Leads to Reduced FX Formation and Decreased Rac Activity
Next, we sought to elucidate the mechanism by which α-pv regulates cell shape, lamellipodia formation, and actin rearrangement in ECs. Stabilization and persistence of the lamellipodia depend on the assembly of FXs at the leading edge of the lamellipodia.33 First, we investigate whether FX proteins localize to JAIL and found that like α-pv, vinculin also localized at the leading edge of certain JAIL (Figure 7A). Vinculin immunostaining also showed reduced levels of vinculin at cell–cell borders, as well as at the leading edges of lamellipodia in α-pv–deficient cells compared with control cells (Figure 7B). Therefore, we next investigated the assembly of FXs in α-pv–deficient cells. The analysis of control and α-pv–deficient HUVECs immunostained for paxillin, a marker for FXs and FAs, showed that while control cells displayed multiple FXs along the leading edges in the proximity of FAs, α-pv–deficient cells displayed few FXs and paxillin was restricted to FA-like structures (Figure 7C). At FXs, paxillin is usually phosphorylated on the tyrosine residue (Y) 11834. Consistent with reduced levels of FXs in gelatin-stimulated HUVECs lacking α-pv, phosphorylation of paxillin at Y118 was significantly reduced in these cells (Figure 7D). Importantly, whole-mount immunostaining of retinas also showed reduced paxillin phosphorylation in α-pviΔEC retinas compared with control retinas, confirming that integrin signaling is also affected in vivo (Figure 7E).
Figure 7.
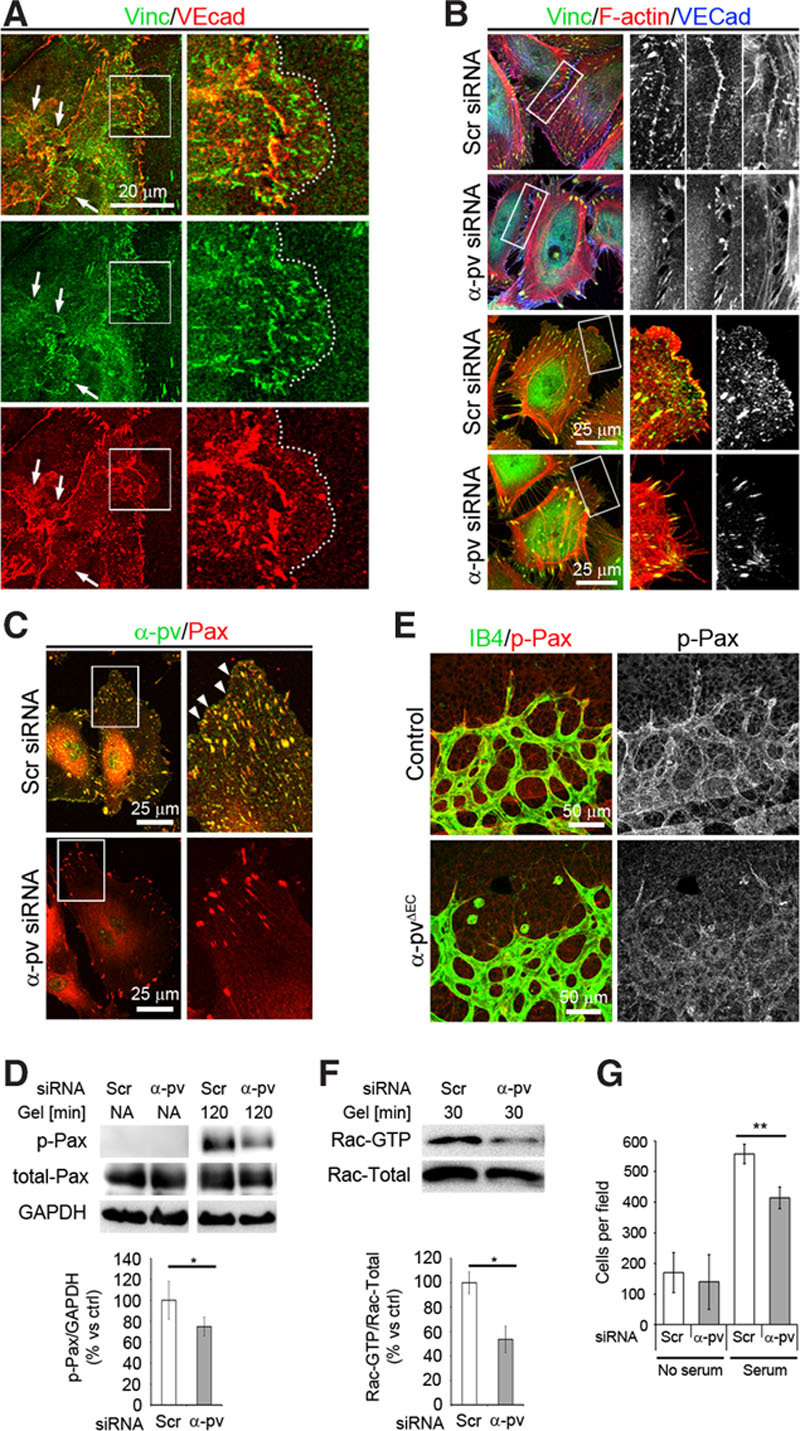
Depletion of α-parvin (α-pv) impairs formation of focal complexes (FXs) and decreased Rac activity. A, Double immunostaining of vinculin and VE-cadherin showing vinculin localization at leading edges of junction-associated intermittent lamellipodia (JAIL). Arrows point to JAIL. Dotted lines highlight the edge of a JAIL. B, Triple-fluorescent labeling for vinculin, F-actin, and VE-cadherin of control and α-pv–depleted human umbilical vein endothelial cells (HUVECs) cultured on gelatin. C, Double-fluorescent labeling for α-pv and paxillin of control and α-pv–depleted HUVECs cultured on gelatin for 12 hours. Arrowheads point to FXs. D (top), Phosphorylation of paxillin (Tyr118) was determined in nonadherent control and α-pv–depleted cells, and after 120 minutes adherence to gelatin. D (bottom), Quantification of paxillin phosphorylation; data represent relative mean values±SEM from 3 independent experiments. P value is 0.026. E, P7.5 control and α-pviΔEC retinas labeled for isolectin-B4 (IB4) and phospho-paxillin (Tyr118). F (top), α-pv–depleted HUVECs showed decreased Rac activity after 30 minutes on gelatin. F (bottom), Quantification of Rac-GTP vs total Rac; data represent relative mean values±SEM from 3 independent experiments. G, Quantification of chemotactic migration using serum as a chemoattractant (24 hours). Medium with 0.5% of serum was used to assess the baseline migration. EC indicates endothelial cell; and siRNA, small interfering RNA. ns P>0.05, *P≤0.05, **P≤0.01.
Rac activity is essential for lamellipodial extension. To test whether loss of α-pv affects Rac activity, we measured Rac activation in control and α-pv–deficient cells after 30 minutes on gelatin and found reduced Rac activity in α-pv–deficient cells compared with control cells (Figure 7F). Finally, we evaluated whether loss of α-pv impaired cell migration of ECs by performing a modified Boyden-chamber assay and found a significantly decreased migration of α-pv–deficient cells compared with control cells (Figure 7G).
Discussion
The deletion of α-pv in ECs in mice results in hemorrhages and decreased vascular density, ultimately culminating in late embryonic lethality. Postnatal EC-specific deletion of α-pv leads to retinal hypovascularization because of reduced vessel sprouting and excessive vessel regression. In the absence of endothelial α-pv, vessels display impaired cell–cell junction morphology, increased intercellular spaces between ECs, reduced EC proliferation, and increased EC apoptosis. In vitro, α-pv–deficient ECs show reduced AJ stability, impaired monolayer formation, and reduced cell motility, associated with decreased formation of integrin-mediated cell–ECM adhesion structures, reduced JAIL formation, and altered actin cytoskeleton.
The formation and maintenance of a functional vascular network require coordinated vessel sprouting and pruning and dynamic cell–cell contact remodeling between ECs to ensure vessel formation and stability.1,9 Vessel sprouting involves the specification of tip and stalk cells and the subsequent elongation of the stalk. The selection of tip and stalk cells is regulated by the Dll4/Notch signaling pathway. The promotion of Dll4 expression by vascular endothelial growth factor-A on certain ECs leads to their specification into tip cells, and these then activate Notch in the neighboring cells, which defines them as stalk cells.1 The expression of Dll4 in ECs is also promoted by β1 integrins, thereby limiting the formation of tip cells and preventing excessive vessel sprouting.17 β1 integrins are also essential for adhesion and migration of ECs,14 and integrin-mediated FA formation and actin rearrangement are essential for normal postnatal vessel sprouting and vascular plexus formation.35 Consistent with these fundamental functions, deletion of β1 integrins in ECs leads to early embryonic lethality because of angiogenic defects.15,16 Here, we show that deletion of the adaptor protein α-pv in ECs, which links integrins to the actin cytoskeleton, leads to decreased vascular density because of reduced vessel sprouting and excessive vessel regression. The reduced vessel sprouting together with the normal filopodia formation in α-pviΔEC retinas indicates that α-pv is not required for the integrin-mediated regulation of endothelial tip-cell specification, but it is essential for the elongation of endothelial sprouts. Vessel elongation depends on cell proliferation and cell intercalation.1,9 Our results show that α-pv positively regulates proliferation of ECs in vivo. Moreover, the heterogeneity in vessel diameter and the immature nature of the vascular plexus in α-pviΔEC retinas match with an inability of ECs to effectively intercalate. Cell intercalation involves collective cell migration, which requires dynamic regulation of integrin-mediated cell–ECM adhesions and VE-cadherin–mediated cell–cell junctions.9 α-pv–deficient ECs display reduced integrin-mediated cell–ECM adhesion structures, altered cell–cell junctions, and reduced cell migration. Together, these findings indicate that α-pv controls vessel sprouting by regulating integrin-mediated processes required for the elongation of endothelial sprouts.
To maintain the integrity of newly formed vessels, ECs establish cell–cell junctions and induce the recruitment of mural cells to the vessel wall.5,27 Impaired remodeling of cell–cell junctions leads to vessel regression and ultimately to vessel rupture and hemorrhages.5,36 We observed hemorrhages in α-pvΔEC embryos and excessive vessel regression in α-pviΔEC retinas, despite apparently normal mural cell coverage of the vessels, indicating an EC autonomous role for α-pv in vessel stabilization. In the absence of endothelial α-pv, vessels display fragmented VE-cadherin junctions and increased incidence of intercellular spaces between ECs. Thus, endothelial α-pv seems to stabilize developing vessels by maintaining the integrity of VE-cadherin junctions. This is in agreement with recent observations showing that β1 integrins and thereby cell–ECM adhesion controls blood vessel stability by preserving cell–cell junction integrity.37 In vitro analysis of α-pv–deficient ECs shows that α-pv is required for the stabilization of AJs, the formation of reticular junctions, and monolayer formation and integrity. The dynamic rearrangement of endothelial AJs is regulated by a variety of stimuli that often act by inducing the reorganization of the actin cytoskeleton.8 Stable AJs are connected with cortical actin.28,29 Integrins regulate integrity of cell–cell junctions by controlling local rearrangement of the actin cytoskeleton at the cell–cell contacts.18,19 Our results show that α-pv associates with the cortical actin at cell–cell borders, and α-pv–deficient cells display impaired organization of cortical actin filaments, indicating that the effects of α-pv deficiency on AJs stability are mediated by defects of actin cytoskeleton rearrangement. Altogether these findings suggest that loss of α-pv affect cell–cell junction integrity and vessel stability by perturbing integrin-mediated signaling and actin rearrangement.
Recently, it has been shown that JAIL, actin-driven lamellipodia that develop at small VE-cadherin gaps within continuous AJs, are required for the maintenance of stable AJs and for the integrity of endothelial monolayers in vitro.11 Our results show that α-pv localizes to the leading edge of JAIL and that it is required for proper JAIL formation. This suggests that loss of α-pv might impair monolayer integrity also by decreasing JAIL development and thereby VE-cadherin dynamics. Lamellipodia protrusions are driven by Arp2/3-mediated actin polymerization, which is regulated by several actin-binding and regulatory proteins. For instance, the small Rho GTPase Rac induces lamellipodial extension via activation of the Arp2/3-complex. Rac activity is also essential for JAIL formation and monolayer integrity.38 Vinculin, which associates with integrin-mediated adhesions and VE-cadherin–mediated junctions, is required for Rac-depend lamellipodia formation.5,10,39 Vinculin binds and recruits the Arp2/3-complex to cell membrane, thereby coupling the actin machinery to the adhesion sites and enabling lamellipodia protrusion.39 Our results show that vinculin localizes to certain JAIL and that loss of α-pv leads to reduced levels of vinculin at cell membranes. Moreover, α-pv–deficient ECs also show decreased Rac activity. Thus, the absence of α-pv might affect JAIL formation/stability by perturbing Rac activity. Further experiments are needed to understand molecularly the role of integrins and cell–ECM adhesion in JAIL and to define the importance of JAIL in vascular development and vessel stability in vivo.
In conclusion, we show that α-pv is critical for sprouting angiogenesis and is required for the stability of developing blood vessels.
Acknowledgments
We thank Reinhard Fässler (Max Planck Institute of Biochemistry) for providing the α-pv floxed mice and Anne-Marieke van Stalborch (Sanquin Research) for cloning and live-cell imaging experiments.
Sources of Funding
B. Pitter and E. Montanez are supported by DFG MO2562/1-1. H.-J. Schnittler is supported by DFG INST2105/24-1 and SCH430/6-2. This work was supported by the Bioimaging Network of the Ludwig-Maximilians University Munich (LMU innovative) and the Munich Cluster for Systems Neurology (SyNergy).
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- α-pv
- α-parvin
- AJ
- adherens junction
- E
- embryonic day
- EC
- endothelial cell
- ECM
- extracellular matrix
- EGFP
- enhanced green fluorescent protein
- FA
- focal adhesion
- FX
- focal complex
- HUVEC
- human umbilical vein endothelial cell
- IB4
- isolectin-B4
- JAIL
- junction-associated intermittent lamellipodia
- P
- postnatal day
- siRNA
- small interfering RNA
In March 2015, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.68 days.
Current address for H.G.: Integrative Vascular Biology, Max-Delbrück-Center for Molecular Medicine, Berlin-Buch, Germany.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.117.305818/-/DC1.
Novelty and Significance
What Is Known?
α-parvin (α-pv) is an adaptor protein that localizes to focal adhesions and facilitates the interaction of integrins with the actin cytoskeleton.
α-pv is essential for cardiovascular development in mice.
What New Information Does This Article Contribute?
α-pv positively regulates angiogenesis and is required for the stability of developing blood vessels.
α-pv is required for maintenance of cell–cell junction integrity in endothelial cells (ECs).
Deletion of α-pv in mice leads to embryonic lethality because of cardiovascular defects. However, the precise role of α-pv in ECs in vivo is not known. In this study, we deleted the α-pv gene specifically in ECs of mice to study its role in angiogenesis. We show here for the first time that endothelial α-pv controls sprouting angiogenesis and stability of developing vessels. The deletion of α-pv in ECs in mice leads to impaired angiogenesis because of both reduced vessel sprouting and excessive vessel regression. Our results also show for the first time that α-pv is indispensable for the integrity of cell–cell junctions in ECs. Together, these findings provide new insights on the role α-pv in vascular development.
References
- 1.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 2.De Smet F, Segura I, De Bock K, Hohensinner PJ, Carmeliet P. Mechanisms of vessel branching: filopodia on endothelial tip cells lead the way. Arterioscler Thromb Vasc Biol. 2009;29:639–649. doi: 10.1161/ATVBAHA.109.185165. doi: 10.1161/ATVBAHA.109.185165. [DOI] [PubMed] [Google Scholar]
- 3.Fraccaroli A, Franco CA, Rognoni E, Neto F, Rehberg M, Aszodi A, Wedlich-Söldner R, Pohl U, Gerhardt H, Montanez E. Visualization of endothelial actin cytoskeleton in the mouse retina. PLoS One. 2012;7:e47488. doi: 10.1371/journal.pone.0047488. doi: 10.1371/journal.pone.0047488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441–454. doi: 10.1016/j.devcel.2013.08.020. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 6.Zaidel-Bar R. Cadherin adhesome at a glance. J Cell Sci. 2013;126:373–378. doi: 10.1242/jcs.111559. doi: 10.1242/jcs.111559. [DOI] [PubMed] [Google Scholar]
- 7.Oldenburg J, De Rooij J. Mechanical control of the endothelial barrier. Cell Tissue Res. 2014;355:545–55. doi: 10.1007/s00441-013-1792-6. [DOI] [PubMed] [Google Scholar]
- 8.Huveneers S, de Rooij J. Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci. 2013;126:403–413. doi: 10.1242/jcs.109447. doi: 10.1242/jcs.109447. [DOI] [PubMed] [Google Scholar]
- 9.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, Weström S, Claesson-Welsh L, Vestweber D, Gerhardt H. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16:309–321. doi: 10.1038/ncb2926. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- 10.Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol. 2012;196:641–652. doi: 10.1083/jcb.201108120. doi: 10.1083/jcb.201108120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abu Taha A, Taha M, Seebach J, Schnittler HJ. ARP2/3-mediated junction-associated lamellipodia control VE-cadherin-based cell junction dynamics and maintain monolayer integrity. Mol Biol Cell. 2014;25:245–256. doi: 10.1091/mbc.E13-07-0404. doi: 10.1091/mbc.E13-07-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolfenson H, Lavelin I, Geiger B. Dynamic regulation of the structure and functions of integrin adhesions. Dev Cell. 2013;24:447–458. doi: 10.1016/j.devcel.2013.02.012. doi: 10.1016/j.devcel.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hynes RO. Cell-matrix adhesion in vascular development. J Thromb Haemost. 2007;5(Suppl 1):32–40. doi: 10.1111/j.1538-7836.2007.02569.x. doi: 10.1111/j.1538-7836.2007.02569.x. [DOI] [PubMed] [Google Scholar]
- 14.Malan D, Wenzel D, Schmidt A, Geisen C, Raible A, Bölck B, Fleischmann BK, Bloch W. Endothelial beta1 integrins regulate sprouting and network formation during vascular development. Development. 2010;137:993–1002. doi: 10.1242/dev.045377. doi: 10.1242/dev.045377. [DOI] [PubMed] [Google Scholar]
- 15.Carlson TR, Hu H, Braren R, Kim YH, Wang RA. Cell-autonomous requirement for beta1 integrin in endothelial cell adhesion, migration and survival during angiogenesis in mice. Development. 2008;135:2193–2202. doi: 10.1242/dev.016378. doi: 10.1242/dev.016378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanjore H, Zeisberg EM, Gerami-Naini B, Kalluri R. Beta1 integrin expression on endothelial cells is required for angiogenesis but not for vasculogenesis. Dev Dyn. 2008;237:75–82. doi: 10.1002/dvdy.21385. doi: 10.1002/dvdy.21385. [DOI] [PubMed] [Google Scholar]
- 17.Stenzel D, Franco CA, Estrach S, Mettouchi A, Sauvaget D, Rosewell I, Schertel A, Armer H, Domogatskaya A, Rodin S, Tryggvason K, Collinson L, Sorokin L, Gerhardt H. Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 2011;12:1135–1143. doi: 10.1038/embor.2011.194. doi: 10.1038/embor.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Jin G, Miao H, Li JY, Usami S, Chien S. Integrins regulate VE-cadherin and catenins: dependence of this regulation on Src, but not on Ras. Proc Natl Acad Sci U S A. 2006;103:1774–1779. doi: 10.1073/pnas.0510774103. doi: 10.1073/pnas.0510774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su G, Atakilit A, Li JT, Wu N, Bhattacharya M, Zhu J, Shieh JE, Li E, Chen R, Sun S, Su CP, Sheppard D. Absence of integrin αvβ3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation. Am J Respir Crit Care Med. 2012;185:58–66. doi: 10.1164/rccm.201108-1381OC. doi: 10.1164/rccm.201108-1381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Legate KR, Montañez E, Kudlacek O, Fässler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- 21.Montanez E, Wickström SA, Altstätter J, Chu H, Fässler R. Alpha-parvin controls vascular mural cell recruitment to vessel wall by regulating RhoA/ROCK signalling. EMBO J. 2009;28:3132–3144. doi: 10.1038/emboj.2009.295. doi: 10.1038/emboj.2009.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, Barberis A, Benjamin LE, Mäkinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 24.Seebach J, Donnert G, Kronstein R, Werth S, Wojciak-Stothard B, Falzarano D, Mrowietz C, Hell SW, Schnittler HJ. Regulation of endothelial barrier function during flow-induced conversion to an arterial phenotype. Cardiovasc Res. 2007;75:596–607. doi: 10.1016/j.cardiores.2007.04.017. doi: 10.1016/j.cardiores.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5:1518–1534. doi: 10.1038/nprot.2010.113. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
- 26.Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–1815. doi: 10.1016/S0002-9440(10)63540-7. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 28.van Geemen D, Smeets MW, van Stalborch AM, Woerdeman LA, Daemen MJ, Hordijk PL, Huveneers S. F-actin-anchored focal adhesions distinguish endothelial phenotypes of human arteries and veins. Arterioscler Thromb Vasc Biol. 2014;34:2059–2067. doi: 10.1161/ATVBAHA.114.304180. doi: 10.1161/ATVBAHA.114.304180. [DOI] [PubMed] [Google Scholar]
- 29.Geyer H, Geyer R, Odenthal-Schnittler M, Schnittler HJ. Characterization of human vascular endothelial cadherin glycans. Glycobiology. 1999;9:915–925. doi: 10.1093/glycob/9.9.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernández-Martín L, Marcos-Ramiro B, Bigarella CL, Graupera M, Cain RJ, Reglero-Real N, Jiménez A, Cernuda-Morollón E, Correas I, Cox S, Ridley AJ, Millán J. Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol. 2012;32:e90–102. doi: 10.1161/ATVBAHA.112.252080. doi: 10.1161/ATVBAHA.112.252080. [DOI] [PubMed] [Google Scholar]
- 31.Mitra P, Keese CR, Giaever I. Electric measurements can be used to monitor the attachment and spreading of cells in tissue culture. Biotechniques. 1991;11:504–510. [PubMed] [Google Scholar]
- 32.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 34.Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007;120:137–148. doi: 10.1242/jcs.03314. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
- 35.Raimondi C, Fantin A, Lampropoulou A, Denti L, Chikh A, Ruhrberg C. Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J Exp Med. 2014;211:1167–1183. doi: 10.1084/jem.20132330. doi: 10.1084/jem.20132330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phng LK, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, Ondr JK, Rao S, Lang RA, Thurston G, Gerhardt H. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell. 2009;16:70–82. doi: 10.1016/j.devcel.2008.12.009. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, Frye M, Zeuschner D, Nakayama M, Vestweber D, Adams RH. Integrin β1 controls VE-cadherin localization and blood vessel stability. Nat Commun. 2015;6:6429. doi: 10.1038/ncomms7429. doi: 10.1038/ncomms7429. [DOI] [PubMed] [Google Scholar]
- 38.Breslin JW, Zhang XE, Worthylake RA, Souza-Smith FM. Involvement of local lamellipodia in endothelial barrier function. PLoS One. 2015;10:e0117970. doi: 10.1371/journal.pone.0117970. doi: 10.1371/journal.pone.0117970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeMali KA, Barlow CA, Burridge K. Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J Cell Biol. 2002;159:881–891. doi: 10.1083/jcb.200206043. doi: 10.1083/jcb.200206043. [DOI] [PMC free article] [PubMed] [Google Scholar]