

Abstract

A wide variety of alkoxymethyltrifluoroborate substrates were prepared via SN2 displacement of potassium bromomethyltrifluoroborate with various alkoxides in excellent yields. These alkoxymethyltrifluoroborates were effectively cross-coupled with several aryl chlorides. This method provides a unique dissonant disconnect that allows greater flexibility in the design of new and improved synthetic pathways.
Ethers are prevalent in natural products and compounds of pharmacological interest. Ethers are also one of the most widely used protecting groups for alcohols in organic synthesis.1 There are numerous ways to incorporate ether linkages into organic molecules. However, retrosynthetic analyses of such molecules tend to focus on two consonant disconnects employing either alkoxides or oxonium ions. Alkoxides are capable of reacting with various electrophiles to form C–O bonds (eq 1), while oxonium ions react with soft nucleophiles to form C–C bonds (eq 2).
| (1) |
| (2) |
| (3) |
A dissonant disconnect employing nucleophilic alkoxymethylating agents would provide a nontraditional approach to the construction of the alkoxymethyl linkage in straightchain ethers and heterocycles (eq 3). This strategic disconnect might prove useful for the synthesis of protected aryl- and heteroaryl alcohols in a single-step process, thereby avoiding a multistep process consisting of carbonylation/carboxylation, reduction, and protection (Scheme 1).
Scheme 1.
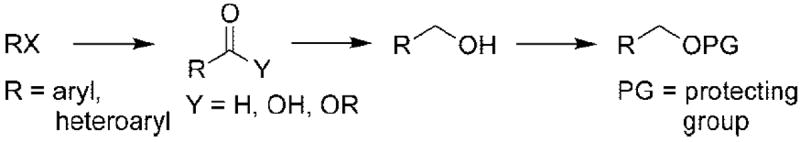
The preparation of numerous alkoxymethyl anions or their equivalents has been reported,2 but the majority of these synthons has found very little use in organic synthesis owing to their limited substrate scope, tedious preparation, and low functional group tolerance.
Relevant to the discussion herein, alkoxymethylstannanes have been used as hydroxymethylating agents in palladium catalyzed cross-coupling reactions, but to a limited extent (eqs 4-6).3 One possible reason for the minimal use of these otherwise useful synthons is the inherent toxicity and difficulty in purification that organostannanes are well-known to possess.
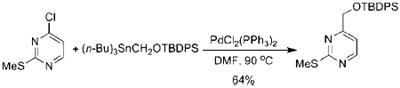 |
(4) |
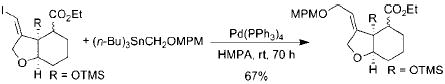 |
(5) |
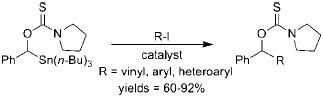 |
(6) |
The corresponding alkoxymethylboronate esters are known,4 but to the best of our knowledge, these have never been used in cross-coupling reactions. Furthermore, the α-alkoxy substituents are reported to sensitize the boronates to air oxidation, thereby making them more difficult to handle.4
We envisioned that these limitations could be easily avoided through the use of potassium organotrifluoroborates. Organotrifluoroborates have several advantages over organostannanes: they are relatively nontoxic, air- and moisturestable crystalline solids, atom-economic, and easily prepared by the addition of inexpensive KHF2 to various organoboron intermediates.
In 2006, our group published the preparation of various potassium organotrifluoroborates, including alkoxymethyltrifluoroborates, via the direct nucleophilic substitution of potassium halomethyltrifluoroborates.5 Soon after, Tanaka et al. patented the preparation and cross-coupling of alkoxymethyltrifluoroborates with various halides.6 These organotrifluoroborates were prepared from the corresponding alkoxymethylstannanes in a cumbersome and atom-inefficient multistep process. Additionally, the desired products were isolated in low to moderate yields (eq 7). The cross-coupling reactions themselves were substrate dependent and capricious, with the products being obtained in highly variable yields (eq 8).
Herein, we report a more practical and convenient synthesis of potassium alkoxymethyltrifluoroborates and the Suzuki–Miyaura cross-coupling of these agents with various aryl- and heteroaryl halides.
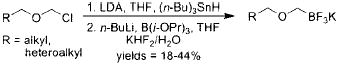 |
(7) |
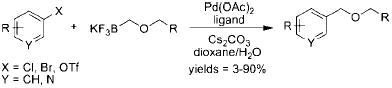 |
(8) |
Potassium alkoxymethyltrifluoroborates can be routinely and easily prepared via SN2 displacement of potassium bromomethyltrifluoroborate (1) with various alkoxides. To access large quantities of the SN2 precursor 1 (Table 1), we first optimized and scaled our previous conditions until we could routinely produce 100 g batches of highly pure compound.7 With 1 in hand, we optimized the SN2 reaction and found that 3 equiv of the alkoxides was needed for an efficient reaction. Because the alkoxymethyltrifluoroborates had low solubility in organic solvents (e.g., acetone and acetonitrile), separation from the inorganic byproducts was difficult and the desired products were at first isolated in low yields. This obstacle was circumvented through the use of continuous Soxhlet extraction. Using this technique, the desired alkoxymethyltrifluoroborates were obtained in excellent yields (Table 1). Of special note: some of the alkoxides employed may be viewed as hydroxyl protecting groups, which are stable to the conditions of a Suzuki–Miyaura cross-coupling but easily removed later in a synthetic sequence (Table 1, entries 1–5). It is also worth noting that silanols, including TIPSOH and TESOH, were not good nucleophiles for alkylation, presumably the direct result of the inductively stabilized and thus less reactive oxyanion.
Table 1.
Preparation of Potassium Alkoxymethyltrifluoroboratesa
|
| |||||
|---|---|---|---|---|---|
| entry | nucleophile | reaction time (h) | product | isolated yield (%) | |
| 1 |
|
3 |
|
2a | 98 |
| 2 |
|
5 |
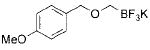
|
2b | 97 |
| 3 |
|
11 |
|
2c | 91b |
| 4 |
|
7 |
|
2d | 99 |
| 5 |
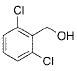
|
5 |
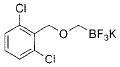
|
2e | 75 |
| 6 |
|
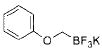
24 |
|
2f | 95 |
| 7 |
|
6 |
|
2g | 86 |
| 8 | MeONa | 14 |
|
2h | 91b |
| 9 |
|
3 |
|
2i | 84 |
| 10 |
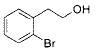
|
5 |
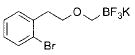
|
2j | 98 |
| 11 |
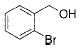
|
5 |
|
2k | 92 |
Conditions: ROH (3 equiv), NaH (3 equiv), THF (0.2 M), 0 °C–rt.
Commercially available alkoxide was used.
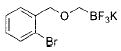
With the requisite alkoxymethyltrifluoroborates in hand, we next examined the Suzuki–Miyaura cross-coupling of these compounds. Toward this end, we chose 4-bromobenzonitrile and benzyloxymethyltrifluoroborate 2a as our model electrophile and nucleophile, respectively. We screened a wide variety of catalyst/ligandcombinations, solvents, bases, and temperatures.8 Even though several ligands afforded promising results (Table 2), RuPhos9proved to be the most general ligand for the cross-coupling reactions (Table 2, entry 1). The best coupling conditions were determined to be 3 mol % of Pd(OAc)2, 6 mol % of RuPhos, and 10:1 dioxane/H2O, using Cs2CO3 as the base.
Table 2.
Cross-Coupling of 2a and 4-Bromobenzonitrile with Diverse Ligandsa
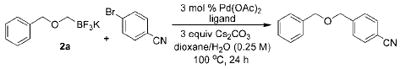
| ||||
|---|---|---|---|---|
| entry | ligand | abbreviation | ligand loading (%) | isolated yield (%) |
| 1 |
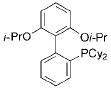
|
RuPhos | 6 | 76 |
| 2 |
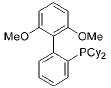
|
SPhos | 6 | 58 |
| 3 |
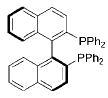
|
[(S)-BINAP] | 3 | 76 |
| 4 |
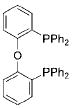
|
DPEPhos | 3 | 68 |
| 5 |
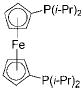
|
FcP(i-Pr)2 | 3 | 73 |
All reactions were carried out using 0.5 mmol of 4-bromobenzonitrile and 0.55 mmol of 2a.
During the course of this investigation, our group demonstrated that potassium N,N-dialkylaminomethyltrifluoroborates were effective cross-coupling partners with various aryl- and heteroaryl chlorides.10 Encouraged by these results, we sought to determine whether our optimized conditions for the cross-coupling of alkoxymethyltrifluoroborates with aryl bromides would also be applicable to aryl chlorides. To our surprise, we obtained comparable or better results with these electrophiles. Consequently, we utilized the more stable and less expensive aryl chlorides as the coupling partner throughout the remainder of the study.
Initially, we investigated the cross-coupling of potassium benzyloxymethyltrifluoroborate (2a) with electron-rich, electron-neutral, and electron-poor aryl chlorides (Table 3) using the optimized conditions. The desired alkoxymethylated products were isolated in good yield in all cases studied. Additionally, esters, pyrroles, and nitriles were tolerated. Even sterically hindered electrophiles coupled in good yield (Table 3, entries 2, 3, and 6). Surprisingly, electron-rich products, 3b–e were obtained in yields comparable to that of the electron-poor product 3g (Table 3, entries 2–5 and 7).
Table 3.
Cross-Coupling of Potassium Benzyloxytrifluoroborate (2a) with Various Aryl Chloridesa
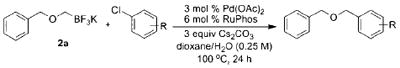
| ||||
|---|---|---|---|---|
| entry | aryl chloride | product | isolated yield (%) | |
| 1 |
|
|
3a | 74 |
| 2 |
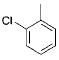
|
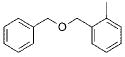
|
3b | 86 |
| 3 |
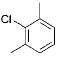
|
|
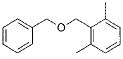
3c | 72 |
| 4 |
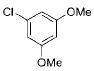
|
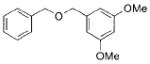
|
3d | 75 |
| 5 |
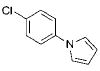
|
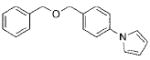
|
3e | 85 |
| 6 |
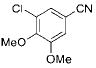
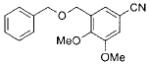
|
|
3f | 73 |
| 7 |
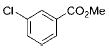
|
|
3g | 72 |
All reactions were carried out using 0.5 mmol of aryl chloride and 0.55 mmol of 2a.
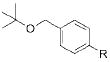
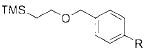
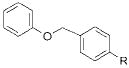
To demonstrate the scope of this method further, we crosscoupled various alkoxymethyltrifluoroborates with 4-chlorobenzonitrile (Table 4). Most alkoxymethyltrifluoroborates were found to couple effectively,11 providing the respective products 4a–d,g in good yield (Table 4, entries 1–4 and 6). Unfortunately, phenoxymethyltrifluoroborate (2f) gave the desired product in only moderate yield (Table 4, entry 5). 4-Chloroanisole was a more sensitive substrate as several alkoxymethyltrifluoroborates (2c–d,f) did not couple effectively (Table 4, entries 3–5). However, we were able to cross-couple 2a,b,g in good to moderate yields (Table 4, entries 1, 2, and 6).
Table 4.
Cross-Coupling of Aryl Chlorides with Various Potassium Alkoxymethyltrifluoroboratesa
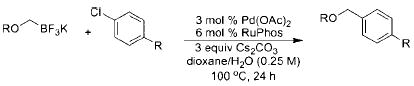
| |||||
|---|---|---|---|---|---|
| entry | nucleophile | product | isolated yield (%) | ||
| 1 |
|
2a |
|
R = CN 4a | 75 |
| OME4a’ | 77 | ||||
| 2 |
|
2b |
|
R = CN 4b | 72 |
| OME4b’ | 74 | ||||
| 3 |
|
2c |
|
R = CN 4c | 67 |
| 4 |
|
2d |
|
R = CN 4d | 80 |
| 5 |
|
2f |
|
R = CN 4f | 48 |
| 6 |
|
2g |
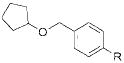
|
R = CN 4g | 67 |
| OME4g’ | 60 | ||||
All reactions were carried out using 0.5mmol of aryl chloride and 0.55 mmol of alkoxymethyltrifluoroborate.
The scope of the cross-coupling was expanded to heteroaryl chlorides. Although we screened numerous heteroaryl chlorides, the couplings of these substrates proved to be a challenging endeavor because either the products were obtained in low yields or the starting material was recovered. General conditions for the cross-coupling of heteroaryl chlorides were not found in these preliminary screening efforts. However, we were nonetheless able to couple several heteroaryl halides (Table 5) using our previously optimized conditions. Heteroaryl chlorides 5a,b coupled and provided the products in moderate yield (Table 5, entries 1 and 2). Nitrogen-containing heteroaryl chlorides were especially difficult to cross-couple. However, when we switched to bromide electrophiles, we were able to couple 3-bromopyridine (5c) and 3-bromopyrimidine (5d) in 70% and 51% yields, respectively (Table 5, entries 3 and 4).
Table 5.
Cross-Coupling of 2d with Heteroaryl Halidesa
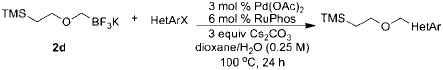
| |||||
|---|---|---|---|---|---|
| entry | HetArX | product | isolated yield (%) | ||
| 1 | 5a |
|
6a |
|
50 |
| 2 | 5b |
|
6b |
|
54 |
| 3 | 5c |
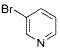
|
6c |
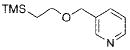
|
70 |
| 4 | 5d |
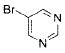
|
6d |
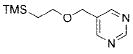
|
51 |
| 5 | 5e |
|
6e |
|
40 |
All reactions were carried out using 0.5 mmol of heteroaryl halide and 0.55 mmol of 2d.
In summary, we have prepared a variety of potassium alkoxymethyltrifluoroborates via SN2 reactions from potassium bromomethyltrifluoroborate with alkoxides. Under optimized conditions, alkoxymethyltrifluoroborates were successfully coupled with various aryl chlorides in good yields. Unfortunately, these optimized conditions lacked generality for the cross-coupling of heteroaryl halides, but several heteroaryl chlorides and bromides coupled in moderate to good yields. This method represents a fundamentally unique dissonant disconnect that allows greater flexibility than those protocols reported in previous synthetic approaches to aryl- and heteroaryl ethers.
Supplementary Material
Acknowledgments
The authors thank NIH (R01 GM-081376), Merck Research Laboratories, and Amgen for their generous support of our program. Johnson Matthey and Frontier Scientific are acknowledged for their donation of palladium catalysts. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data. We thank Dr. Jungyeob Ham (Korea Institute of Science and Technology) for his seminal contributions to this project. We also thank Paul Gormisky (University of Pennsylvania) for his help with the preparation of potassium bromomethyltrifluoroborate.
Footnotes
Supporting Information Available: Experimental procedures, compound characterization data, and NMR spectra for all compounds prepared in this investigation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Greene TW, Wuts PGM. ProtectiVe Groups in Organic Synthesis. John Wiley; New York: 1999. [Google Scholar]
- 2.Tamao K, Ishida N, Ito Y, Kumada M. Org Synth. 1990;8:315–320., and references cited therein. Hutchinson DK, Fuchs PL. J Am Chem Soc. 1987;109:4930–4939.
- 3.(a) Majeed AJ, Antonsen O, Benneche T, Undheim K. Tetrahedron. 1989;45:993–1006. [Google Scholar]; (b) Ferezou JP, Julia M, Li Y, Liu LW, Pancrazi A. Synlett. 1991:53–56. [Google Scholar]; (c) Kosugi M, Sumiya T, Ohhashi K, Sano H, Migita T. Chem Lett. 1985:997–998. [Google Scholar]; (d) Falck JR, Patel PK, Bandyopadhyay A. J Am Chem Soc. 2007;129:790–793. doi: 10.1021/ja064948q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matteson DS. Sci Synth. 2004;6:607–622. [Google Scholar]
- 5.Molander GA, Ham J. Org Lett. 2006;8:2031–2034. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka K, Inoue S, Ito D, Murai N, Kaburagi Y, Shirotori S, Suzuki S, Ohashi Y. Chem Abstr. 2006 Sep 21;145:356920. JP304894. [Google Scholar]
- 7.See Supporting Information for the improved procedure to synthesize potassium bromomethyltrifluoroborate.
- 8.Variables changed in coupling optimization included: catalyst/ligand combinations [PdCl2/PPh3, Pd(TFA)2/PPh3, Pd(OAc)2/PPh3, Pd(OAc)2/XPhos, Pd(OAc)2/DavePhos, Pd(OAc)2/(o-tolyl)3P, Pd(OAc)2/dppe, Pd(OAc)2/dppb, PdCl2(dppf) and Peppsi]. Catalyst/ligand ratios: (2/4 mol % and 5/10 mol %); solvents [THF, toluene, cyclopentyl methyl ether (CPME), and MeOH]; and bases (Cs2CO3, K2CO3, CsOH, and NEt3).
- 9.Milne JE, Buchwald SL. J Am Chem Soc. 2004;126:13028–13032. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]
- 10.Molander GA, Gormisky PE, Sandrock DL. J Org Chem. 2008;73:2052–2057. doi: 10.1021/jo800183q. [DOI] [PubMed] [Google Scholar]
- 11.The cross-coupling of 2h with 4-chlorobenzonitrile and 4-chloroanisole gave the products in low yield (34% and 41%, respectively). The cross-coupling of 2i with 4-chlorobenzonitrile gave the desired product along with two alkene-isomerized products in 78% yield. Attempted intramolecular cyclizations of 2j and 2k were unsuccessful.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.