

Abstract
Background
Ischemia-reperfusion (I/R) injury is a prime antigen-independent inflammatory factor in the dysfunction of liver transplants. The precise contribution of T cells in the mechanism of I/R injury remains to be elucidated. As the CD154-CD40 co-stimulation pathway provides essential second signal in the initiation and maintenance of T-cell–dependent immune responses, this study was designed to assess the role of CD154 signaling in the pathophysiology of liver I/R injury.
Methods
A mouse model of partial 90-min warm hepatic ischemia followed by 6 hr of reperfusion was used. Three animal groups were studied: (1) wild-type (WT) mice treated with Ad-(-gal versus Ad-CD40 immunoglobulin; (2) untreated WT versus CD154 (MR1) monoclonal antibody-treated WT mice; and (3) untreated WT versus CD154 knockout mice.
Results
The disruption of CD154 signaling in all three animal groups ameliorated otherwise fulminant liver injury, as evidenced by depressed serum glutamic oxaloacetic transaminase levels, compared with controls. These beneficial effects were accompanied by depressed hepatic T-cell sequestration, local decrease of vascular endothelial growth factor expression, inhibition of tumor necrosis factor-( and T-helper type 1 cytokine production, and induction of antiapoptotic (Bcl-2/Bcl-xl) but depression of proapoptotic (caspase-3) proteins.
Conclusions
By using in parallel a gene therapy approach, pharmacologic blockade, and genetically targeted mice, these findings document the benefits of disrupting CD154 to selectively modulate inflammatory responses in liver I/R injury. This study reinforces the key role of CD154-CD40 T-cell co-stimulation in the pathophysiology of liver I/R injury.
Keywords: Ischemia-reperfusion injury, Vascular endothelial growth factor, CD154-CD40, Gene transfer
Ischemia-reperfusion (I/R) injury, an antigen-independent inflammatory component of organ “harvesting,” remains an important problem in clinical transplantation. In the case of liver, primary and delayed hepatic nonfunction caused by I/R injury may continue to both early and late liver transplant outcomes (1). Although studies on both “warm” and “cold” components of hepatic I/R injury have provided some etiologic factors, including activation of Kupffer cells, release of proinflammatory cytokines, increased expression of vascular cell adhesion molecules, and neutrophil accumulation (2), the exact mechanisms and mediators involved remain unclear.
It has been postulated that prolonged ischemia results in an allograft becoming more susceptible to T-cell immune responses (3). Indeed, evidence is mounting on the importance of T cells in mediating both short- and long-term organ ischemic insult (4, 5). The contributory role of lymphocytes in hepatic I/R injury is likely a multifactorial one. The demonstration that systemic immunosuppression (cyclosporine A/FK506) attenuates hepatocellular damage implies a pathophysiologic role of T lymphocytes (6), data supported by our own findings in T-cell–deficient mice (7) and in rats in which treatment with FTY720 prevented I/R injury in parallel with redistribution of T cells from host peripheral blood into lymph nodes (8).
The CD154-CD40 co-stimulation provides the essential second signal in the initiation and maintenance of T-cell–dependent responses (9). Indeed, the efficacy of CD154-targeted therapy to prevent rejection and to induce tolerance in some transplant models has been established (10–12). Much less is known about the role of CD154 signaling in antigen-independent inflammatory responses triggered by I/R injury. Indeed, we have shown that disruption of CD154 co-stimulation or treatment with CD154 monoclonal antibody (mAb) prevented hepatic I/R injury in mice (7), whereas gene transfer of CD40 immunoglobulin (Ig) protected rat livers from I/R injury (13). This study was aimed at delineating mechanisms of cytoprotection against “warm” hepatic I/R injury in three distinct mouse models: (1) gene therapy-mediated local CD154 blockade (Ad-CD40Ig); (2) antibody-induced systemic CD154 blockade (MR1 mAb); and (3) genetically targeted CD154 absence (CD154 knockout [KO] mice).
MATERIALS AND METHODS
Animals
Male B6/129 wild-type (WT) mice and CD154 KO mice (8–12 weeks of age) were used (The Jackson Laboratory, Bar Harbor, ME). Animals were maintained under conditions approved by the University of Southern California, Los Angeles Chancellor’s Animal Research Committee. All animals were housed in microisolator cages in pathogen-free facilities and according to National Institutes of Health guidelines.
Generation of Recombinant Adenovirus
Generation of recombinant adenovirus (Ad)-CD40Ig was previously described (13). Briefly, the mouse CD40Ig cDNA was placed under the transcriptional control of a murine cytomegalovirus promoter. Ad-CD40Ig was constructed by homologous recombination in 293 cells with dl324 viral DNA, which was Ad5-derived, E1- and E3-deleted adenovirus. Purified Ad-CD40Ig was amplified in 293 cells grown in SMEM suspension medium (GIBCO, Grand Island, NY) with 10% newborn calf serum (GIBCO). The titration of the virus was analyzed by plaque assay. Virus stocks of 2.5 ×1010 plaque-forming units/mL were stored at −80°C. The generation of Ad-encoding Escherichia coli (-galactosidase (Ad(-gal) was described (14).
Hepatic I/R injury Model
A warm hepatic I/R injury model was used, as described (7). Briefly, mice were injected with heparin (100 (g/kg) and an atraumatic clip was used to interrupt the arterial-portal venous blood supply to the cephalad liver lobes. After 90 min of partial hepatic ischemia, the clip was removed, initiating reperfusion. Animals were killed at 6 hr; liver tissue and blood samples were harvested for analysis. The extent and severity of hepatic I/R injury were assessed in groups (n=4–6) of (1) WT mice that were injected intravenously with Ad-CD40Ig or Ad-(-gal (2.5 ×109 plaque-forming units) at day −2; (2) WT recipients that were pretreated with CD154 mAb (MR1, 0.25 mg/mouse intraperitoneally at day −2 and day −1); and (3): CD154 KO mice. Sham WT controls underwent the same procedures but without vascular occlusion.
Hepatocyte Function Assay
Serum glutamic-oxaloacetic transaminase (SGOT) levels, an indicator of hepatocyte function, were measured in blood samples obtained at 6 hr postreperfusion. The SGOT measuring was performed using an autoanalyzer manufactured by ANTECH Diagnostics (Los Angeles, CA).
Immunohistochemical Analysis
Liver tissue samples were embedded in Tissue Tec OCT compound (Miles, Elkhart, IN), snap-frozen in liquid nitrogen, and stored at −70°C. Cryostat sections (5 (m) were fixed in acetone and then endogenous peroxidase activity was inhibited with 0.3% H2O2 in phosphate-buffered saline. Primary rat antibody against mouse CD3+ T cells (17A2) was added at optimal dilutions (BD Biosciences Pharmingen). Bound primary antibody was detected using biotinylated anti-rat IgG and streptavidin peroxidase-conjugated complexes (DAKO, Carpinteria, CA). Negative controls included sections in which the primary antibody was replaced with dilution buffer or normal mouse serum. The sections were evaluated blindly by counting the labeled cells in triplicate within 10 high-power fields per section.
RNA Extraction-Competitive Template Reverse-Transcriptase Polymerase Chain Reaction
To study cytokine gene expression patterns, we used competitive template reverse-transcriptase polymerase chain reaction (PCR), as described (13). Briefly, total RNA was extracted from frozen liver tissue samples using RNAse Mini Kit (Qiagen, Inc., Chatsworth, CA), and RNA concentration was determined by a spectrophotometer. Five micrograms of RNA was reverse-transcribed using oligo(dT) primers and superscript reverse transcriptase (GIBCO). According to the varying contents of specific cDNA and amplification efficiencies, PCR was performed by different cycle numbers at the annealing temperature that was optimized empirically for each primer pair: 40, 60°C (interleukin [IL]-2); 35, 63°C (interferon [IFN]-(); 37, 60°C (tumor necrosis factor [TNF]-(); 35, 60°C (vascular endothelial growth factor [VEGF]); and 35, 63°C ((-actin), respectively. PCR products were analyzed in ethidium bromide-stained 2% agarose gel, and scanned for density using Kodak Digital Science 1D Analysis software (Version 2.0; Eastman Kodak, Rochester, NY). To compare the relative level of each gene in different samples, all samples were normalized against the respective β-actin wild-type cDNA to competitive template DNA ratio.
Western Blot Analysis
Protein was extracted from tissues with protein lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate, and 1% triton X-100; pH 7.2). Proteins (30 (g/sample) in SDS-loading buffer (50 mM Tris, 10% glycerol, 1% SDS) were subjected to 12% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA). The gel was then stained with Coomassie blue to document protein loading. The membrane was blocked with 3% dry milk and 0.1% Tween 20 in phosphate-buffered saline. Polyclonal rabbit anti-mouse Bcl-2/Bcl-xl and anti-mouse caspase-3 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were used. The membranes were incubated with antibodies and then developed according to the Amersham enhanced chemiluminescence protocol. Relative quantities of Bcl-2, Bcl-xl, and caspase-3 were determined by densitometer and expressed in absorbance units (AU) (Kodak Digital Science 1D Analysis Software; Eastman Kodak).
Statistical Analysis
All data are expressed as mean±SD. Statistical comparisons between groups were analyzed by Student’s t test. All differences were considered statistically significant at a value of P<0.05.
RESULTS
CD154-CD40 T-Cell Signaling Is Required in the Mechanism of Hepatic I/R Injury
Ninety minutes of hepatic warm ischemia followed by 6 hr of reperfusion significantly increased SGOT levels from 32±10 IU/L in sham-operated controls to 2,283±238 IU/L in the WT group (Fig. 1) (P<0.0001). However, treatment of WT mice with Ad-CD40Ig but not with Ad-(-gal markedly decreased SGOT levels (465±90 and 2,485±478 IU/L, respectively; P<0.005). The disruption of CD154 signaling with CD154 (MR1) mAb in WT mice subjected to hepatic I/R injury or its absence in CD154 KO mice prevented hepatocellular damage, as evidenced by diminished SGOT (Fig. 1) (151±21 and 181±51 IU/L, respectively; P<0005) and preservation of liver histology (not shown). To analyze mechanisms by which CD154-CD40 co-stimulation promotes hepatic I/R injury, we contrasted (1) WT mice treated with Ad(-gal versus Ad-CD40 Ig; (2) untreated WT versus CD154 (MR1) mAb-treated WT mice; and (3) untreated WT versus CD154 KO mice.
FIGURE 1.

SGOT levels in mice that underwent 90 min of hepatic warm ischemia followed by 6 hr of reperfusion. Groups included sham-operated controls; T-cell–competent untreated WT mice; WT mice that were pretreated with Ad-CD40Ig, Ad-(-gal (2.5 × 109 plaque-forming units at day −2), or CD154 mAb (MR1; 0.25 mg at days −2 and −1); and CD154-deficient (KO) mice. There were four to six animals in each group. Means and SD are shown.
Disruption of CD154-CD40 Signaling Prevents T-Cell Infiltration
As shown in Figure 2, the hepatic I/R injury was accompanied by significantly increased sequestration of T cells in untreated and Ad-(-gal–treated WT mice, as compared with sham-operated controls (11.3±2.1. and 11.6±2.8 vs. 1.4±0.8, respectively; P<0.0001). In contrast, treatment of WT mice with Ad-CD40-Ig, CD154 mAb, or CD154 KO mice resulted in diminished T-cell levels (3.2±1.3, 3.3±1.9, and 2.5±1.6; P<0.0001, as compared with WT and Ad-(-gal–treated controls).
FIGURE 2.

T-cell (CD3+) infiltration in murine livers undergoing I/R injury with or without CD154-CD40 co-stimulation blockade. Data are representative of three recipients per group (immunoperoxidase labeling of cryostat sections, hematoxylin counterstained; magnification ×400). For details, see Materials and Methods and Figure 1.
Disruption of CD154-CD40 Signaling Depresses VEGF
As shown in Figure 3, warm hepatic ischemia followed by reperfusion significantly increased hepatic expression of mRNA coding for VEGF in WT and Ad-(-gal–treated controls as compared with sham-operated mice (2.37±0.17 and 2.45±0.18 vs. 0.32±0.11, respectively; P<0.005). In marked contrast, WT mice treated with Ad-CD40 Ig or CD154 mAb and CD154 KO mice showed markedly decreased VEGF levels (0.82±0.15, 0.85±0.12, and 0.71±0.15, respectively) as compared with WT (P<0.01) and Ad-(-gal (P<0.01) controls.
FIGURE 3.

Reverse-transcriptase PCR-assisted detection of mRNA coding for VEGF in hepatic ischemic lobes at 6 hr of reperfusion after 90 min of warm ischemia. For details, see Materials and Methods and Figure 1. There were four to six animals in each group. Means and SD are shown.
Disruption of CD154-CD40 Signaling Decreases TNF-( and T-Helper Type 1 Cytokines
Next, we used competitive-template reverse-transcriptase PCR to analyze cytokine mRNA expression in the ischemic liver lobes. As shown in Figure 4, WT and Ad-(-gal–treated mice revealed markedly increased levels of TNF-( (2.35±0.19 and 2.24±0.12 vs. 0.21±0.03, respectively; P<0.005), IL-2 (2.41±0.16 and 2.51±0.23 vs. 0.21±0.03, respectively; P<0.005), and IFN-( (2.08±0.09 and 2.04±0.09 vs. 0.21±0.04, respectively; P<0.005), as compared with sham-operated controls. In contrast, WT mice treated with Ad-CD40Ig or CD154 mAb and CD154 KO mice showed decreased hepatic expression of mRNA coding for TNF-( (0.86±0.1, 0.88±0.14, and 0.79±0.16, respectively), IL-2 (0.72±0.15, 0.78±0.16, and 0.7±0.14, respectively), and IFN-( (0.84±0.14, 0.82±0.15, and 0.77±0.16, respectively), as compared with WT (P<0.05) and Ad-(-gal (P<0.05) controls.
FIGURE 4.

Reverse-transcriptase PCR-assisted detection of mRNA coding for TNF-( and Th1 (IL-2 and IFN-() cytokines in hepatic ischemic lobes at 6 hr of reperfusion after 90 min of warm ischemia. For details, see Materials and Methods and Figure 1. There were four to six animals in each group. Means and SD are shown.
Disruption of CD154-CD40 Signaling Increases Antiapoptotic Molecules
We used Western blots to evaluate the expression of antiapoptotic Bcl-2 and Bcl-xl gene products in the ischemic liver lobes. As shown in Figure 5, the expression of antiapoptotic Bcl-2 and Bcl-xl (AU) was strongly increased in WT mice treated with Ad-CD40Ig (1.6–1.8 AU and 1.7–1.9 AU, respectively) or CD154 mAb (1.8–2.0 AU and 1.7–1.9 AU, respectively) and in CD154 KO mice (1.7–1.9 AU and 1.6–1.8 AU, respectively), as compared with WT and Ad-(-gal controls (0.1–0.3 AU and 0.1–0.2 AU, respectively). In contrast, the expression of proapoptotic caspase-3 was inhibited in WT mice treated with Ad-CD40Ig (0.2–0.4 AU) or CD154 mAb (0.3–0.4 AU) and in CD154 KO mice (0.2–0.5 AU), as compared with WT and Ad-(-gal controls (1.7–1.9 AU and 1.9–2.1 AU, respectively).
FIGURE 5.
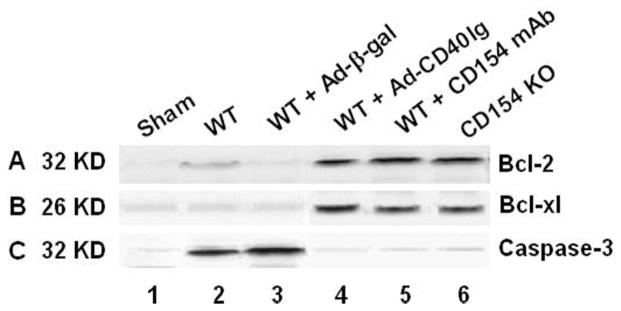
Western blot analysis of antiapoptotic (Bcl-2/Bcl-xl) gene products in mouse livers after 90 min of warm ischemia followed by 6 hr of reperfusion. The protein expression was probed by using antibodies against mouse Bcl-2 (A), Bcl-xl (B), and caspase-3 (C). (lane 1) Sham-operated control; (lane 2) WT mice; (lane 3) WT mice treated with Ad-(-gal; (lane 4) WT mice treated with Ad-CD40Ig; (lane 5) WT mice treated with CD154 mAb; (lane 6) CD154 KO mice. Data shown are representative of three separate experiments.
DISCUSSION
The results of this study document the key role of the CD154-CD40 co-stimulation pathway in the mechanism of antigen-independent inflammatory responses triggered by hepatic I/R injury. Indeed, gene therapy-mediated prolonged local CD154 blockade (Ad-CD40 Ig), antibody-induced systemic CD154 blockade (MR1 mAb), and genetically targeted CD154 absence (CD154 KO mice) all ameliorated otherwise fulminant injury in a warm mouse liver ischemia model followed by reperfusion. These beneficial effects resulting from disruption of CD154-CD40 signaling were accompanied by (1) diminished intrahepatic T-cell sequestration; (2) decrease of VEGF expression; (3) inhibition of TNF-( and T-helper (Th) type 1 cytokine production; and (4) induction of anti-apoptotic (Bcl-2/Bcl-xl) and depression of proapoptotic (caspase-3) proteins. Thus, by using gene therapy, pharmacologic blockade, and genetically targeted mice in parallel, these findings document the benefits of disrupting the CD154 signaling that otherwise modulates inflammatory pathways in liver I/R injury.
VEGF, the most potent angiogenesis factor described to date, is a 45-kDa glycoprotein expressed by monocytes-macrophages, endothelial cells, activated T cells, and a variety of other cell types (15, 16). Indeed, VEGF is involved in an array of delayed-type hypersensitivity and chronic inflammatory disorders. Of interest are recent findings that CD154-CD40 interactions are the most potent for VEGF expression in endothelial cells and monocytes in vitro (17), and that the proangiogenic effect of CD154 in vivo is VEGF dependent (18). As the implication of VEGF, a hepatocyte product that acts on sinusoidal endothelial cells in the mechanism of I/R injury, remains to be elucidated, we asked whether 90 min of warm ischemia will affect its expression in our I/R injury model. Indeed, hepatic mRNA levels coding for VEGF increased sharply at 6 hr of reperfusion, a period of marked postischemic intrahepatic T-cell and macrophage sequestration (7). This is consistent with a robust and rapid increase of VEGF mRNA expression in rat hepatocyte cultures preexposed to periods of cold ischemia followed by warm reoxygenation (19). Interestingly, cold preservation alone did not trigger VEGF secretion. These in vitro findings support the role of VEGF in the mechanism of hepatic I/R injury, consistent with recent reports that in vivo myocardial ischemia significantly augmented VEGF levels in pig hearts (20), whereas anti-VEGF antibody treatment prevented ischemia-induced neovascularization in mice (21) and decreased the degree of I/R injury in cold-preserved rat liver grafts (22). Indeed, in our own studies, mice conditioned with anti-VEGF antibodies became resistant against otherwise fulminant warm hepatic I/R injury (J. W. Kupiec-Weglinski, 2005).
In agreement with the role of CD154-CD40 co-stimulation in T-cell function (9–12), the interruption of CD154 signaling not only depressed intrahepatic T-cell infiltration but also diminished activation of liver-infiltrating mononuclear cells, as measured by local Th1-dependent tissue inflammation. IFN-( can activate Kupffer cells and lead to a further recruitment and activation of neutrophils (23), consistent with our finding of decreased IFN-( mRNA and neutrophil infiltrate in Ad-CD40Ig–transfected cold-stored rat liver grafts (13). Moreover, we have shown that Th1-type inflammatory responses in mouse livers undergoing warm I/R injury are STAT4-dependent (24). Conversely, the benefits of inducing Th2-type responses in liver I/R injury models (25, 26) are in agreement with STAT4 disruption-mediated cytoprotection (24), or activation of STAT6, a key negative regulator of the hepatic I/R injury response (27). In addition to depressing the Th1-type cytokine program, CD154 blockade also diminished local expression of TNF-(, the key effector molecule in liver I/R injury. Our studies in this murine model of hepatic I/R injury have documented that TNF-( induction is IRF-3 (but not MyD88) dependent (28), implying that multiple cell types might be involved in the pathogenesis of liver inflammation. One possible scenario of TNF-( induction is that IFN-inducible gene products (e.g., inducible protein-10) recruit or activate intrahepatic lymphocytes to produce the majority of TNF-(. This may provide a putative link between innate immune activation by liver I/R injury and adaptive immune responses in the absence of exogenous antigens.
The disruption of CD154-CD40 co-stimulation triggered increased expression of antiapoptotic (Bcl-2/Bcl-xl) yet diminished expression of proapoptotic (caspase 3) proteins. These “cytoprotective” molecules associate with the Th2-enriched microenvironment and remain increased exclusively in I/R injury-resistant livers (2, 29). Not surprisingly, gene therapy-induced antiapoptotic Bcl-2 (30) or Bag-1 (31) prevented hepatic I/R injury sequelae, whereas the blockade of apoptosis with a caspase inhibitor ameliorated I/R injury in liver grafts (32). Thus, disruption of CD154 signaling might interact with a number of pathways that mediate I/R injury cytotoxicity (1) by stabilizing antiapoptotic proteins and preventing cell death by reactive oxygen species; (2) by inhibiting postmitochondrial apoptosis and influencing apoptosome formation (33); and (3) by activating a positive feedback between cytoprotective genes and type-2 cytokines to amplify anti-inflammatory and antiapoptotic capacity, possibly by means of p38 MAPK (34) or STAT6-dependent signaling (24).
CONCLUSION
By selectively modulating host inflammatory responses, the disruption of CD154-CD40 T-cell co-stimulation after gene therapy, pharmacologic blockade, or genetic engineering exerted potent cytoprotection against antigen-independent hepatic I/R injury. This study reinforces the key role of CD154 signaling in the pathophysiology of liver I/R injury.
Acknowledgments
This work was supported by National Institutes of Health grants RO1 DK062357, AI23847, and AI42223 (J.W.K.W.) and by the Dumont Research Foundation.
Footnotes
Selected as one of the top abstracts for presentation at the XX International Congress of The Transplantation Society (ICTS) held in Vienna, September 5–10th, 2004; the invited manuscript was peer-reviewed and accepted for publication in the special issue dedicated to the XX Congress.
References
- 1.Farmer DG, Amersi F, Kupiec-Weglinski JW, et al. Current status of ischemia and reperfusion injury in the liver. Transplant Rev. 2000;14:106. [Google Scholar]
- 2.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury: A fresh look. Exp Mol Pathol. 2003;74:86. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 3.Bulkley GB, Oshima A, Bailey RW. Pathophysiology of hepatic ischemia in cardiogenic shock. Am J Surg. 1996;151:87. doi: 10.1016/0002-9610(86)90017-6. [DOI] [PubMed] [Google Scholar]
- 4.Zwacka RM, Zhang Y, Halldorson J, et al. CD4+ T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100:279. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burne MJ, Daniels F, Ghamdour AE, et al. Identification of the CD4+ T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest. 2001;108:1283. doi: 10.1172/JCI12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki S, Toledo-Pereyra LH, Rodrigyez FJ, et al. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury: Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Shen XD, Ke B, Zhai Y, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74:315. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 8.Anselmo DM, Amersi FF, Shen X-D, et al. FTY720 pretreatment reduces warm hepatic ischemia reperfusion injury through inhibition of T-lymphocyte infiltration. Am J Transplant. 2002;2:843–849. doi: 10.1034/j.1600-6143.2002.20906.x. [DOI] [PubMed] [Google Scholar]
- 9.Sayegh MH, Turka LA. The role of T-cell costimulatory activation pathways in transplant rejection. N Engl J Med. 1998;338:1813. doi: 10.1056/NEJM199806183382506. [DOI] [PubMed] [Google Scholar]
- 10.Parker DC, Greiner DL, Phillips NE, et al. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA. 1995;92:9560. doi: 10.1073/pnas.92.21.9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen CP, Alexander DZ, Hollenbaugh D, et al. CD40-gp39 interactions play a critical role during allograft rejection: Suppression of allo-graft rejection by blockade of the CD40-gp39 pathway. Transplantation. 1996;61:4. doi: 10.1097/00007890-199601150-00002. [DOI] [PubMed] [Google Scholar]
- 12.Kirk AD, Harlan DM, Armstrong NN, et al. CTLA4Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA. 1997;94:8789. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke B, Shen XD, Gao F, et al. Gene therapy for liver transplantation using adenoviral vectors: CD40-CD154 blockade by gene transfer of CD40Ig protects rat livers from cold ischemia and reperfusion injury. Mol Ther. 2004;9:38. doi: 10.1016/j.ymthe.2003.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolls J, Peppel K, Silva M, et al. Prolonged and effective blockade of tumor necrosis factor activity through adenovirus-mediated gene transfer. Proc Natl Acad Sci USA. 1994;91:215. doi: 10.1073/pnas.91.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown LF, Detmar M, Claffey JA, et al. Vascular permeability factor/vascular endothelial growth factor: A multifunctional angiogenic cytokine. EXS. 1997;79:233. doi: 10.1007/978-3-0348-9006-9_10. [DOI] [PubMed] [Google Scholar]
- 16.Freeman MR, Schneck FX, Gagnon ML, et al. Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: A potential role for T cells in angiogenesis. Cancer Res. 1995;55:4140. [PubMed] [Google Scholar]
- 17.Melter M, Reinders ME, Sho M, et al. Ligation of CD40 induces the expression of vascular endothelial growth factor by endothelial cells and monocytes and promotes angiogenesis in vivo. Blood. 2000;96:3801. [PubMed] [Google Scholar]
- 18.Reinders ME, Sho M, Robertson SW, et al. Proangiogenic function of CD40 ligand-CD40 interactions. J Immunol. 2003;171:1534. doi: 10.4049/jimmunol.171.3.1534. [DOI] [PubMed] [Google Scholar]
- 19.Archambault AJ, Sirois MG, Bernatchez PN, et al. Endothelial growth factor production by isolated rat hepatocytes after cold ischemia-warm reoxygenation. Liver Transpl. 2001;11:988. doi: 10.1053/jlts.2001.28444. [DOI] [PubMed] [Google Scholar]
- 20.Banai S, Shweiki D, Pinson A, et al. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: Implications for coronary angiogenesis. Cardiovasc Res. 1994;28:1176. doi: 10.1093/cvr/28.8.1176. [DOI] [PubMed] [Google Scholar]
- 21.Michel F, Ambroisine ML, Duriez M, et al. Aldosterone enhances ischemia-induced neovascularization through angiotensin II-dependent pathway. Circulation. 2004;109:1933. doi: 10.1161/01.CIR.0000127112.36796.9B. [DOI] [PubMed] [Google Scholar]
- 22.Boros P, Tarcsafalvi A, Wang L, et al. Intrahepatic expression and release of vascular endothelial growth factor following orthotopic liver transplantation in the rat. Transplantation. 2001;72:805. doi: 10.1097/00007890-200109150-00011. [DOI] [PubMed] [Google Scholar]
- 23.Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. J Mol Med. 1999;77:577. doi: 10.1007/s001099900029. [DOI] [PubMed] [Google Scholar]
- 24.Shen XD, Ke B, Zhai Y, et al. Stat4 and Stat6 signaling in hepatic ischemia/reperfusion injury: HO-1 dependence of STAT4 disruption-mediated cytoprotection. Hepatology. 2003;37:296. doi: 10.1053/jhep.2003.50066. [DOI] [PubMed] [Google Scholar]
- 25.Ke B, Shen XD, Lassman CR, et al. Interleukin-13 gene transfer protects rat livers from antigen-independent injury induced by ischemia and reperfusion. Transplantation. 2003;75:1118. doi: 10.1097/01.TP.0000062861.80771.D5. [DOI] [PubMed] [Google Scholar]
- 26.Ke B, Shen XD, Lassman CR, et al. Cytoprotective and antiapoptotic effects of IL-13 in hepatic cold ischemia/reperfusion injury are heme oxygenase-1 dependent. Am J Transplant. 2003;3:1076. doi: 10.1034/j.1600-6143.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 27.Ke B, Shen XD, Gao F, et al. Interleukin 13 gene transfer in liver ischemia and reperfusion injury: Role of Stat6 and TLR4 pathways in cytoprotection. Hum Gene Ther. 2004;15:691. doi: 10.1089/1043034041361244. [DOI] [PubMed] [Google Scholar]
- 28.Zhai Y, Shen XD, O’Connell R, et al. TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IRF3-dependent, MyD88-independent pathway. J Immunol. 2004;173:7115. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 29.Tsuchihashi S, Fondevila C, Kupiec-Weglinski JW. Heme oxygenase-1 and heat shock proteins in ischemia/reperfusion injury. Curr Opin Organ Transplant. 2004;9:145. [PubMed] [Google Scholar]
- 30.Bilbao G, Contreras JL, Eckhoff DE, et al. Reduction of ischemia-reperfusion injury of the liver by in vivo adenovirus-mediated gene transfer of the antiapoptotic Bcl-2 gene. Ann Surg. 1999;230:185. doi: 10.1097/00000658-199908000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawitzki B, Amersi F, Ritter T, et al. Upregulation of Bag-1 by ex-vivo gene transfer protects rat livers from ischemia/reperfusion injury. Hum Gene Ther. 2002;13:1495. doi: 10.1089/10430340260185120. [DOI] [PubMed] [Google Scholar]
- 32.Natori S, Selzner M, Valentino KL, et al. Apoptosis of sinusoidal endothelial cells occurs during liver preservation injury by a caspase-dependent mechanism. Transplantation. 1999;68:89. doi: 10.1097/00007890-199907150-00018. [DOI] [PubMed] [Google Scholar]
- 33.Antoku K, Maser RS, Scully WJ, et al. Isolation of Bcl-2 binding proteins that exhibit homology with BAG-1 and suppressor of death domains protein. Biochem Biophys Res Commun. 2001;286:1003. doi: 10.1006/bbrc.2001.5512. [DOI] [PubMed] [Google Scholar]
- 34.Amersi F, Shen XD, Anselmo D, et al. Ex-vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology. 2002;35:815. doi: 10.1053/jhep.2002.32467. [DOI] [PubMed] [Google Scholar]