

Abstract
Fluorescence of environment-sensitive dyes is widely applied to monitor local structure and solvation dynamics of biomolecules. It has been shown that, in comparison with a parent indole fluorophore, fluorescence of 2-(2′-pyridyl)-5-methylindole (5M-PyIn-0) and 2-[2′-(4′,6′-dimethylpyrimidyl)]-indole (DMPmIn-0) is remarkably sensitive to hydrogen bonding with protic partners. Strong fluorescence, observed for these compounds in nonpolar and polar aprotic solvents, is efficiently quenched in aqueous solution. This study demonstrates that 5M-PyIn-0 and DMPmIn-0, which are almost nonemitting in aqueous solution, become highly fluorescent upon titrating with phospholipid vesicles. The fluorescence enhancement is accompanied by a significant blue shift of emission maximum. The Gibbs free energy of membrane partitioning, measured by the increase in the steady-state fluorescence intensities during transfer from an aqueous environment to a lipid bilayer, is very favorable for both compounds, being in a range from −7.1 to −8.0 kcal/mol and depending only slightly on lipid composition of the membrane. The fluorescence enhancement upon membrane partitioning is indicative of the loss of the specific hydrogen-bonding interactions between the excited fluorophore and water molecules, causing efficient fluorescence quenching in bulk water. This conclusion is supported by atomistic molecular dynamics (MD) simulations, demonstrating that both 5M-PyIn-0 and DMPmIn-0 bind rapidly and partition deeply into a lipid bilayer. MD simulations also show a rapid, nanosecond-scale decrease in the probability of solute–solvent hydrogen bonding during passive diffusion of the probe molecules from bulk water into a lipid bilayer. At equilibrium conditions, both 5M-PyIn-0 and DMPmIn-0 prefer deep localization within the hydrophobic, water-free region of the bilayer. A free energy profile of penetration across a bilayer estimated using MD umbrella sampling shows that both indole derivatives favor residence in a rather wide potential energy well located 10–15 Å from the bilayer center.
Introduction
The photophysical properties of an excited molecule are known to be very sensitive to the interplay of various interactions of a fluorophore with its environment. In particular, hydrogen bonding with protic solvents may lead to significant changes in the excited state behavior of heteroazaaromatic fluorophores. The formation of solute–solvent hydrogen bonds plays an important role in many fundamental photochemical reactions, such as excited state proton transfer. It has been proposed that biprotonic phototautomerization, occurring in hydrogen-bonded base pairs, could provide a mechanism for the initial step in ultraviolet mutagenic effects in DNA.1 The excited state multiple proton relay along a hydrogen-bonded proton wire is believed to contribute to the emitting mechanism of green fluorescent proteins.2–4 Therefore, knowledge of a mechanism of proton conductivity and reorganization of the hydrogen-bonded networks is important for our understanding of light-driven processes occurring in biological systems.
We have been carrying out detailed studies of fluorescence properties of a series of bifunctional fluorophores that reveal an unusual fluorescence behavior in the presence of protic solvents.5,6 High quantum yield fluorescence, which is characteristic for these compounds in nonpolar and polar aprotic solvents, becomes dramatically quenched in alcohols or aqueous solutions.7 A common feature of the studied bifunctional fluorophores is that they are composed of a hydrogen-bond-donating moiety (pyrrole/indole) and a proton-accepting unit (pyridine/pyrimidine/quinoline) linked by a single C–C bond. It has been shown that in these systems, the solvent-induced fluorescence quenching depends on the structure of a bifunctional chromophore, requiring a certain topology of the proton donor (the NH group) and the acceptor (pyridine-type nitrogen) groups.
We have also demonstrated that in addition to topological criteria, the photophysics of bifunctional hydrogen-bond donor/acceptor indole derivatives is conformationally dependent.8–10 The fluorescence quenching of the lowest excited state of the fluorophore in protic solvents is thought to occur in cyclic, hydrogen-bonded solute–solvent complexes via two competing mechanisms: (i) dual proton transfer and (ii) enhanced internal S0 ← S1 conversion to the ground state. Recent experimental and theoretical studies demonstrate that excited state proton transfer in 7-(2-pyridyl)indole, in which a hydrogen bond between the donor and acceptor units can be formed intramolecularly, is accompanied by torsion twisting of the indole and pyridyl moieties in the excited tautomer form.11 For isomeric structures of 2-, 3-, and 7-(4′-pyridyl)indoles, which are not able to form an intramolecular hydrogen bond, the alternative mechanism of the fluorescence quenching by hydroxylic solvents has been considered.12 It has been suggested that the origin of the fluorescence quenching might involve excited state protonation of the pyridine nitrogen atom, followed by twisting of the pyridyl group, leading to a low-energy structure.
In addition to bifunctional compounds, connected by a single C–C bond, their structural analogues possessing a rigid planar structure, in which donor and acceptor units are joined by an aromatic moiety, reveal an interesting phenomenon of dual emission in the presence of protic partner molecules.13–16 It has been shown that, in such bifunctional compounds, solvent-assisted excited state tautomerization may lead to the formation of a fluorescent tautomer, emitting red-shifted fluorescence.6,14,17 This phenomenon has been first reported by Kasha and co-workers in the dimer of 7-azaindole (7AI),1 which has now become one of the best-known systems in this field. Since then, the remarkable sensitivity of a bifunctional fluorophore to its state of solvation has attracted considerable attention. The chromophoric moiety of 7AI has been applied to the development of a family of nonnatural amino acids, such as 7-azatryptophan, which is considered as a promising alternative to tryptophan as an optical probe of protein structure and dynamics.18 Due to high sensitivity to its microenvironments, bifunctional hydrogen bond donor–acceptor heterocyclic probes are often applied for studying solvation and hydrogen bonding dynamics in micelle–water interfaces,19–21 microemulsions,22 and in water nanopools.23,24 When studying solute–solvent hydrogen bonding in heterogeneous environments, such as micelles and biomembranes, it becomes critical to know the distribution and precise location of a fluorophore.25
One of the powerful supplementary tools for experimental spectroscopic studies of binding, distribution, and transport of small organic compounds and ions in lipid membranes is molecular dynamics (MD) simulations.26–29 MD studies have long been used to explore binding, permeation, and accumulation of biologically relevant drugs30,31 and volatile anesthetics, such as lidocaine,32 halothane,33,34 adamantanes,35,36 and furocoumarins37 in membranes. A series of constraint MD simulations have been carried out to evaluate the preferred location and free energy profiles of small solutes in lipid bilayers.38 Rather limited attention has been devoted to the study of a partitioning behavior of aromatic and heteroaromatic fluorescent molecules. A solidstate 2H NMR study combined with all-atom MD simulations has been applied to determine the preferred alignment, position, and mobility of pyrene in POPC39 and DPPC40,41 bilayers. The well-known fluorescent probe 1,6-diphenyl-1,3,5-hexatriene (DPH), which is frequently employed for studying rotational and lateral diffusion processes in lipid membranes, has been explored using both spectroscopy tools and MD simulations.41–43 In addition, MD simulations in combination with differential scanning calorimetry and time-resolved fluorescence anisotropy studies has recently been applied for studying the structural and dynamics properties of 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) acyl tail-labeled phosphatidylcholine (NBD-PC) in a DPPC bilayer.44,45 Coarse-grained MD simulations of the interaction of styryl-type voltage-sensitive dyes in biomembranes have been reported.46 The role of lipid tail anchoring of large fluorescent chromophores, such as Texas-Red and rhodamine, and their interactions with the bilayer has also been addressed.47,48 Alignment, localization, and partition free energies of indole in a POPC bilayer have been explored by both constrained and unconstrained MD samplings.49,50
The purpose of the present work is to study membrane binding and a partitioning behavior of two fluorescent, environment-sensitive derivatives of 2-substituted-indole; namely, 5M-PyIn-0 and DMPmIn-0, shown in Scheme 1A. The fluorescence titration of aqueous solutions of these compounds with lipid vesicles allows us to estimate the macroscopic measurable quantities such as spectral shifts in emission and the Gibbs free energy of partitioning from aqueous solution into a lipid membrane. The fluorescence studies were complemented by atomistic MD simulations of binding, distribution and preferred localization of the studied compounds in a POPC bilayer so that the solute–membrane interactions were addressed at the molecular level. We simulated how multiple molecules of 5M-PyIn-0 and DMPmIn-0, randomly placed in bulk aqueous solution, could spontaneously bind and insert into a bilayer. Recently, we applied similar MD methodology for studying the equilibrium distribution of structurally similar fluorescent azaaromatic probes, such as 7-azaindole, pyrroloquinoline, and its analogues in a DPPC bilayer.51 We have shown that variation in structure of the azaaromatics with an increase in the number of aromatic rings allowed us to tune the distribution and favorable probe localization across the water/bilayer interface. In this study, we applied a joint refinement of unconstrained MD sampling of passive partitioning with umbrella sampling and potential of mean constrained force calculations of favorable localization and alignment of the two indole derivatives in a lipid bilayer. The calculated free energy of partitioning is directly compared with results from experimental fluorescence titration data.
SCHEME 1.

Structures of 2-(2′-Pyridyl)-5-methyl-indole (5M-PyIn-0) and 2-[2′-(4′,6′-Dimethylpyrimidyl)]-indole (DMPmIn-0) (A); Parent Compound 2′-Pyridylindole (PyIn-0) and Its Hydrogen-Bonded Complexes with Protic Solvent Molecules: (B) Structure of “Cyclic” and “Noncyclic” Hydrogen-Bonded Complexes of PyIn-0 with Solvent; (C) Hydrogen-Bonded Induced syn–anti Rotamerization in PyIn-0
Materials and Methods
Materials
Palmitoyloleoylphosphatidylcholine (POPC) and palmitoyloleoylphosphatidylglycerol (POPG) were obtained from Avanti Polar Lipids (Alabaster, AL). Large unilamellar vesicles (LUV) of 0.1 μm diameter were prepared by extrusion52,53 using the following molar mixtures of POPC and POPG: 1:3 (25POPC/75POPG) and 3:1 (75POPC/25POPG). Lipid concentrations of stock solutions were determined according to the procedure of Bartlett.54 The synthesis and purification of 2-(2′-pyridyl)-5-methylindole and 2-[2′-(4′,6′-dimethylpyrimidyl)]-indole have been described elsewhere.7,55,56
Steady-State and Time-Resolved Fluorescence Measurements
Fluorescence was measured using a SPEX Fluorolog FL3-22 steady-state fluorescence spectrometer (Jobin Yvon, Edison, NJ) equipped with double-grating excitation and emission monochromators. The measurements were made in a 2 × 10 mm cuvette oriented perpendicular to the excitation beam and maintained at 25 °C using a Peltier device from Quantum Northwest (Spokane, WA). Fluorescence decays were measured with a time-resolved fluorescence spectrometer, FluoTime 200 (PicoQuant, Berlin, Germany), using a standard time-correlated single-photon counting scheme. Samples were excited at 310 nm by a subnanosecond pulsed LED laser diode PLS 310 (PicoQuant, Berlin, Germany) with a repetition rate of 10 MHz. Fluorescence emission was detected at 385 nm, selected by a Sciencetech model 9030 monochromator, using a PMA-182 photomultiplier (PicoQuant, Berlin, Germany).
Molecular Dynamics Simulation Setup
A lipid vesicle was modeled using a phospholipid bilayer consisting of 128 lipid molecules of POPC (64 per leaflet) surrounded by 4553 water molecules. A bilayer was simulated at the fully hydrated state with a lipid-to-water ratio equal of 1:35. In the case of POPC molecules, all carbon atoms of CH2 and CH3 groups with nonpolar hydrogen atoms were treated as united atoms. A MD force field of a POPC bilayer was adopted from ref 57. The force filed for 5M-PyIn-0 and DMPmIn-0 was developed, and its reliability was tested as described previously.7,13,48,58 The bond length and angle parameters for 5M-PyIn-0 and DMPmIn-0 were optimized by density functional theory calculations at the B3LYP/cc-pVDZ level and adopted for the GROMACS force field. Rotation around the C–C bond connecting the indole and pyridine/pyrimidine rings was modeled using the periodic Ryckaert–Bellemans dihedral potential.59 Partial charges needed for Coulomb interactions were derived from the B3LYP/ccpVDZ electron densities by fitting the electrostatic potential to point (ESP) charges. The simple point charge (SPC) model60 was used for water.
All MD simulations were carried out at a constant number of particles; constant pressure, P = 1 atm; and constant temperature, T = 323 K (NPT ensemble). Three-dimensional periodic boundary conditions were applied with the z axis lying along a direction normal to the bilayer. The pressure was controlled semi-isotropically, so that the x–y and z dimensions of the simulation box were allowed to fluctuate independently from each other, keeping the total pressure constant. Thus, during MD simulations, the membrane area and thickness were free to adjust under the NPT condition. The reference temperature and pressure were kept constant using the Berendsen weak coupling scheme61 with a coupling constant of τT = 0.1 ps for the temperature coupling and τP(x–y) = τP(z) = 1.0 ps for the pressure coupling.
Electrostatic interactions were simulated with the particle mesh Ewald (PME)62 approach using the long-range cutoff of 1.4 nm. The cutoff distance of Lennard-Jones interactions was also equal to 1.4 nm. All bond lengths in POPC, 5M-PyIn-0, and DMPmIn-0 were kept constant using the LINCS routine.63 The MD integration time step was 2 fs. The unconstraind MD simulations were carried out using the GROMACS set of programs, version 3.3.3.64 Umbrella sampling was used to calculate the free energy profile for partitioning in a bilayer using GROMACS, version 4.0.5.65 A harmonic restraint of 1500 kJ mol−1 nm−2 was applied to the distance between the center of mass of a probe and the center of mass of a POPC bilayer. A total of 25 restraint points were sampled in the direction normal to the bilayer. Each simulation was run for 2 ns. The potential of mean force was calculated from the biased distributions using the weighted histogram analysis method.66 Molecular graphics and visualization were performed using gOpenMol 3.067 and VMD 1.8.668 software packages.
Results and Discussion
Photophysical Properties in Organic Solvents and in Water
The electronic absorption spectra and the photophysical properties of 5M-PyIn-0 and DMPmIn-0 in organic solvents have been described elsewhere.7,8 The photophysical properties have also been reported for PyIn-0 (see Scheme 1B),5,7 the parent compound of 5M-PyIn-0, in which the 5-methyl group is missing. Here, we briefly summarize these properties and study the fluorescence behavior of 5M-PyIn-0 and DMPmIn-0 in aqueous solution.
The first absorption band of the electronic spectra of the two indoles is characterized by a distinctive vibronic structure in a nonpolar solvent, n-hexane. A loss of the vibronic structure, accompanied by a slight blue shift, was observed upon passing to polar protic solvents, such as acetonitrile.7 In alcohol solutions, a red spectral shift was found, indicating the formation of an intermolecular hydrogen bond.5,7,8 Similar to electronic absorption, fluorescence spectra of PyIn-0, 5M-PyIn-0, and DMPmIn-0 revealed a loss of the characteristic vibronic structure upon passing from nonpolar n-hexane to aprotic, polar solvents, such as acetonitrile. The largest differences in the photophysical behavior, however, were observed upon passing to alcohol solutions. In all the studied compounds, the emission in alcohols was weaker than those in aprotic solvents, both polar and nonpolar. The relevant fluorescence properties are summarized in Table 1.
TABLE 1.
Photophysical Parameters of 5M-PyIn-0 and DMPmIn-0 in Organic Solvents and in 50 mM Sodium Phosphate Buffer at pH 8 in the Absence and in the Presence of 4 mM POPC LUV
The quenching of fluorescence in alcohols was very strong for DMPmIn-0, so that upon passing from acetonitrile to 1-propanol, the fluorescence quantum yield (φfl) was decreased by a factor 35, changing from 0.7 to 0.002, (Table 1). 5M-PyIn-0 revealed a smaller quantum yield decrease by a factor of ~4.5.8 We have shown that, in a protic environment, bifunctional compounds are able to form various kinds of hydrogen-bonded complexes with protic solvent molecules.6–9,58 Among these species, a certain population of hydrogen-bonded complexes exists, in which both donor and acceptor sites become cyclically, doubly hydrogen-bonded by a protic solvent molecule (see Scheme 1B). We have also demonstrated that the formation of such a cyclic, properly solvated, solute–solvent complex plays a critical role in controlling the excited state behavior of a bifunctional chromophore.6–17,69–71
In contrast to the case of electronic absorption, no similarity was observed in the pattern of emission spectra of 5M-PyIn-0 and DMPmIn-0 in organic solvents. A considerable red shift, observed in the former upon changing solvents from polar aprotic acetonitrile to polar protic 1-propanol, was not reproduced in the latter (Table 1). This effect has been explained by conformational changes leading to syn-anti rotamerization in PyIn-0 and in 5M-PyIn-0, which was believed to be due to specific interactions with alcohol molecules, as shown in Scheme 1C.7 The solvent-induced rotamerization allows us to explain the difference in the photophysics and φfl of 5M-PyIn-0 and DMPmIn-0 in alcohols. The former can exist in both syn and anti rotamers, among which, only the syn rotamer is efficiently quenched by the specific interactions with protic solvents. In contrast, in the latter, the syn and anti forms are identical, resulting in the strong fluorescence quenching of both rotamers in alcohol solutions. We have shown that the relative populations of the syn/anti forms in PyIn-0 can be varied by solvent polarity and hydrogen bonding to solvent molecules.7,8 The solvent-induced syn-anti rotamerization, which was originally found in the parent chromophore PyIn-0,7 has also been described for other structurally similar pyridyl-indole derivatives in alcohols.8,9
Upon passing from alcohols to aqueous solution, both 5M-PyIn-0 and DMPmIn-0 showed a further decrease in fluorescence quantum yield and a red shift of emission maxima (Table 1). In contrast to the photophysics in alcohols, no significant difference in fluorescence spectra of 5M-PyIn-0 and DMPmIn-0 was observed in bulk water. The compounds are characterized by very similar, low-intensity fluorescence with a maximum around 420–425 nm.
Fluorescence Titration with Lipid Vesicles
The two probe molecules are almost nonfluorescent in buffer solution at pH 8 (Table 1). To study the affinity of 5M-PyIn-0 and DMPmIn-0 for membranes, we did titration of their buffer solutions containing a fixed probe content with lipid vesicles. Three molar percentages of the lipid composition of LUV were studied: pure POPC and 75:25 and 25:75 molar ratios of POPC/POPG. The studied indole derivatives exhibited a remarkable enhancement of fluorescence emission upon titrating with lipid vesicles (Figure 1). At lipid saturation conditions, the increase in the fluorescence intensity was observed to be in a range from ~60-to 90-fold, depending on the probe and the composition of LUV. Examples of the fluorescence spectra of 5M-PyIn-0 (part A) and DMPmIn-0 (part B) recorded upon titrating with LUV composed of 25POPC/75POPG are shown in Figure 1.
Figure 1.

Examples of changes in the fluorescence spectra of 5M-PyIn-0 (A) and DMPmIn-0 (B) occurring upon titration with lipid vesicles. The enhancement of fluorescence is observed upon partitioning of the probe molecules in a lipid membrane. This enhancement is also accompanied by a blue shift of the emission maximum. The examples are shown for LUV composed of 25POPC/75POPG (see text). Concentration of the probes was 5–7 × 10−6 M, whereas the LUV concentration in the solution was varied from C = 0 to 2 mM. All the fluorescence spectra were measured in 50 mM sodium phosphate buffer at pH 8 with excitation at 313 nm. The spectra were corrected by subtracting scattering and background fluorescence.
Upon the membrane interaction, the fluorescence of 5M-PyIn-0 and DMPmIn-0 was enhanced, and this effect was also accompanied by a blue shift of the emission peaks (Figure 1A, B). In Figure 2, the normalized fluorescence spectra of 5M-PyIn-0 are shown in the absence and in the presence of different concentrations of LUV(25POPC/75POPG). It can be seen that the emission peaks were shifted from 418 to 382 nm upon saturating the buffer solution with LUV. The enhancement and the blue shift of the fluorescence spectra of 5M-PyIn-0 and DMPmIn-0 upon interaction with LUV may be explained by transfer of the probe molecules from polar aqueous solution to a hydrophobic environment of a lipid membrane. The strong fluorescence in LUV may be attributed to the loss of cyclic hydrogen bonding between the fluorophore and water molecules; this specific hydrogen bonding is anticipated to be a reason for the efficient fluorescence quenching in bulk water (Scheme 1B, C). In addition, the fluorescence enhancement may also be indicative of restriction of the internal mobility of 5M-PyIn-0 and DMPmIn-0 fluorophores, which is expected upon deep partitioning in the confined acyl tails region of the membrane.
Figure 2.

A blue shift of the maxima of the fluorescence spectra of 5M-PyIn-0, observed upon titration with lipid vesicles, indicates partitioning of probe molecules from the aqueous solution into a hydrophobic membrane environment. The fluorescence spectra were normalized for the same intensity (see Figure 1 for more details). The spectra were recorded in 50 mM sodium phosphate buffer at pH 8 for different LUV concentrations: (1) no LUV and (2) 0.01, (3) 0.1, (4) 0.5, and (5) 2.5 mM, respectively.
Our earlier observations have provided no clear evidence that the syn-anti rotamerization exists in PyIn-0 in bulk water. However, this rotamerization was still observed in acetonitrile solution of PyIn-0 to which small amounts of water were gradually added.7 To elucidate the origin of the observed fluorescence quenching in the studied indole derivatives and the presence of the anti form in 5M-PyIn-0 in bulk aqueous solution, we performed a series of time-resolved fluorescence experiments using a time-correlated single-photon counting setup. The measurements of fluorescence lifetimes also provide the most definitive method to distinguish between static and dynamics mechanisms of the fluorescence quenching, which might occur in 5M-PyIn-0 and DMPmIn-0 in bulk water.
Figure 3 shows fluorescence decays measured for both indole derivatives in sodium phosphate buffer at pH 8 in the absence and in the presence of POPC vesicles. The most interesting observation is that a fluorescence decay of 5M-PyIn-0 had no monoexponential character in solution in the absence of lipid vesicles (Figure 3A). The present results show that the fluorescence decay can be well fitted using two decay components.
Figure 3.

Fluorescence decays of 5M-PyIn-0 and DMPmIn-0 in sodium phosphate buffer at pH 8 were measured in the absence and in the presence of POPC vesicles. (A) 5M-PyIn-0, vesicle concentration and double exponential fitting parameters are given. The longer-lived component of curves 2–6 was fixed to 3.1 ns during the deconvolution fitting; 1: no LUV, A1 = 0.94, τ1 < 0.1 ns, A2 = 0.06, τ2 = 3.1 ns. 2: 0.01 mM LUV, A1 = 0.83, τ1 = 0.77 ns, A2 = 0.17, τ2 = 3.1 ns. 3: 0.03 mM LUV, A1 = 0.80, τ1 = 0.78 ns, A2 = 0.20, τ2 = 3.1 ns. 4: 0.07 mM LUV, A1 = 0.80, τ1 = 0.82 ns, A2 = 0.20, τ2 = 3.1 ns. 5: 0.14 mM LUV, A1 = 0.78, τ1 = 0.84 ns, A2 = 0.22, τ2 = 3.1 ns. 6: 1.3 mM LUV, A1 = 0.76, τ1 = 0.88 ns, A2 = 0.24, τ2 = 3.1 ns. (B) DMPmIn-0; fluorescence decays 2–6 were well fitted using a single lifetime of 1.25 ns. 1: no LUV, τ<0.1 ns. 2: 0.015 mM. 3: 0.03 mM. 4: 0.05 mM. 5: 0.075 mM. 6: 1.23 mM.
We have demonstrated that in the case of PyIn-0 in hydrogen-bonding solvents such as alcohols, the two lifetime components correspond to the presence of two, syn and anti, rotameric forms, as shown in Scheme 1C.7,8 The syn species are able to form cyclic complexes with solvent molecules, which become rapidly deactivated via enhanced S0 ← S1 internal conversion. The longer-lived decay should therefore be assigned to the anti rotamer, not capable of cyclic hydrogen bonding with solvent molecules. The fitting of the fluorescence decays of 5M-PyIn-0 using two components shows that the first one is the short-lived with a lifetime shorter than 100 ps, whereas the second one is found to be 3.1 ns (see the caption for Figure 3A for more details).
It is interesting that the fluorescence lifetime of the anti form of PyIn-0 in 1-propanol was also found to be about 3.2 ns.7 The second, long-lived component, assigned to the anti rotameric form of 5M-PyIn-0, was also observed in aqueous solution in the presence of lipid vesicles. This component of 3.1 ns was therefore fixed during the biexponential fitting analysis of curves 2–6, shown in Figure 3A. The first component, shorter than 100 ps in phosphate buffer, became ~800 ps upon partitioning into POPC vesicles, whereas the second, long-lived lifetime was still found to be 3.1 ns. The present results corroborate the above model for the simultaneous presence of the syn and anti rotamers of 5M-PyIn-0. Both of these rotamers are thought to be buried deeply into the water-free region of a lipid bilayer, and they therefore exist in a hydrogen-bond-free form.
In contrast to 5M-PyIn-0, DMPmIn-0 showed no long-lived fluorescence in phosphate buffer at pH 8 (Figure 3B). In the absence of LUV, the fluorescence decay of DMPmIn-0 could not be resolved accurately with time resolution of our lifetime spectrometer (<50 ps). In DMPmIn-0 both the syn and anti forms are identical, and both forms are therefore capable of efficient quenching due to cyclic hydrogen bonding with water molecules. As can be seen from decays 2–6 in Figure 3B, the long-lived monoexpenential fluorescence appears upon partitioning of DMPmIn-0 into POPC vesicles. The fluorescence decays of DMPmIn-0 in the presence of different concentrations of LUV could be well fitted to the single component of 1.25 ns. This component can be assigned to the hydrogen-bond-free fraction of DMPmIn-0 deeply partitioned in the POPC bilayer. Such fluorescence behavior can be explained on the basis of the static mechanism of fluorescence quenching of DMPmIn-0 by water molecules. This result is in accordance with our general observation that cyclic hydrogen-bonded complexes between bifunctional indole derivatives and solvent molecules are already formed in the ground electronic state, S0.6–9
The Gibbs Free Energy of Partitioning in a Lipid Bilayer
In Figure 4, the relative fluorescence enhancement observed upon titration with LUV, composed of the three different molar percentages of the POPC and POPG lipids, is shown for both 5M-PyIn-0 and DMPmIn-0. In addition, the effect of the presence of cholesterol in POPC vesicles on the probe partition was also considered. It can be noted that for the same vesicle composition, DMPmIn-0 showed the larger-fold increase in the fluorescence emission compared with that of 5M-PyIn-0. In the case of LUV(25POPC/75POPG), the membrane partitioning results in the almost identical 70-fold increase in fluorescence for both 5M-PyIn-0 and DMPmIn-0 (Figure 4A, B). The largest difference was observed for the partition of 5M-PyIn-0 and DMPmIn-0 in pure POPC vesicles. DMPmIn-0 was characterized by an ~100-fold increase in fluorescence, whereas in the case of 5M-PyIn-0, only a 70-fold increase was observed (Figure 4A, B). This finding also corroborates our model of the presence of a certain population of the anti form of 5M-PyIn-0 in solution, the fluorescence of which is not quenched by specific hydrogen bonding with bulk water molecules.
Figure 4.

Relative fluorescence intensities of 5M-PyIn-0 (A) and DMPmIn-0 (B) in the 50 mM sodium phosphate buffer at pH 8 are plotted as a function of the concentration of lipid vesicles. Relative intensities (Ii/I0, Ii is the fluorescence intensity in the presence of LUV, and I0 is the intensity in the buffer solution with no LUV) were determined at fixed wavelengths of 389 and 379 nm for 5M-PyIn-0 and DMPmIn-0, respectively (see Figure 1 for more details). The fluorescence intensities were corrected by subtracting the scattering and background fluorescence. The lipid compositions of the vesicles POPC, 70POPC/30Chol, 75POPC/25POPG, and 25POPC/75POPG are shown by green circles (○), olive top triangles (△), red squares (□), and blue bottom triangles (▽), respectively. Titration profiles were fitted to eq 1 to determine the partition coefficient Kp.
The apparent partition coefficient Kp was determined by fitting fluorescence intensities, Ii, measured at the fixed wavelengths, to eq 1,
| (1) |
where I0 is the initial fluorescence signal in water in the absence of LUV, which was normalized to unity, Imax is the maximal fluorescence increase on the complete partition, [L] is the molar concentration of lipid, [W] is the molar concentration of water (55.3 M), and Kp is the mole fraction partition coefficient.72 The Gibbs free energy, ΔG, of transfer from water to a lipid membrane was calculated from the mole fraction partition coefficients, Kp, using eq 2:
| (2) |
Kp and ΔG of partitioning of 5M-PyIn-0 and DMPmIn-0 in the lipid vesicles are summarized in Table 2.
TABLE 2.
Partition Coefficient, Kp, and the Gibbs Free Energy ΔG of Transfer of 5M-PyIn-0 and DMPmIn-0 from Buffer Solution to a Lipid Vesiclea
| LUV | 5M-PyIn-0
|
DMPmIn-0
|
||
|---|---|---|---|---|
| Kp × 10−4 | ΔG, kcal/mol | Kp × 10−4 | ΔG, kcal/mol | |
| POPC | 43.4 ± 1.5 | −7.7 ± 0.1 | 32.4 ± 0.8 | −7.5 ± 0.1 |
| POPC (at 323K) | 60.7 ± 0.9 | −7.9 ± 0.1 | 22.9 ± 0.9 | −7.3 ± 0.1 |
| 70POPC/30Chol | 77.1 ± 1.5 | −8.0 ± 0.1 | 27.6 ± 1.1 | −7.4 ± 0.1 |
| 75POPC/25POPG | 43.5 ± 2.1 | −7.7 ± 0.1 | 21.7 ± 0.9 | −7.3 ± 0.1 |
| 25POPC/75POPG | 25.4 ± 0.8 | −7.4 ± 0.1 | 16.2 ± 0.3 | −7.1 ± 0.1 |
Measured in 50 mM sodium phosphate buffer at pH 8 at T = 298 K; for POPC vesicles, Kp and ΔG were also determined at T = 323 K.
The Gibbs free energy of partitioning is found to be of similar magnitude for both 5M-PyIn-0 and DMPmIn-0 derivatives. ΔG depends only slightly on lipid compositions of the membranes, and it varies in a range from −7.1 to −8.0 kcal/mol. The partition of the two indoles to LUV composed of POPC lipids also has weak temperature dependences, as shown for Kp and ΔG obtained at T = 323 K (Table 2). For both indole derivatives, the highest values of ΔG are observed for electro-neutral POPC vesicles (Table 2). The presence of cholesterol and the increase in content of polar anionic POPG lipids results in a small decrease in ΔG. These results indicate that the partition of the indole derivatives is driven mainly by the hydrophobic effect so that a membrane dipole potential plays a smaller role in the partition thermodynamics. Most of the fluorescence titrations of 5M-PyIn-0 and DMPmIn-0 with LUV were performed in the phosphate buffer at pH 8. We also verified the membrane interaction of the two indoles with LUV at pH 5. The fluorescence titration data at pH 5 (not shown) looked very similar to those measured at pH 8.
MD Simulations of Partitioning in a POPC Bilayer
To study the distribution and favorable localization of 5M-PyIn-0 and DMPmIn-0 in a lipid membrane, we first applied unconstrained atomistic MD simulations based on passive distribution of probe molecules between bulk water and a POPC bilayer. We placed eight molecules of each probe (four molecules per each bilayer leaflet) at random locations and orientations in bulk solution at the vicinity of 5–8 Å from a bilayer surface. To remove high energy contacts, the system was equilibrated at NPT conditions for 2 ns. After the initial equilibration period, interactions between probe molecules and a bilayer were studied using a series of unconstrained MD simulations with a total simulation time of 50 ns. The MD sampling started from self-diffusion of the probe molecules from bulk solution toward the bilayer interface, which was followed by probe binding, partitioning, and localization in the bilayer. The initial configuration of POPC/5M-PyIn-0 system at t = 0 ns is shown in Figure 5 (left). It is known that unconstrained MD simulations of passive diffusion and partitioning of probes in a bilayer are generally a difficult task due to convergence problems and limited sampling times. Despite this, it has also been shown that passive thermally driven distributions of small probes between bulk solution and a bilayer usually converge at a time scale of ~50 ns.39,50,51,73
Figure 5.
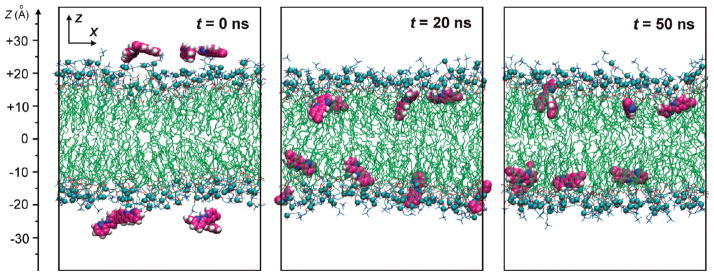
MD simulations of partitioning dynamics of 5M-PyIn-0 in a POPC bilayer are shown for different simulation periods. Snapshots of the MD system consisting of a hydrated POPC bilayer and eight molecules of 5M-PyIn-0, with four molecules on the top and bottom leaflet of the bilayer, are taken after t = 0, 20, and 50 ns of MD sampling. The molecules of 5M-PyIn-0 are drawn in van der Waals representation. Phosphorus atoms of POPC molecules are shown by cyan balls. For clarity, water molecules are not shown.
Figure 5 shows snapshots of the partitioning dynamics of 5M-PyIn-0 in a POPC bilayer at different sampling times. It is seen that all eight probe molecules already become bound to the interfacial region of the POPC bilayer after the first 20 ns of the MD sampling (Figure 5, center). The bound probe molecules orient themselves by a long molecular axis lying roughly parallel to the bilayer surface. After an additional 30 ns of MD simulations, the probe molecules become buried deeper into the hydrophobic region of the bilayer, as shown in Figure 5 (right).
To ensure that the MD sampling converges to an equilibrium distribution of the probes in a bilayer, we also traced the partitioning kinetics of the probes during the whole sampling period. Movement of center-of-mass (COM) of each of eight probe molecules along the bilayer normal z was monitored as a function of simulation time, t. The MD trajectories of the COM distances of 5M-Pyin-0 and DMPmIn-0 calculated from the center of a POPC bilayer are presented in Figure 6.
Figure 6.

Kinetics of partitioning of 5M-PyIn-0 (A) and DMPmIn-0 (B) in a POPC bilayer was monitored by COM of the probe molecules. The COM distances were calculated from the bilayer center along the z-axis for each of the eight probe molecules as a function of MD simulation time. For clarity, the COM distances for the four molecules on the top and bottom leaflet of the bilayer are black, green, red, and yellow and schematically superimposed on a bilayer snapshot.
As can be seen, both 5M-PyIn-0 and DMPmIn-0 become bound to the bilayer on a time scale of a few nanoseconds. Some of the eight probe molecules spontaneously bind to the bilayer after 1–2 ns, whereas the others did so after 12–15 ns. After 20 ns of unconstrained MD sampling, all the probe molecules were located in the bilayer interface so that no appreciable amount of the population was found in bulk water outside the bilayer. After the rapid binding period, there was a slow equilibration and partitioning of the probes deeply inside the bilayer. The large-scale fluctuations of the COM distances, which are observed at t = 20–50 ns, indicate that the probe molecules were still quite mobile in the bilayer environment. At the end of the sampling period, the COM trajectories of all the probes converged to the same bilayer region located at distances of 12–13 Å from the bilayer center. It can also be noted that during the whole 50-ns simulation period, there were no events for which the probe crosses from the interfacial region of one leaflet to the interfacial region of another leaflet.
Probe Partitioning Kinetics Monitored by Hydrogen Bonding with Water
The presence of hydrogen bonds between the probe and water molecules was established using two geometry criteria: (1) the distance between the donor and acceptor is ri ≤ rHB = 3.5 Å; (2) the angle made by the donor, the hydrogen, and the acceptor atom is αi ≤ αHB = 0° ± 30°. Four kinds of a hydrogen bond formed between the indole derivatives and either water or lipid molecules were considered (Table 3). To investigate the role of hydrogen bonding in the fluorescence behavior of the partitioned probe, we first considered the formation of hydrogen bonds between a probe and water molecules. Each of the indole derivatives is able to form two kinds of a hydrogen bond with water molecules. In the first case, the indole NH group was chosen as a hydrogen bond donor, and an oxygen atom of water molecules was selected as a hydrogen bond acceptor. In the second case, either the pyridine or pyrimidine-type nitrogen atom was chosen as a hydrogen bond acceptor, whereas the OH group of water molecules acted as a hydrogen bond donor. The hydrogen bonds were analyzed for all possible syn and anti rotamerical forms of 5M-PyIn-0 and DMPmIn-0, whereas for illustration purposes, the probe water hydrogen bonds for their anti forms are shown schematically in Figure 7.
TABLE 3.
Parameters of Hydrogen Bonds Formed between 5M-PyIn-0, DMPmIn-0 and Either Water or POPC Molecules of a Bilayer Estimated at Different MD Sampling Times
| hydrogen bond type | time (ns) | 5M-PyIn-0
|
DMPmIn-0
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Fa (%) | rHBd (Å) | fwhmc (Å) | αHBd (deg) | Fa (%) | rHBd (Å) | fwhmc (Å) | αHBd (deg) | ||
| NH· · ·O (water) | 0–1 ns | 89.6 | 2.88 | 0.50 | 11.5 | 90.2 | 2.78 | 0.30 | 9.5 |
| 20–22 ns | 5.5 | 2.98 | 0.45 | 19.5 | 13.3 | 2.93 | 0.35 | 17.5 | |
| 45–50 ns | 3.1 | 3.21 | 0.44 | 21.5 | 1.1 | 3.18 | 0.51 | 26.5 | |
| N· · ·H–O (water) | 0–1 ns | 68.5 | 3.00 | 0.50 | 12.5 | 86.3 | 2.97 | 0.37 | 9.8 |
| 20–22 ns | 7.6 | 3.12 | 0.52 | 14.6 | 7.5 | 3.00 | 0.40 | 13.5 | |
| 45–50 ns | 5.0 | 3.15 | 0.51 | 16.5 | 2.9 | 2.98 | 0.44 | 14.5 | |
| NH· · ·O=P=O (phosphate) | 20–22 ns | 25.5 | 2.58 | 0.46 | 7.5 | 23.4 | 2.48 | 0.37 | 8.3 |
| 45–50 ns | 12.1 | 2.63 | 0.44 | 6.8 | 18.3 | 2.46 | 0.38 | 6.7 | |
| NH· · ·O=C (carbonyl) | 20–22 ns | 23.8 | 2.64 | 0.43 | 7.5 | 35.6 | 2.58 | 0.40 | 7.4 |
| 45–50 ns | 11.8 | 2.67 | 0.41 | 6.5 | 37.2 | 2.56 | 0.42 | 6.8 | |
The relative frequency of the occurrence of hydrogen bonds.
Average H-bond donor–acceptor distance.
Full width at half-maximum of the distribution of the H-bond donor–acceptor distances.
Average H-bond donor–acceptor angle.
Figure 7.
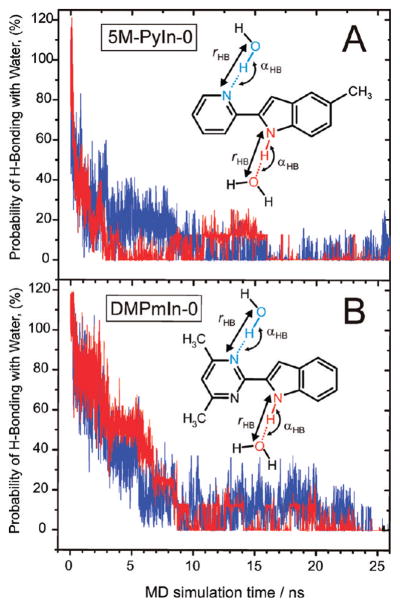
Time evolution of hydrogen-bonding interactions between either 5M-PyIn-0 (A) or DMPmIn-0 (B) and water molecules is shown as a function of MD simulation time. Probability of the formation of a hydrogen bond is calculated and averaged for all the eight probe molecules (see Figure 5 for more details). A rapid decrease in the probability of hydrogen bonding between probe and water molecules indicates rapid transfer of the probe molecules from bulk water to a hydrophobic, water-free region of a POPC bilayer. Two kinds of hydrogen bonds, which are thought to contribute to the fluorescence quenching of the probe in water, were monitored during MD sampling: (i) a H-bond between NH of the indole moiety and water oxygen atoms (red) and between the nitrogen atom of pyridine/pyrimidine rings and water oxygens (OH) (blue).
The probability (PHB) for the formation of a hydrogen bond between a probe and water molecules was estimated as the average number of hydrogen bonds in each 100 ps time frame over the whole period of the MD sampling. In Figure 7, the time evolution of PHB is shown for the first 25 ns of the MD sampling. At t = 0 ns, all molecules of 5M-PyIn-0 and DMPmIn-0 exist in a form of hydrogen-bonded complexes with water molecules. PHB also indicates the formation of bifurcated hydrogen bonds, for which PHB is formally higher than 100% (Figure 7). Upon partitioning of the probe molecules into the bilayer, PHB shows a rapid decrease from almost complete hydrogen bonding (100%) at t = 0 ns to values below 10% at t = 25 ns. In addition, as can be seen from Table 3, the hydrogen bonding of the probe molecules with phosphate and carbonyl oxygen atoms of POPC molecules plays a minor role in overall intermolecular interactions with the lipid bilayer. Upon the passive partitioning from the bulk water to the bilayer, the hydrogen bonding with the phosphate and carbonyl groups reaches the maximum values at t = 20–22 ns, after which the frequency of occurrence of this type of hydrogen bond is found to be in the range of 11–12%.
The results of the above hydrogen bonding behavior are in good agreement with the results of the partitioning dynamics monitored with the COM distances, as shown in Figure 6A, B. The partition of the probe molecules deep in the hydrophobic, water-free region of the POPC bilayer results in a significant decrease in PHB of probe water hydrogen bonding. The small values of PHB for the partitioned forms of the probes are consistent with the loss of the fluorescence quenching and the strong enhancement of the emission of the studied compounds that are observed experimentally upon titration with lipid vesicles (Figures 1 and 3). It is evident that the decrease in overall PHB of probe water hydrogen bonding results in the decrease of populations of both cyclic and noncyclically hydrogen-bonded species of 5M-PyIn-0 and DMPmIn-0 in the bilayer (Scheme 1B C), leading therefore to overall fluorescence enhancement.
Favorable Localization of a Fluorophore in a Bilayer
We explored the equilibrium distribution of 5M-PyIn-0 and DMPmIn-0 across the POPC bilayer by averaging their mass densities. The density distribution was averaged for all atoms of the probe molecules over the last 40 ns of the MD sampling trajectories. To estimate the static structure of the bilayer, we also averaged mass densities of the whole bilayer and certain functional groups of its lipid molecules, such as carbonyl, choline, phosphate groups, and acyl chains. The position of selected groups of atoms was averaged with respect to the bilayer normal (z) for the last 40 ns of MD sampling. For symmetry reasons, all the mass density profiles were also averaged over the two bilayer leaflets. The distribution of the mass densities of 5M-PyIn-0 and DMPmIn-0 in the POPC bilayer are shown in Figure 8.
Figure 8.

Mass density distributions of 5M-PyIn-0 (A) and DMPmIn-0 (B) across a POPC bilayer. The density distribution of water (red solid line) and a whole POPC bilayer (black solid) and its individual components are shown, as are (1, green solid) alkyl chains, (2, cyan dash) carbonyls, (3, blue dot) choline moieties, (4, orange dash-dot) phosphate groups. To improve visualization, the mass density of 5M-PyIn-0 and DMPmIn-0 is scaled by a factor of 10. All the distribution profiles are averaged for the last 40 ns of the MD sampling period. The distributions are plotted with respect to the center of the z axis of the MD simulation box (see Figure 5 for more details).
As can be seen, both probes are distributed in the region of the POPC bilayer corresponding to its hydrocarbon core. The peak distribution of 5M-PyIn-0 is observed at 12.2 Å from the bilayer center. This region of the bilayer is located below the carbonyl groups of the lipid molecules. The major population of DMPmIn-0 is localized at 13.1 Å from the bilayer center. Figure 8 also shows that there is a minor population of DMPmIn-0 molecules distributed throughout the interfacial region of the bilayer.
The mass densities shown in Figure 8 correspond to the overall statistical distribution of the probes in the bilayer. To explore favorable orientation of the aromatic rings of the probe molecules in the bilayer, we calculated the distribution of angles between a vector normal to the plane of the indole ring and the bilayer normal z (not shown). We found that for the initial sampling period, t = 0–10 ns, all probe molecules have a uniform distribution with no preferential alignment of the probes in bulk water. At the end of the sampling period at t = 40–50 ns, all the molecules demonstrate a tendency to align the plane of the indole ring parallel with the lipid tails. The effect of the fluorophore alignment in the organized membrane environment has already been reported for other aromatic probes, such as DPH,42 pyrroloquinoline,51 pyrene,39 and tryptophan.50
Free Energy Profile of Lipid Bilayer Penetration
The free energy profile of penetration of 5M-PyIn-0 and DMPmIn-0 into a POPC bilayer was estimated using the method of potential of mean constraint force (PMF).74 The PMFs symmetrized for two bilayer leaflets are shown in Figure 9. As can be noted, despite the similar chemical nature of the two indole derivatives, their partitioning behavior is modulated by structural modification of the fluorophore. The replacement of the pyridine ring in 5M-PyIn-0 by pyrimidine in DMPmIn-0 results in the shift of the partition free energy profile toward the more polar interfacial region of the bilayer.
Figure 9.

Potential of mean constraint force for 5M-PyIn-0 (1) and DMPmIn-0 (2) in a POPC bilayer. The PMFs are calculated for the center of mass of the probe molecule relative to the center of mass of the bilayer. The PMFs are set to zero in the bulk water phase. For visualization clarity, the PMFs are schematically superimposed on a bilayer snapshot.
It is seen that both 5M-PyIn-0 and DMPmIn-0 have deep global minima of the Gibbs free energy located in the interfacial region of the bilayer (10–15 Å). 5M-PyIn-0 has the deepest negative minimum of the partition free energy of −8.6 kcal/mol at 9.7 Å from the bilayer center (Figure 9). The second deepest local minimum is −7.9 kcal/mol, located at 14.6 Å. DMPmIn-0 also has two free energy minima of −8.1 and −7.1 kcal/mol located at 14.1 and 19.0 Å, respectively, from the bilayer center. The overall agreement of the calculated ΔG with experiment is very good. The comparison of the calculated ΔG with the corresponding experimental values reported in Table 1 shows that the former is overestimated by 0.5–1.0 kcal/mol only.
Figure 9 also demonstrates that the PMF for both indole derivatives shows unfavorable free energies in the center of the bilayer. This explains why no significant population of probe molecules has been observed in this region during the MD sampling of the unconstrained passive partitioning (Figure 6). In addition, during 50 ns of the MD sampling, there were no events in which the indole probes crossed the bilayer from one leaflet to the other. At the first assumption, it might be that 5M-PyIn-0 and DMPmIn-0 composed of the hydrophobic moieties should be found in the most hydrophobic part of the bilayer; that is, the middle of the membrane. On the other hand, the presence of such large, rigid aromatic molecules in the middle of membrane would force the lipid acyl chains to arrange themselves around the molecule. It would reduce the mobility of the chain ends, and hence, it would reduce the whole entropy of the membrane, which is highly unfavorable. The appearance of the energy maximum of the PMF profile at the center of the bilayer has been reported for other small amphipathic solutes.31,38,39,49,50,73,76
The interfacial preference of the studied indole derivatives is consistent with the observation of preferred localization of indole and tryptophan in a lipid bilayer.49,50,75,76 Both 5M-PyIn-0 and DMPmIn-0 have amphipathic character, and therefore, they favorably localize in the region of the bilayer containing the interface between the hydrophobic lipid tails and the polar carbonyl and phosphate groups. This region of the bilayer is also characterized by penetration of water molecules. As a result, for both 5M-PyIn-0 and DMPmIn-0, there is an evident trend for the polar imino NH fragment of the indole to face out of the bilayer toward the bulk water phase. The polar NH group of the indole moiety, which is also capable of hydrogen bonding, tends to drive the probe toward polar environments of the lipid headgroups, while at the same time, the apolar aromatic rings drive the molecules into the hydrophobic lipid tail region of the bilayer. Similar arguments are applied to explain the preference of natural aromatic amino acids, such as tyrosine, tryptophan, and phenylalanine for the membrane interface.76 Umbrella sampling simulations of partitioning of these aromatic amino acids in a DOPC bilayer have shown that they all have partition free energy minima 12–14 Å from the bilayer center.
Summary and Conclusions
We have investigated the fluorescence properties of the two indole derivatives, 2-(2′-pyridyl)-5-methyl-indole and 2-[2′-(4′,6′-dimethylpyrimidyl)]-indole, in the lipid membrane using steady-state and time-resolved emission spectroscopy. We have previously demonstrated that the fluorescence of 5M-PyIn-0 and DMPmIn-0 is strongly sensitive to changes in their local environment and can be altered by hydrogen bonding with protic solvents.6 The results presented in this paper demonstrate that the very weak fluorescence, measured for 5M-PyIn-0 and DMPmIn-0 in neutral aqueous solution, was enhanced up to 60–90 times upon titrating with lipid membranes. It was found that upon membrane interaction, their fluorescence quantum yield can be restored from values of 0.005–0.01, measured in aqueous solution, to those of 0.4–0.45, which are typical for their fluorescence in nonpolar solvents such as n-hexane.
In addition, the enhancement of the fluorescence intensity was accompanied by a blue shift in the emission peaks. These observations of spectral changes in intensities and the position of emission maxima are consistent with the transfer of 5M-PyIn-0 and DMPmIn-0 from polar aqueous solution to a nonpolar, hydrophobic environment of the lipid bilayer. The partitioning of the probe molecules deeply into the water-free region of the membrane results in the loss of specific hydrogen bonding with water molecules, which is seen as the strong fluorescence enhancement. To estimate the thermodynamics of membrane interaction of the studied compounds, we measured the Gibbs free energy (ΔG) of transfer from an aqueous environment to a lipid bilayer by monitoring the increase in the steady-state fluorescence intensities. For both 5M-PyIn-0 and DMPmIn-0, ΔG of membrane partitioning was found to be in the range from −7.1 to −8.0 kcal/mol, depending only slightly on the lipid composition of a bilayer. A rather weak dependence of ΔG on the amount of zwitterionic/anionic lipids in the membrane suggests that membrane partitioning of 5M-PyIn-0 and DMPmIn-0 was largely driven by hydrophobic effects.
Our experimental fluorescence study was combined with a series of atomistic classical MD simulations of partitioning of 5M-PyIn-0 and DMPmIn-0 between bulk aqueous solution and a POPC bilayer. Two MD methods were used to determine the favorable localization of the probes in a bilayer. The first method involves the free passive diffusion and distribution of probe molecules between bulk water and a lipid bilayer. The second method utilizes biased MD sampling of penetration of a probe across a bilayer. The first set of unconstrained MD simulations revealed that passive diffusion and partitioning of the probe molecules to the POPC bilayer occur in the time scale of 10–20 ns. At the end of the 50-ns MD sampling, both 5M-PyIn-0 and DMPmIn-0 were found to be localized deeply in the hydrocarbon core of the bilayer. The deep localization of the fluorophore in the hydrophobic, water-free environment of the bilayer results in a dramatic decrease in the probability of the formation of specific hydrogen-bonded complexes between the probe and water molecules. We suggest that the loss of the specific hydrogen bonding of 5M-PyIn-0 and DMPmIn-0 with bulk water molecules can explain the experimental observation of the strong increase in fluorescence of their membrane-bound form. Using the MD umbrella sampling and the potential of mean constrained force calculations, we found that both 5M-PyIn-0 and DMPmIn-0 have deep free energy minima located at distances of 10–14 Å from the center of the POPC bilayer. The good predictions obtained for these two free and biased, MD sampling methods indicate that the combined simulation approach presented here provides a converged view on the preferred localization of 5M-PyIn-0 and DMPmIn-0 in a bilayer.
Our results may have potentially important implications for developing highly sensitive fluorescence probes for membrane studies and membrane protein analysis. In contrast to traditional probes, which are sensitive to changes occurring in the whole solvation shell surrounding an excited dye, the emission signal of 5M-PyIn-0 and DMPmIn-0 may be altered by interactions with a few protic solvent molecules, opening up the possibility for fluorescence monitoring of water content in a membrane up to a level of single water molecules.
Acknowledgments
F.W. and R.P.T. thank the Robert A. Welch Foundation (E-621) and the National Science Foundation (CHE-0714751) for financial support. A.S.L. acknowledges support from NIH grant GM-069783.
Abbreviations used
- 5M-PyIn-0
2-(2′-pyridyl)-5-methylindole
- PyIn-0
2-(2′-pyridyl)-indole
- DMPmIn-0
2-[2′-(4′,6′-dimethylpyrimidyl)]-indole
- LUV
large unilamellar vesicles
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphatidylcholine
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol
- 75POPC/25POPG, 25POPC/75POPG
mixtures of POPC and POPG that contain a molar percentage of the corresponding lipid specified by the number
- Chol
cholesterol
- 70POPC/30Chol
a mixture that contain a molar percentage of 70:30 of POPC and cholesterol
- MD
molecular dynamics
References and Notes
- 1.Taylor CL, El-Bayoumi MA, Kasha M. Proc Natl Acad Sci USA. 1969;63:253–260. doi: 10.1073/pnas.63.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimmer M. Chem Rev. 2002;102:759–781. doi: 10.1021/cr010142r. [DOI] [PubMed] [Google Scholar]
- 3.Pakhomov AA, Martynov VI. Chem Biol. 2008;15:755–764. doi: 10.1016/j.chembiol.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Vendrell O, Gelabert R, Moreno M, Lluch JM. J Phys Chem B. 2008;112:13443–13452. doi: 10.1021/jp805049c. [DOI] [PubMed] [Google Scholar]
- 5.Herbich J, Hung CY, Thummel RP, Waluk J. J Am Chem Soc. 1996;118:3508–3518. [Google Scholar]
- 6.Waluk J. Acc Chem Res. 2003;36:832–838. doi: 10.1021/ar0200549. [DOI] [PubMed] [Google Scholar]
- 7.Kyrychenko A, Herbich J, Wu F, Thummel RP, Waluk J. J Am Chem Soc. 2000;122:2818–2827. [Google Scholar]
- 8.Kijak M, Petkova I, Toczek M, Wiosna-Sałyga G, Zielińska A, Herbich J, Thummel RP, Waluk J. Acta Phys Pol, A. 2007;112:S105–S120. [Google Scholar]
- 9.Petkova I, Mudadu MS, Singh A, Thummel RP, van Stokkum IHM, Buma WJ, Waluk J. J Phys Chem A. 2007;111:11400–11409. doi: 10.1021/jp0735841. [DOI] [PubMed] [Google Scholar]
- 10.Wiosna-Sałyga G, Nosenko Y, Kijak M, Thummel RP, Brutschy B, Waluk J. J Phys Chem A. 2010;114:3270–3279. doi: 10.1021/jp909409d. [DOI] [PubMed] [Google Scholar]
- 11.Nosenko Y, Wiosna-Sałyga G, Kunitski M, Petkova I, Singh A, Buma WJ, Thummel RP, Brutschy B, Waluk J. Angew Chem, Int Ed. 2008;47:6037–6040. doi: 10.1002/anie.200801350. [DOI] [PubMed] [Google Scholar]
- 12.Vetokhina V, Kijak M, Wiosna-Sałyga G, Thummel RP, Herbich J, Waluk J. Photochem Photobiol Sci. 2010;9:923–930. doi: 10.1039/c0pp00043d. [DOI] [PubMed] [Google Scholar]
- 13.Kyrychenko A, Waluk J. Challenges and Advances in Computational Chemistry and Physics. In: Paneth P, Dybala-Defratyka A, editors. Kinetics and Dynamics: from Nano- to Bio-Scale. Vol. 12. Springer Science+Business Media B.V; New York: 2010. pp. 35–75. [Google Scholar]
- 14.Kyrychenko A, Herbich J, Izydorzak M, Gil M, Dobkowski J, Wu FY, Thummel RP, Waluk J. Isr J Chem. 1999;39:309–318. [Google Scholar]
- 15.Nosenko Y, Kunitski M, Riehn C, Thummel RP, Kyrychenko A, Herbich J, Waluk J, Brutschy B. J Phys Chem A. 2008;112:1150–1156. doi: 10.1021/jp076839j. [DOI] [PubMed] [Google Scholar]
- 16.Kyrychenko A, Herbich J, Izydorzak M, Wu F, Thummel RP, Waluk J. J Am Chem Soc. 1999;121:11179–11188. [Google Scholar]
- 17.Kijak M, Zielińska A, Thummel RP, Herbich J, Waluk J. Chem Phys Lett. 2002;366:329–335. [Google Scholar]
- 18.Smirnov AV, English DS, Rich RL, Lane J, Teyton L, Schwabacher AW, Luo S, Thornburg RW, Petrich JW. J Phys Chem B. 1997;101:2758–2769. [Google Scholar]
- 19.Kelepouris L, Blanchard GJ. J Phys Chem B. 2002;106:6600–6608. [Google Scholar]
- 20.Kelepouris L, Blanchard GJ. J Phys Chem B. 2003;107:1079–1087. [Google Scholar]
- 21.Mukherjee TK, Ahuja P, Koner AL, Datta A. J Phys Chem A. 2005;109:12567–12573. doi: 10.1021/jp051574z. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee TK, Panda D, Datta A. J Phys Chem A. 2005;109:18895–18901. doi: 10.1021/jp052917w. [DOI] [PubMed] [Google Scholar]
- 23.Kwon OH, Jang DJ. J Phys Chem B. 2005;109:8049–8052. doi: 10.1021/jp050743c. [DOI] [PubMed] [Google Scholar]
- 24.Kwon OH, Jang DJ. J Phys Chem B. 2005;109:20479–20484. doi: 10.1021/jp053187v. [DOI] [PubMed] [Google Scholar]
- 25.Demchenko AP, Mély Y, Duportail G, Klymchenko AS. Biophys J. 2010;96:3461–3470. doi: 10.1016/j.bpj.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tieleman DP. Proc Aust Physiol Soc. 2006;37:3715–27. [Google Scholar]
- 27.Berkowitz ML, Bostick DL, Pandit S. Chem Rev. 2006;106:1527–1539. doi: 10.1021/cr0403638. [DOI] [PubMed] [Google Scholar]
- 28.Vácha R, Siu SWI, Petrov M, Böckmann RA, Jurkiewicz P, Barucha-Kraszewska J, Hof M, Berkowitz ML, Jungwirth P. J Phys Chem B. 2009;113:7235–7243. doi: 10.1021/jp809974e. [DOI] [PubMed] [Google Scholar]
- 29.Vácha R, Jurkiewicz P, Petrov M, Berkowitz ML, Böckmann RA, Barucha-Kraszewska J, Hof M, Jungwirth P. J Phys Chem B. 2010;114:9504–9509. doi: 10.1021/jp102389k. [DOI] [PubMed] [Google Scholar]
- 30.Xiang TX, Anderson BD. Adv Drug Delivery Rev. 2006;58:1357–1378. doi: 10.1016/j.addr.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Boggara MB, Krishnamoorti R. Biophys J. 2010;98:586–595. doi: 10.1016/j.bpj.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Högberg CJ, Maliniak A, Lyubartsev AP. Biophys Chem. 2007;125:416–424. doi: 10.1016/j.bpc.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Koubi L, Tarek M, Klein ML, Scharf D. Biophys J. 2000;78:800–811. doi: 10.1016/S0006-3495(00)76637-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koubi L, Saiz L, Tarek M, Scharf D, Klein ML. J Phys Chem B. 2003;107:14500–14508. [Google Scholar]
- 35.Li C, Yi M, Hu J, Zhou HX, Cross TA. Biophys J. 2008;94:1295–1302. doi: 10.1529/biophysj.107.112482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chew CF, Guy A, Biggin PC. Biophys J. 2008;95:5627–5636. doi: 10.1529/biophysj.108.139477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.dos Santos DJVA, Eriksson LA. Biophys J. 2006;91:2464–2474. doi: 10.1529/biophysj.105.077156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacCallum J, Tieleman DP. J Am Chem Soc. 2005;128:125–130. doi: 10.1021/ja0535099. [DOI] [PubMed] [Google Scholar]
- 39.Hoff B, Strandberg E, Ulrich AS, Tieleman DP, Posten C. Biophys J. 2005;88:1818–1827. doi: 10.1529/biophysj.104.052399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Repáková J, Holopainen JM, Karttunen M, Vattulainen I. J Phys Chem B. 2006;110:15403–15410. doi: 10.1021/jp061300r. [DOI] [PubMed] [Google Scholar]
- 41.Čurdová J, Čapková P, Plášek J, Repáková J, Vattulainen I. J Phys Chem B. 2007;111:3640–3650. doi: 10.1021/jp065956w. [DOI] [PubMed] [Google Scholar]
- 42.Repáková J, Čapková P, Holopainen JM, Vattulainen I. J Phys Chem B. 2004;108:13438–13448. doi: 10.1021/jp908533x. [DOI] [PubMed] [Google Scholar]
- 43.Fraňová M, Repáková J, Čapková P, Holopainen JM, Vattulainen I. J Phys Chem B. 2010;114:2704–2711. doi: 10.1021/jp908533x. [DOI] [PubMed] [Google Scholar]
- 44.Loura LMS, Ramalho JPP. Biochim Biophys Acta. 2007;1768:467–478. doi: 10.1016/j.bbamem.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 45.Loura LMS, Fernandes F, Fernandes AC, Ramalho JPP. Biochim Biophys Acta. 2008;1778:491–501. doi: 10.1016/j.bbamem.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 46.Hinner MJ, Marrink SJ, de Vries AH. J Phys Chem B. 2009;113:15807–15819. doi: 10.1021/jp907981y. [DOI] [PubMed] [Google Scholar]
- 47.Skaug MJ, Longo ML, Faller R. J Phys Chem B. 2009;113:8758–8766. doi: 10.1021/jp902877y. [DOI] [PubMed] [Google Scholar]
- 48.Kyrychenko A. Chem Phys Lett. 2010;485:95–99. [Google Scholar]
- 49.Grossfield A, Woolf TB. Langmuir. 2002;18:198–210. [Google Scholar]
- 50.Norman KE, Nymeyer H. Biophys J. 2006;91:2046–2054. doi: 10.1529/biophysj.105.080275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kyrychenko A, Waluk J. Biophys Chem. 2008;136:128–135. doi: 10.1016/j.bpc.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Hope MJ, Bally MB, Mayer LD, Janoff AS, Cullis PR. Chem Phys Lipids. 1986;40:89–107. doi: 10.1016/0009-3084(86)90077-0. [DOI] [PubMed] [Google Scholar]
- 53.Mayer LD, Hope MJ, Cullis PR. Biochim Biophys Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 54.Bartlett GR. J Biol Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 55.Thummel RP, Hegde V. J Org Chem. 1989;54:1720–1725. [Google Scholar]
- 56.Wu F, Hardesty J, Thummel RP. J Org Chem. 1998;63:4055–4061. [Google Scholar]
- 57.Berger O, Edholm O, Jähnig F. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kyrychenko A, Stepanenko Y, Waluk J. J Phys Chem A. 2000;104:9542–9555. [Google Scholar]
- 59.van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol E, Meulenhoff PJ, Tieleman DP, Sijbers ALTM, Feenstra KA, van Drunen R, Berendsen HJC. Gromacs User Manual version 3.3. 2005 www.gromacs.org.
- 60.Hermans J, Berendsen HJC, van Gunsteren WF, Postma JPM. Biopolymers. 1984;23:1513–1518. [Google Scholar]
- 61.Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 62.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 63.Hees B, Bekker H, Berendsen HJC, Fraaije JGEM. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 64.Lindahl E, Hess B, van der Spoel D. J Mol Model. 2001;7:306–317. [Google Scholar]
- 65.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 66.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- 67.Laaksonen L. Centre for Scientific Computing, ESPOO; Finland: 1997. [Google Scholar]
- 68.Humphrey W, Dalke A, Schulten KJ. Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 69.Kyrychenko A, Waluk J. J Phys Chem A. 2006;110:11958–11967. doi: 10.1021/jp063426u. [DOI] [PubMed] [Google Scholar]
- 70.Nosenko Y, Kunitski M, Thummel RP, Kyrychenko A, Herbich J, Waluk J, Riehn C, Brutschy B. J Am Chem Soc. 2006;128:10000–10001. doi: 10.1021/ja063093e. [DOI] [PubMed] [Google Scholar]
- 71.Nosenko Y, Kyrychenko A, Thummel RP, Waluk J, Brutschy B, Herbich J. Phys Chem Chem Phys. 2007;9:3276–3285. doi: 10.1039/b703908e. [DOI] [PubMed] [Google Scholar]
- 72.White SH, Wimley WC, Ladokhin AS, Hristova K. Methods Enzymol. 1998;295:62–87. doi: 10.1016/s0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- 73.Vijayan R, Biggin PC. Biophys J: Biophys Lett. 2008;95:L45–L47. doi: 10.1529/biophysj.108.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marrink SJ, Berendsen HJC. J Phys Chem. 1994;98:4155–4168. [Google Scholar]
- 75.Yau WM, Wimley WC, Gawrisch K, White SH. Biochem. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 76.MacCallum JL, Bennett WFD, Tieleman DP. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]