

Abstract
Lung cancer is the most common cause of cancer deaths worldwide. The two broad histological subtypes of lung cancer are small-cell lung cancer (SCLC), which is the cause of 15% of cases, and non-small-cell lung cancer (NSCLC), which accounts for 85% of cases and includes adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma. Although NSCLC and SCLC are commonly thought to be different diseases owing to their distinct biology and genomic abnormalities, the idea that these malignant disorders might share common cells of origin has been gaining support. This idea has been supported by the unexpected findings that a subset of NSCLCs with mutated EGFR return as SCLC when resistance to EGFR tyrosine kinase inhibitors develops. Additionally, other case reports have described the coexistence of NSCLC and SCLC, further challenging the commonly accepted view of their distinct lineages. Here, we summarise the published clinical observations and biology underlying tumours with combined SCLC and NSCLC histology and cancers that transform from adenocarcinoma to SCLC. We also discuss pre-clinical studies pointing to common potential cells of origin, and speculate how the distinct paths of differentiation are determined by the genomics of each disease.
Introduction
Lung cancer is the leading cause of cancer death in both men and women worldwide. WHO classifies lung cancer into two broad histological subtypes: non-small-cell lung cancer (NSCLC) which is the cause of about 85% of cases, and small-cell lung cancer (SCLC), which accounts for the remaining 15%. NSCLC is further subdivided into adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma. The approval by the US Food and Drug Administration to introduce targeted therapies against EGFR, such as erlotinib and afatinib, has revolutionised the treatment of a subset of adenocarcinomas that have EGFR-activating mutations;1–6 however, acquired resistance develops after about 12 months.7,8 Repeat biopsy samples from patients with EGFR-mutant cancers have shown that several mechanisms bring about the acquired resistance, including the rare but consistent observation of histological transformation from adenocarcinoma to SCLC.9,10 Transformation to SCLC suggests that both adenocarcinoma and SCLC arise from a common cell type. Here, we explore the cellular and molecular relationship of adenocarcinoma to SCLC by discussing the clinical experience of combined-histology tumours and transformation from adenocarcinoma to SCLC.
Combined SCLC and NSCLC histology
Treatment strategies for lung cancer are based on the assumption that an individual patient's cancer is purely of one subtype.11,12 For example, limited-stage SCLC and localised NSCLC are both potentially curable diseases, but their treatment strategies differ substantially.11,13,14 Surgery has been largely abandoned as a treatment modality in patients with limited-stage SCLC, which is now treated with concurrent chemotherapy and radiotherapy.11 By contrast, patients with localised stage I or II NSCLC are treated mainly with surgery possibly followed by adjuvant chemotherapy, although radiotherapy is typically not part of the strategy.15 In advanced stage lung cancer, chemo therapy is the mainstay of treatment for both NSCLC and SCLC but the drugs used are distinct for each subtype. The initial response to chemotherapy is much greater for patients with extensive-stage SCLC than patients with metastatic adeno carcinoma,11,12 which suggests inherent differences in tumour biology. For example, pemetrexed is one of the most active agents in meta static adeno-carcinoma,12 but it is fairly inactive in extensive-stage SCLC.16 This difference could result from the high levels of thymidylate synthase in patients with SCLC,17 which can cause resistance to pemetrexed.18
In practice, combined-histology lung cancers can make treatment decisions difficult. SCLC with a large-cell component accounts for about 10% of SCLC cases; at present, these patients are given standard SCLC chemo-therapy regimens.11,14 Two large case series have investigated the frequency of tumours with combined SCLC and NSCLC histology.19,20 In the first study,19 176 SCLC tumours were analysed and 17 (10%) tumours also had an NSCLC component. Seven of these patients who had originally been diagnosed with NSCLC and given chemotherapy or radiotherapy were found to have combined histology at surgery or autopsy. Additionally, in autopsies of 40 patients originally diagnosed with SCLC, two were reported to have only adenocarcinoma. In the second case series,20 the histology of 429 SCLC tumours was studied; nine (2%) tumours contained NSCLC (six adenocarcinoma, three squamous-cell carcinoma). Repeat biopsy samples from six of these nine patients showed one dominant histology (three SCLC, two adenocarcinoma, one squamous-cell carcinoma). Although one possible ex planation for these results is the transformation from one phenotype to another, another possibility is that core biopsy samples or fine-needle aspirates used to make the initial diagnosis did not provide sufficient pathological material to determine the presence of combined histology to be identified at diagnosis. Whether the results from these autopsy studies represent transformation or com bined-histology tumours, these striking observations pose several practical questions that could affect clinical practice. First, in localised, early-stage, combined-histology tumours, how does the presence of adenocarcinoma affect the decision on whether to offer surgery? Alternatively, should patients with tumours that have an SCLC component always be given concurrent chemotherapy and radio therapy? Furthermore, in an advanced-stage setting, if tumours do not respond as initially expected or if the responses among two or more different lesions are discordant, is repeat biopsy indicated to rule out a dominant histology that was not identified at initial diagnosis?
EGFR-mutant adenocarcinoma and transformation to SCLC
Three EGFR inhibitors—erlotinib, gefitinib, and afatinib—are now used worldwide for first-line treatment of lung cancers that have EGFR-activating mutations, and their use has greatly changed clinical practice. However, within an average time of 12 months,7 resistance develops, and samples from repeat biopsies have shown several distinct mechanisms of acquired resistance to EGFR inhibitors. The most common resistance mechanism is a Thr790Met mutation in EGFR, which increases the affinity of the receptor for ATP and allows for continued EGFR signalling in the presence of the inhibitor.21 Thr790Met mutations are reported in 50–60% of samples from resistant tumours.9,10 Mechanisms that bypass the requirement for EGFR signalling, such as MET and HER2 amplification, could account for another 15–20% of resistance to EGFR inhibitors.9,10,22
Another mechanism of resistance to EGFR inhibitors is histological transformation of EGFR-mutant adeno-carcinoma to SCLC. This process was first described in 2006 in a 45-year-old woman with EGFR-mutant adenocarcinoma who had never smoked; she was originally diagnosed with adenocarcinoma and given erlotinib.23 The tumour responded for 18 months, but at the time of progression, the repeat biopsy sample showed SCLC with the original EGFR exon 19 deletion mutation. Since this first case, several other case series have been reported of EGFR-mutant adenocarcinoma transforming to SCLC as a mechanism of resistance to EGFR-inhibitor treatment (table 1).9,10,23–28 In all reported cases, small-cell tumours were identified by morphology and positive immunohistochemical staining for synaptophysin, chromogranin, or NCAM. In a case series in which repeat biopsies were done when resistance developed in patients given EGFR inhibitors,9 transformation to SCLC was reported in 14% of cases. In another case series, trans formation to SCLC was apparent in less than 5% of repeat biopsy samples.10 Genomic sequencing of EGFR from both the original and repeat biopsy samples at the time of resistance shows that every transformed SCLC tumour sample retained its original EGFR-activating mutation (table 1),9 which suggests that these were not independent de-novo cancers, but a transformed phenotype as a mechanism of resistance to treatment instead. Another hypothesis for this observation is that patients had tumours with combined histology at the time of initial diagnosis, which was not apparent on the diagnostic biopsy sample, and then the SCLC component became dominant as the adenocarcinoma component was successfully treated with the EGFR inhibitor. However, the clinical course of the reported cases has not typically been consistent with this hypothesis because most patients responded well to EGFR inhibitors for several months to years (table 1)9 and clinically had greater tumour growth at the time when SCLC was diagnosed. If tumour heterogeneity accounted for the development of SCLC in these patients, a less dramatic response to EGFR inhibitors and acquired resistance at an earlier time would be expected.
Table 1.
Reported cases of transformation from EGFR-mutant adenocarcinoma to SCLC as a mechanism of resistance to EGFR inhibition
| Age (years) | Sex | Smoking history (pack years) | Tumour histology | EGFR mutation in initial sample | Treatment | Duration of TKI treatment before transformation (months) | EGFR mutation in SCLC | EGFR protein expression in SCLC | Treatment of SCLC with TKI alone | |
|---|---|---|---|---|---|---|---|---|---|---|
| Zakowski and colleagues23 | 45 | F | 0 | Adenocarcinoma | Not tested | Erlotinib | 18 | Exon 19 deletion | Not tested | Gefitinib (no response) |
| Sequist and colleagues9 | ||||||||||
| Patient 1 | 40 | F | 0 | Adenocarcinoma | Exon 19 deletion | Erlotinib | 38 | Exon 19 deletion | Not tested | No |
| Patient 2 | 67 | F | 0 | Adenocarcinoma | Leu858Arg | Erlotinib | 22 | L858R | Not tested | No |
| Patient 3 | 54 | F | 25 | Adenocarcinoma | Exon 19 deletion | Erlotinib | 35 | Exon 19 deletion | Not tested | No |
| Patient 4 | 56 | F | 0 | Adenocarcinoma | Leu858Arg | Erlotinib | 14 | L858R | Not tested | No |
| Patient 5 | 61 | F | 80 | Adenocarcinoma | Leu858Arg | Erlotinib | 18 | L858R | Not tested | No |
| van Riel and colleagues24 | 42 | F | 0 | Adenocarcinoma | Not tested | Erlotinib | 11 | Exon 19 deletion | Not tested | No |
| Morinaga and colleagues25 | 46 | F | 0 | Adenocarcinoma | Not tested | Gefitinib | 10 | Exon 19 deletion | Not tested | No |
| Norkowski and colleagues26 | ||||||||||
| Patient 1 | 60 | F | 0 | Adenocarcinoma | Exon 19 deletion | Erlotinib | 31 | E782K | Not tested | No |
| Patient 2 | 50 | F | 0 | Adenocarcinoma | Exon 19 deletion | Erlotinib | 14 | Exon 19 deletion | Not tested | No |
| Watanabe and colleagues27 | 52 | F | 0 | Adenocarcinoma | Exon 19 deletion | Erlotinib | 11 | Exon 19 deletion | Not tested | No |
| Popat and colleagues28 | 46 | F | 0 | Adenocarcinoma | Exon 19 deletion | Erlotinib | 12 | Exon 19 deletion | Not tested | Afatinib (no response) |
Rows are individual patients. TKI=tyrosine kinase inhibitor. SCLC=small-cell lung cancer.
Does transformation to SCLC result solely from EGFR inhibition, or does EGFR-mutant adenocarcinoma have the potential to transform into SCLC? Furthermore, are adenocarcinomas that do not have EGFR mutations similarly able to transform to SCLC? Synchronous development of adenocarcinoma and SCLC was noted in EGFR-mutant tumours before treatment with an EGFR inhibitor.26 This observation suggests that the development of SCLC in EGFR-mutant cancers is not exclusively the result of EGFR inhibition. Furthermore, this series26 included two cases of adenocarcinoma that transformed to SCLC in tumours that did not have EGFR driver mutations in the original biopsy sample; this finding suggests that transformation can occur independently of EGFR mutational status. This was unlikely to be the result of false-negative EGFR testing because in both cases the specimens tested were resected tumours and one patient was reported to have a KRAS mutation. In a case series reported in 1986, before the discovery of EGFR-activating mutations, when resistance to conventional chemotherapy or radiotherapy developed, about 5% of patients originally diagnosed with NSCLC had SCLC at relapse.19 Whether the tumours of these patients did have EGFR-activating mutations is unknown, but the patients did show transformation to SCLC independently of EGFR inhibition. In their 2011 study, Sequist and colleagues9 found no cases of transformation to SCLC in 79 surgical specimens from patients with stage III NSCLC who received chemotherapy and radiotherapy,9 which suggests that EGFR-wild-type NSCLC is less likely to transform to SCLC than EGFR-mutant NSCLC. Larger studies will be needed to identify the precise frequencies of SCLC transformation in EGFR-mutant and EGFR-wild-type NSCLC, but the information available at present suggests that transformation to SCLC is more common in EGFR-mutant cancers treated with EGFR inhibitors than in wild-type cancers.
EGFR mutations in de-novo SCLC
Two large case series29,30 have investigated whether EGFR mutations occur de novo in classic SCLC (table 2).26,29–35 In the series reported by Tatematsu and colleagues,30 five of 122 patients had EGFR-mutated SCLC. These patients had a history of heavy smoking (average of 30 pack-years), but a significantly lower pack-year smoking history than that of patients who did not have EGFR mutations. Three of the five patients with EGFR-mutant SCLC had combined small-cell and adeno carcinoma histology. Two of the patients, both of whom had EGFR amplification, were given gefitinib and responded to treatment. One responder had combined SCLC and adenocarcinoma with EGFR amplification reported only in the adenocarcinoma component. The other responder had small-cell histology with EGFR amplification and had a rare Gly719Ala EGFR mutation. Of note is another case report of a patient with de-novo SCLC with an EGFR-activating mutation that did respond to gefitinib.31 Immunohistochemistry showed that this patient had high expression of EGFR. In another large case series from Taiwan,29 76 patients with SCLC were analysed for the presence of EGFR mutations. Two patients had EGFR mutations, who did not have previously known EGFR-mutant adenocarcinoma. One patient was treated with gefitinib and showed no response to treatment.29
Table 2.
Cases of de-novo SCLC with EGFR mutations
| Age (years) |
Sex | Smoking history (pack years) |
Tumour histology |
EGFR mutation in SCLC initial sample |
Treatment with TKI |
Response to TKI | EGFR amplification or protein expression in SCLC |
|
|---|---|---|---|---|---|---|---|---|
| Shiao and colleagues29 | ||||||||
| Patient 1 | 54 | F | 0 | SCLC | Exon 19 deletion | Gefitinib | Progressed | Unknown |
| Patient 2 | 63 | M | Heavy smoker | SCLC | Exon 19 deletion | No | NA | Unknown |
| Tatematsu and colleagues30 | ||||||||
| Patient 1 | 36 | F | 0 | Combined SCLC/adenocarcinoma | Leu858Arg | Gefitinib | Partial response | Amplification of EGFR in adenocarcinoma only |
| Patient 2 | 81 | M | 40 | SCLC | Gly719Ala | Gefitinib | Partial response | Amplification of EGFR in SCLC |
| Patient 3 | 69 | M | 30 | Combined SCLC/adenocarcinoma | Leu858Arg | No | NA | Amplification of EGFR in adenocarcinoma only |
| Patient 4 | 89 | F | 2·5 | SCLC | Leu858Arg | No | NA | No EGFR amplification |
| Patient 5 | 65 | M | 67·5 | Combined SCLC/adenocarcinoma | Exon 19 deletion | No | NA | Amplification of EGFR in adenocarcinoma only |
| Araki, Okamoto, and colleagues31,32 | 72 | F | 0 | SCLC | Exon 19 deletion | Gefitinib | Partial response | Immunohistochemistry positive for EGFR protein expression |
| Norkowski and colleagues26 | ||||||||
| Patient 1 | 62 | M | 0 | Combined SCLC/adenocarcinoma | Gly719Ala and exon 21 deletion found in both SCLC and adenocarcinoma | No | NA | Unknown |
| Patient 2 | 66 | F | 80 | Combined SCLC/adenocarcinoma | Exon 19 deletion in adenocarcinoma component only | No | NA | NA |
| Patient 3 | 65 | F | 45 | Synchronous SCLC and adenocarcinoma | Leu858Arg in adenocarcinoma component only | No | NA | NA |
| Fukui and colleagues33 | 62 | F | 0 | Combined SCLC/adenocarcinoma | Leu858Arg in both SCLC and adenocarcinoma | No | NA | Unknown |
| Lu and colleagues34 | ||||||||
| Patient 1 | 62 | F | 0 | Combined SCLC/adenocarcinoma | Exon 19 deletion in both SCLC and adenocarcinoma | No | NA | Unknown |
| Patient 2 | 61 | M | Smoker | Combined SCLC/squamous-cell carcinoma | Exon 19 deletion in SCLC | No | NA | Unknown |
| Kurahara and colleagues35 | 65 | F | 0 | SCLC | Leu858Arg | No | NA | Unknown |
Rows are individual patients. SCLC=small-cell lung cancer. TKI=tyrosine kinase inhibitor. NA=not applicable.
In a phase 2 trial,36 responses to gefitinib were tested in 19 patients with SCLC previously given chemotherapy. The participants were not tested specifically for EGFR mutations.29,30 Only two of the patients had stable disease and there were no partial or complete responses.36 Analysis of EGFR-mutant tumours that transformed to SCLC from adenocarcinoma as a mechanism of acquired resistance shows that, at transformation, the cancers lose protein expression of EGFR and have lower levels of EGFR amplification.37 In the past, SCLC was reported to have lower protein expression of EGFR than NSCLC.38,39 The mechanisms underlying low expression of EGFR in SCLC remain unknown, but this could explain the absence of response to EGFR inhibitors in several case reports.23,28,29 Overall, response to EGFR inhibitors in SCLC patients with EGFR mutations do not seem to match those of EGFR-mutant adenocarcinoma, perhaps because of mechanisms that suppress EGFR protein expression in these tumours. However, a case report by Araki and colleagues31 suggests that patients can have EGFR-mutant SCLC that does express EGFR and responds to EGFR inhibition. In view of individual case reports of patients with rare SCLC with EGFR amplification, larger studies will be needed to correlate protein expression with EGFR inhibition to find out whether other patients with SCLC could benefit from EGFR inhibition.
Genomic analysis of SCLC and adenocarcinoma
Genomic analyses have improved understanding of the pathways that are dysregulated in each specific tumour type. These efforts aim to identify oncogenic driver mutations, copy number changes, and translocations that can be targeted therapeutically. In adenocarcinoma, these sequencing efforts have resulted in the identification of several mutated oncogenic drivers and tumour suppressor genes and have validated the high prevalence of KRAS and EGFR mutations.40 In clinical practice, the standard of care is to genotype every patient with a new diagnosis of metastatic adenocarcinoma for EGFR mutations and ALK translocations, since targeted therapies are now approved in cancers that have these abnormalities.7,41,42 EGFR mutations and ALK translocations are estimated to account for about 30–50% of adenocarcinomas in patients who have never smoked, and the proportion might vary according to race.43,44 Additionally, many centres now undertake multiplexed genetic analyses to identify the presence of other prevalent genetic abnormalities, since therapeutics are under development for those cancers also.45
Two large-scale genome-sequencing projects have been completed in SCLC.46,47 They included whole-genome, transcriptome, exome, and copy-number analyses. Both studies identified a high prevalence of mutations in TP53 and RB1, validating previous human sequencing studies. Indeed, mouse models of SCLC have been developed by conditional inactivation of Tp53 and Rb1.48 Although some findings overlapped between these two studies, several findings were unique to each study, perhaps because of the heterogeneous patient samples in terms of stage and exposure to previous treatment. Known oncogenic drivers reported in adenocarcinoma, such as EGFR or KRAS, were not statistically significantly mutated in either SCLC genome-sequencing study. Of note, MYC amplification was seen in 16% of cases.46 MYCL1 knock-down resulted in decreased cell proliferation in SCLC cell lines,47 which suggests that MYC functions as an oncogenic driver in a subset of small-cell tumours. Activators of ERK signalling, such as EGFR and KRAS, are mutated more commonly in adenocarcinoma. By contrast, RB1 loss seems to be much more common in SCLC.46–48 Together these genomic efforts suggest that fundamental genetic differences drive the development of SCLC and lung adenocarcinoma.
RB1 inactivation as a defining feature of SCLC
Inactivation of RB1 has long been known to have an important role in the tumorigenesis of SCLC. In 2003, a mouse model of SCLC was developed by means of conditional knockout of Tp53 and Rb1.48 Further studies with this model have shown that Rb1 inactivation is essential for the development of SCLC. Mutation or loss of RB1 has been found in 100% of the human SCLC tumours sequenced.46 Furthermore, a proteomic study that compared SCLC and NSCLC cell lines showed that RB1 was quantitatively the most downregulated protein in SCLC compared with NSCLC.17 However, mice with loss of Rb1 alone do not develop small-cell tumours, which suggests that loss of Rb1 is necessary but not sufficient for SCLC tumorigenesis. Analyses of repeat biopsy samples from patients with EGFR-mutant adenocarcinoma that transform to SCLC revealed that 100% of these patients have loss of RB1;37 loss of RB1 seems to be a universal event leading to transformation from adenocarcinoma to SCLC.
Prostate cancer is another example of a cancer in which small-cell differentiation can occur after treatment. In one case series, 90% of small-cell prostate tumours had loss of RB1.49 Furthermore, loss of RB1 selectively occurred in castration-resistant prostate cancer, which suggests that loss of RB1 is a late event in tumorigenesis.50 RB1 loss is well known to be frequent in many non-neuroendocrine tumour types also.51 Why neuroendocrine differentiation occurs only in some tumour types with loss of RB1 is not known. Evidence from animal models supports a role for loss of Rb1 selectively affecting tumours of neuroendocrine origin. For example, Rb1+/− mice form pituitary and thyroid tumours after loss of the wild-type allele.52 Additionally, conditional inactivation of Rb1 in lung tissue results in increased numbers of pulmonary neuroendocrine cells during development.53 The three cancers with the highest frequency of RB1 mutations or deletions are SCLC, prostate cancer, and bladder cancer.51 Interestingly, both prostate and bladder cancers are known to undergo small-cell differentiation.49,54 Additional mechanistic insight into the role of RB1 loss in transformation to SCLC will help elucidate the biology of SCLC, how transformation from adenocarcinoma to SCLC occurs, and possibly help create new therapeutic targets to treat SCLC.
Adenocarcinoma and SCLC cells of origin
The appreciation that some adenocarcinomas and SCLC have the plasticity to switch histologies raises the possibility of the existence of shared cells of origin and greater plasticity than originally appreciated. Historically, SCLC was thought to arise from neuroendocrine cells within the distal part of the conducting airway because SCLC expresses neuroendocrine markers and typically develops in a central location. By contrast, adenocarcinomas are more commonly localised peripherally and were believed to originate from alveolar type II cells. However, studies in mouse models of lung cancer have yielded insights that could explain the shared ancestry of some SCLCs and NSCLCs.
Four studies have used cell-type specific Cre-recombinase conditional knockout mice to identify the cell of origin of SCLC55,56 and KRAS mutant adeno carcinoma57,58 in murine models of lung cancer. Two independent groups crossed Cre-recombinase under the control of cell-type specific promoters with mice that had floxed alleles of Tp53 and Rb1,48 a previously established SCLC model.48 Small-cell tumours were identified by morphological appearance and positive immuno histochemical staining for synaptophysin or NCAM.55,56 These groups noted that the CGRP promoter, which is specifically active in neuroendocrine cells, gave rise to SCLCs.55,56 The proportion of mice in which SCLC formed was similar for those expressing Cre-recombinase under the CGRP promoter or under the CMV promoter, which targets all cells in the lung; this finding suggests that small-cell tumours are mainly derived from neuroendocrine cells.55 Similarly, two additional studies showed that targeted disruption of Tp53 and Rb1 in neuroendocrine cells yields SCLC.56,59 These results were as expected. However, Sutherland and colleagues55 also used Ad5-SPC-Cre selectively to target deletion in alveolar type II cells. Surprisingly, they reported that targeted disruption of Tp53 and Rb1 in alveolar type II cells led to the development of SCLC. By contrast, Park and colleagues56 did not note the formation of SCLC in mice when they used an ER-inducible Ad-SPC-Cre to drive acute deletion of Tp53 and Rb1 on administration of tamoxifen in alveolar type II cells. One explanation for this discrepancy is that the mice developed by Sutherland and colleagues had loss of Tp53 and Rb1 in all alveolar type II cells throughout lung development. Thus, alveolar type II cells might also have the potential to give rise to SCLC, albeit at a much lower frequency than for neuroendocrine cells55 (figure 1). Future studies are needed to establish the capacity for alveolar type II cells to give rise to SCLC and to reconcile the differences between the two mouse studies.55,56
Figure 1. Alveolar type II cells could be a common precursor that can give rise to both adenocarcinoma and small-cell lung cancer (SCLC).
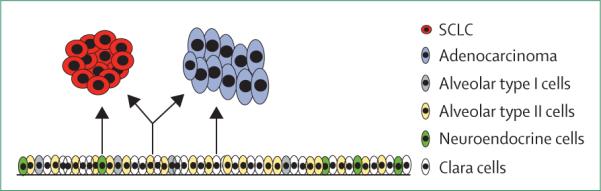
Diagram depicts the cells of origin of adenocarcinoma and SCLC. Neuroendocrine cells and possibly alveolar type II cells can give rise to SCLC (left and centre), whereas clara cells and alveolar type II cells can give rise to adenocarcinoma (right and centre).
Two studies that used the same principles of cell-type specific promoters in mouse models of KRAS mutant adenocarcinoma found that both clara cells and alveolar type II cells could give rise to adenocarcinoma (figure 1).57,58 Furthermore, in transgenic mice, expression of the EGFR L858R mutation under the control of the SPC promoter led to tumorigenesis,60 which suggests that alveolar type II cells can also serve as the cell of origin of EGFR-mutant adenocarcinoma. Together, these studies55,57,58,60 suggest that alveolar type II cells can give rise to both adenocarcinoma and SCLC (figure 1). Thus, alveolar type II cells seem to have the capacity to develop both SCLC55 and EGFR-mutant adenocarcinoma,57,58 and the range of genetic mutations is likely to affect the type of cancer that develops. Thus, EGFR-mutant lung cancers that develop in alveolar type II cells might have the potential to transform into SCLC.
In fact, activation of EGFR signalling could be impor tant for the fully differentiated alveolar-cell phenotype. Transcriptome analyses show that well differentiated alveolar type II cells have high expression of EGFR family members including EGFR, Erbb2, and Erbb3. Alveolar-cell proliferation was inhibited by EGFR-blocking antibodies.61 Furthermore, EGFR knockout mice have low expression of SPC and have defects in alveolarisation; these features support a crucial role for EGFR in the development of alveolar type II cells.62,63 Thus, we speculate that the presence of the EGFR mutation and active EGFR signalling drive both proliferation and differentiation of type II alveolar cells. In fact, this characteristic might be why EGFR-mutant lung cancers tend to be well differentiated adenocarcinomas.64 However, EGFR tyrosine kinase inhibitors might block this proliferation and differentiation. When additional key genetic events such as RB1 inactivation occur, these same alveolar type II cells might subsequently transform to SCLC and become independent of EGFR signalling (figure 2). However, if other paths of resistance develop instead, such as an EGFR Thr790Met gatekeeper mutation, EGFR signalling is restored and the resistant cells resume adenocarcinoma histology.
Figure 2. Hypothetical model depicting the molecular events that lead to transformation from adenocarcinoma to small-cell lung cancer (SCLC).
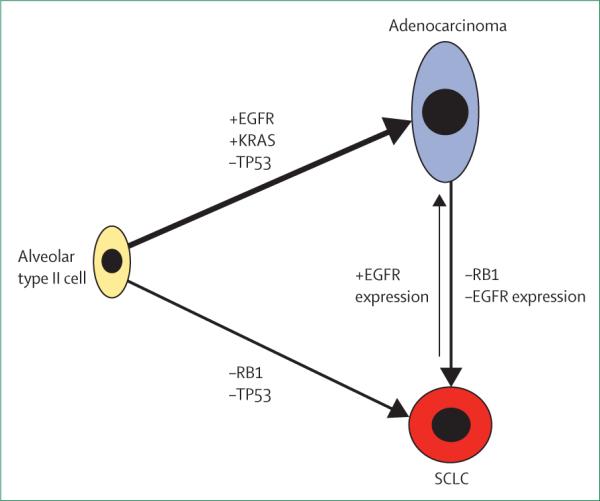
Alveolar type II cells have the potential to form both adenocarcinoma and SCLC depending on the mutational status of key oncogenes and tumour suppressors. Transformation from adenocarcinoma to SCLC involves the loss of RB1 and loss of EGFR protein expression.
Conclusions
Current understanding suggests that both combined-histology tumours and transformation are more common in lung cancers that have EGFR-activating mutations than in EGFR-wild-type tumours. The basis could be that the cell of origin of some EGFR-mutant adenocarcinomas, type II alveolar cells, also have the potential to become SCLC. Many examples used in this review reflect experiences with EGFR-mutant cancer, but the possibility remains that this clinical experience is biased by a greater number of repeat biopsy samples in EGFR-mutant cancers than in non-EGFR-mutant adenocarcinoma because biopsy at progression in NSCLC is not generally indicated. However, other cancers arising from type II alveolar cells could have the capacity to transform into SCLC, and transformation from adenocarcinoma to SCLC has occasionally been observed in lung cancers that do not have EGFR mutations.26 These preliminary observations raise some practical considerations for care of patients. Do specific circumstances exist under which a repeat biopsy should be done on non-EGFR mutant adenocarcinomas to look for transformation to SCLC? The frequency of documented transformation from adeno-carcinoma to SCLC in non-EGFR-mutant cancers is low at present. Future studies might help define which subsets of NSCLC are most prone to SCLC transformation.
We have also discussed case series showing that, although EGFR mutations are identified in SCLC, these patients have had mixed responses to EGFR inhibitors.29 The mixed responses could be due to loss of EGFR expression at the protein level, as shown in transformation to SCLC after development of resistance to an EGFR inhibitor.37 Thus, on the basis of available evidence, we believe that patients with SCLC combined with adenocarcinoma and SCLC transformed from adenocarcinoma should be given standard therapies for SCLC.14 Future studies will be needed to elucidate whether measurement of EGFR protein expression in EGFR-mutant SCLC might identify rare cases that would still respond to EGFR inhibitors.31
Search strategy and selection criteria.
We used the following search terms in PubMed and selected articles on the basis of relevance to transformation from adenocarcinoma to SCLC: “adenocarcinoma to small cell transformation”, “EGFR mutant lung cancer and mechanisms of resistance”, “cell of origin of small cell lung cancer”, “cell of origin of adenocarcinoma, genomics and small cell lung cancer”, and “combined histology small cell lung cancer”. All dates and languages were included in the search.
Acknowledgments
This work was funded by R01 CA137008 (JAE) and T32 CA009172 (MGO).
Footnotes
Contributors
MGO and JAE developed the ideas, reviewed the data, and wrote the review. MJN and LVS reviewed the data, provided intellectual input, and edited the review.
Declaration of interests
JAE reports: grants, consulting fees, travel expenses, and advisory board fees from Novartis; grants, consulting, travel, and advisory board fees from Sanofi-Aventis; grants, consulting fees, and travel expenses from Astra Zeneca; consulting and advisory board fees, and stock from Agios; consulting, advisory board, travel expenses, and stock from Loxo; consulting fees from Cell Signaling Technology; grants, consulting fees, travel expenses, and advisory board fees from GlaxoSmithKline; grants, consulting fees, and travel expenses from Amgen; consulting fees from Aisling; consulting and advisory board fees from G1 Therapeutics; consulting and advisory board fees, and travel expenses from Aveo/Biodesix; consulting and advisory board fees, and travel expenses from Endo; consulting fees from Red Sky; consulting fees from Fstar; consulting and advisory board fees from Jannsen; consulting and advisory board fees, and travel expenses from Piramal; consulting and advisory board fees, and travel expenses from Chugai; consulting and advisory board fees, and travel expenses from Quintiles; consulting fees from Madalon Consulting; consulting fees from Guidepoint Global; consulting fees from Morgan Stanley; consulting fees from Cytomx; consulting and advisory board fees from Pathway Therapeutics; non-compensated consulting for Third Rock Ventures; research grant from Jounce; licensing fees for use of cell lines from Pfizer; licensing fees for use of cell lines from Merrimack; consulting fees and travel expenses from Merck; consulting and advisory board fees, travel expenses, and international patent payment fees from Roche/Ventana/Genetech; and consulting fees and stock from Gatekeeper Pharmaceuticals. JAE has a financial interest in Gatekeeper Pharmaceuticals, a company aimed at development of new therapies to overcome resistance to erlotinib. Gatekeeper Pharmaceuticals has an international patent on an EGFR inhibitor. In addition, JAE has a patent, International Patent Application #PCT/US2008/004804 Filed: 4/11/2008 entitled: “Methods for Treating Cancer Resistant to ERB3 Therapeutics”, with royalties paid from Ventana/Roche to JAE through Partners. JAE's interests were reviewed and managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. LVS reports non-compensated consulting fees from Clovis Oncology, Boehringer Ingelheim, Merrimack Pharmaceuticals, AstraZeneca, Novartis, Taiho, and GlaxoSmithKline. MGO and MJN declare no competing interests.
References
- 1.Maemondo M, Inoue A, Kobayashi K, et al. the North-East Japan Study Group Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–88. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 2.Mitsudomi T, Morita S, Yatabe Y, et al. the West Japan Oncology Group Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–28. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Carcereny E, Gervais R, et al. the Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 4.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 5.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–22. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 6.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–42. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–99. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 9.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–47. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalemkerian GP, Akerley W, Bogner P, et al. the National Comprehensive Cancer Network Small cell lung cancer. J Natl Compr Canc Netw. 2013;11:78–98. doi: 10.6004/jnccn.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–51. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 13.Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366:1385–96. doi: 10.1016/S0140-6736(05)67569-1. [DOI] [PubMed] [Google Scholar]
- 14.van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378:1741–55. doi: 10.1016/S0140-6736(11)60165-7. [DOI] [PubMed] [Google Scholar]
- 15.Goldstraw P, Ball D, Jett JR, et al. Non-small-cell lung cancer. Lancet. 2011;378:1727–40. doi: 10.1016/S0140-6736(10)62101-0. [DOI] [PubMed] [Google Scholar]
- 16.Socinski MA, Smit EF, Lorigan P, et al. Phase III study of pemetrexed plus carboplatin compared with etoposide plus carboplatin in chemotherapy-naive patients with extensive-stage small-cell lung cancer. J Clin Oncol. 2009;27:4787–92. doi: 10.1200/JCO.2009.23.1548. [DOI] [PubMed] [Google Scholar]
- 17.Byers LA, Wang J, Nilsson MB, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozasa H, Oguri T, Uemura T, et al. Significance of thymidylate synthase for resistance to pemetrexed in lung cancer. Cancer Sci. 2010;101:161–66. doi: 10.1111/j.1349-7006.2009.01358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adelstein DJ, Tomashefski JF, Jr, Snow NJ, Horrigan TP, Hines JD. Mixed small cell and non-small cell lung cancer. Chest. 1986;89:699–704. doi: 10.1378/chest.89.5.699. [DOI] [PubMed] [Google Scholar]
- 20.Mangum MD, Greco FA, Hainsworth JD, Hande KR, Johnson DH. Combined small-cell and non-small-cell lung cancer. J Clin Oncol. 1989;7:607–12. doi: 10.1200/JCO.1989.7.5.607. [DOI] [PubMed] [Google Scholar]
- 21.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–75. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 23.Zakowski MF, Ladanyi M, Kris MG, the Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome Group EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355:213–15. doi: 10.1056/NEJMc053610. [DOI] [PubMed] [Google Scholar]
- 24.van Riel S, Thunnissen E, Heideman D, Smit EF, Biesma B. A patient with simultaneously appearing adenocarcinoma and small-cell lung carcinoma harbouring an identical EGFR exon 19 mutation. Ann Oncol. 2012;23:3188–89. doi: 10.1093/annonc/mds525. [DOI] [PubMed] [Google Scholar]
- 25.Morinaga R, Okamoto I, Furuta K, et al. Sequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutation. Lung Cancer. 2007;58:411–13. doi: 10.1016/j.lungcan.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Norkowski E, Ghigna MR, Lacroix L, et al. Small-cell carcinoma in the setting of pulmonary adenocarcinoma: new insights in the era of molecular pathology. J Thorac Oncol. 2013;8:1265–71. doi: 10.1097/JTO.0b013e3182a407fa. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe S, Sone T, Matsui T, et al. Transformation to small-cell lung cancer following treatment with EGFR tyrosine kinase inhibitors in a patient with lung adenocarcinoma. Lung Cancer. 2013;82:370–72. doi: 10.1016/j.lungcan.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Popat S, Wotherspoon A, Nutting CM, Gonzalez D, Nicholson AG, O'Brien M. Transformation to “high grade” neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer. 2013;80:1–4. doi: 10.1016/j.lungcan.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Shiao TH, Chang YL, Yu CJ, et al. Epidermal growth factor receptor mutations in small cell lung cancer: a brief report. J Thorac Oncol. 2011;6:195–98. doi: 10.1097/JTO.0b013e3181f94abb. [DOI] [PubMed] [Google Scholar]
- 30.Tatematsu A, Shimizu J, Murakami Y, et al. Epidermal growth factor receptor mutations in small cell lung cancer. Clin Cancer Res. 2008;14:6092–96. doi: 10.1158/1078-0432.CCR-08-0332. [DOI] [PubMed] [Google Scholar]
- 31.Araki J, Okamoto I, Suto R, Ichikawa Y, Sasaki J. Efficacy of the tyrosine kinase inhibitor gefitinib in a patient with metastatic small cell lung cancer. Lung Cancer. 2005;48:141–44. doi: 10.1016/j.lungcan.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 32.Okamoto I, Araki J, Suto R, Shimada M, Nakagawa K, Fukuoka M. EGFR mutation in gefitinib-responsive small-cell lung cancer. Ann Oncol. 2006;17:1028–29. doi: 10.1093/annonc/mdj114. [DOI] [PubMed] [Google Scholar]
- 33.Fukui T, Tsuta K, Furuta K, et al. Epidermal growth factor receptor mutation status and clinicopathological features of combined small cell carcinoma with adenocarcinoma of the lung. Cancer Sci. 2007;98:1714–19. doi: 10.1111/j.1349-7006.2007.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu HY, Sun WY, Chen B, et al. Epidermal growth factor receptor mutations in small cell lung cancer patients who received surgical resection in China. Neoplasma. 2012;59:100–04. doi: 10.4149/neo_2012_013. [DOI] [PubMed] [Google Scholar]
- 35.Kurahara Y, Kawaguchi T, Tachibana K, et al. Small-cell lung cancer in never-smokers: a case series with information on family history of cancer and environmental tobacco smoke. Clin Lung Cancer. 2012;13:75–79. doi: 10.1016/j.cllc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Moore AM, Einhorn LH, Estes D, et al. Gefitinib in patients with chemo-sensitive and chemo-refractory relapsed small cell cancers: a Hoosier Oncology Group phase II trial. Lung Cancer. 2006;52:93–97. doi: 10.1016/j.lungcan.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Niederst MJ, Sequist LV, Poirier JT, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerny T, Barnes DM, Hasleton P, et al. Expression of epidermal growth factor receptor (EGF-R) in human lung tumours. Br J Cancer. 1986;54:265–69. doi: 10.1038/bjc.1986.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobol RE, Astarita RW, Hofeditz C, et al. Epidermal growth factor receptor expression in human lung carcinomas defined by a monoclonal antibody. J Natl Cancer Inst. 1987;79:403–07. [PubMed] [Google Scholar]
- 40.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–97. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070–80. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ou SH. Lung cancer in never-smokers. Does smoking history matter in the era of molecular diagnostics and targeted therapy? J Clin Pathol. 2013;66:839–46. doi: 10.1136/jclinpath-2012-201296. [DOI] [PubMed] [Google Scholar]
- 45.Sequist LV, Heist RS, Shaw AT, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616–24. doi: 10.1093/annonc/mdr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peifer M, Fernández-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–16. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–89. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 49.Tan HL, Sood A, Rahimi HA, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890–903. doi: 10.1158/1078-0432.CCR-13-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma A, Yeow WS, Ertel A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–92. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 53.Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development. 2004;131:4299–310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- 54.Choong NW, Quevedo JF, Kaur JS. Small cell carcinoma of the urinary bladder. The Mayo Clinic experience. Cancer. 2005;103:1172–78. doi: 10.1002/cncr.20903. [DOI] [PubMed] [Google Scholar]
- 55.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–64. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 56.Park KS, Liang MC, Raiser DM, et al. Characterization of the cell of origin for small cell lung cancer. Cell Cycle. 2011;10:2806–15. doi: 10.4161/cc.10.16.17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mainardi S, Mijimolle N, Francoz S, Vicente-Dueñas C, Sánchez-García I, Barbacid M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc Natl Acad Sci USA. 2014;111:255–60. doi: 10.1073/pnas.1320383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci USA. 2014;111:4952–57. doi: 10.1073/pnas.1319963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song H, Yao E, Lin C, Gacayan R, Chen MH, Chuang PT. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci USA. 2012;109:17531–36. doi: 10.1073/pnas.1207238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin C, Song H, Huang C, et al. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS One. 2012;7:e53817. doi: 10.1371/journal.pone.0053817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507:190–94. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miettinen PJ, Berger JE, Meneses J, et al. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–41. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 63.Miettinen PJ, Warburton D, Bu D, et al. Impaired lung branching morphogenesis in the absence of functional EGF receptor. Dev Biol. 1997;186:224–36. doi: 10.1006/dbio.1997.8593. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y, Xu ML, Zhong HH, Heng WJ, Wu BQ. EGFR mutations are more frequent in well-differentiated than in poor-differentiated lung adenocarcinomas. Pathol Oncol Res. 2008;14:373–79. doi: 10.1007/s12253-008-9113-1. [DOI] [PubMed] [Google Scholar]