

Abstract
Traumatic brain injury (TBI) is associated with a risk for neurodegenerative disease. Some suggest a link between TBI and motor neuron disease (MND), including Amyotrophic Lateral Sclerosis (ALS). To investigate the potential mechanisms linking TBI to MND, we measured motor function and neuropathology following mild-TBI in wild-type and a transgenic model of ALS, G93A mutant mice. Mild-TBI did not alter lifespan of G93A mice or age of onset; however, rotarod performance was impaired in G93A verses wild-type mice. Grip strength was reduced only in G93A mice after mild-TBI. Increased EMG abnormalities and markers of denervation (AchR, Runx1) indicate that mild-TBI may result in peripheral effects that are exaggerated in G93A mice. Markers of inflammation (cell edema, astrogliosis and microgliosis) were detected at 24 and 72 hours in brain and spinal cord in wild-type and G93A mice. Levels of F2-isoprostanes, a marker of oxidative stress, were increased in spinal cord 24 hours post mild-TBI in wild-type mice but not affected by TBI in G93A mice. In summary, our data demonstrate that mild-TBI induces inflammation and oxidative stress and negatively impacts muscle denervation and motor performance, suggesting mild-TBI can potentiate motor neuron pathology and influence development of MND in mice.
Keywords: TBI, amyotrophic lateral sclerosis, mouse model, oxidative stress, spinal cord
INTRODUCTION
Studies suggest that there could be an increased risk for the development of motor neuron disease, including amyotrophic lateral sclerosis (ALS) following trauma such as traumatic brain injury (TBI) (Kurtzke and Beebe, 1980, Gallagher and Sanders, 1987, Chio et al., 1991, Williams et al., 1991, Chen et al., 2007, Schmidt et al., 2008, McKee et al., 2009, McKee et al., 2010, Schmidt et al., 2010, Lehman et al., 2012). ALS is a rapidly progressive, invariably fatal neurological disease characterized by the degeneration of motor neurons in the cerebral motor cortex and spinal cord. Over ninety percent of ALS cases have no clear risk factors and are considered sporadic, while the remaining cases result from genetic mutations and are called familial. Studies of American football players found that brain injuries sustained while playing this sport are associated with a variety of neurological impairment, both short and long-term. (Gessel et al., 2007, Broglio et al., 2009, Funk et al., 2012, Lehman et al., 2012). These findings suggest a possible role for TBI in the development of peripheral neurological pathologies, such as ALS. Additionally, a review of the epidemiological data from 1980 to 2010 concluded that the data currently available do not permit the conclusion that a single instance of head trauma is a risk factor or a cause for ALS (Armon and Nelson, 2012). The inability of research to reach a consensus on the ability of head injury to serve as a risk factor for ALS is possibly due to the heterogeneous nature of TBI (i.e. variance in the location of cortical injury) and the wide range of severity and numbers of repeat head injuries. No animal models of TBI, where head injury location and severity are controlled, have been performed to examine the mechanisms that might underlie the relationship between TBI and motor neuron disease.
TBI is caused by a sudden violent trauma that results in damage to the brain. Mechanical injury to the brain (primary injury) results in overt damage to brain tissue and blood vessels, resulting in cellular edema and disruption of the blood brain barrier (BBB). This is followed by a number of interrelated downstream events including oxidative damage and inflammation (Bains and Hall, 2012). Oxidative damage to lipids, proteins and nucleic acids is generated in response to oxygen radicals produced by mitochondrial and inflammatory pathways (Hall, 2011, Bains and Hall, 2012). In support of this, superoxide radical release has been shown to occur in brain microvasculature almost immediately following brain injury in a fluid percussion animal model of TBI (Kontos and Povlishock, 1986). An inflammatory response to TBI has also been documented within hours to days of injury and can last for many months (Roth et al., 2014). Inflammation is characterized by increases in activated microglial (microgliosis) and astrocyte infiltration (astrogliosis) to the site of injury. It is thought that elevated oxidative damage and inflammation act in a cyclic fashion and propagate secondary injury downstream of the site of TBI (Roth et al., 2014).
Secondary injuries following TBI can be complex and the downstream effects of TBI may contribute to the development of loss of motor coordination (Kamper et al., 2013, Yang et al., 2013a, Yang et al., 2013b, Yu et al., 2013). Although overt injury to the motor cortex, cerebellum, and/or vestibular apparatus can result in motor dysfunction, motor alterations are often present in patients after TBI in the absence of severe injury to these key brain regions. The direct effects of TBI on the brain have been examined; however, the downstream effects of TBI on peripheral motor neurons and the spinal cord are not fully understood (Nagamoto-Combs et al., 2007, Czeiter et al., 2008, Wu et al., 2011). Previous studies have suggested that moderate to severe brain injury can lead to motor dysfunction as a result of the dissemination of oxidative damage and/or inflammatory signals down the motor pathway (Czeiter et al., 2008, Wu et al., 2011, Bains and Hall, 2012). This suggests that some motor dysfunction after TBI may be the result of the propagation of injury to the spinal cord (McFadyen et al., 2009, Wu et al., 2011).
The goal of the present study was to determine the effect of TBI on motor neuron disease. We examined the effect of TBI on disease progression of the G93A mutant mouse model of ALS to uncover potential downstream effects of TBI that might potentiate disease initiation and progression. Gurney et. al. were the first to develop a mouse line that carried a human SOD1 gene that contained a mutation of Gly-93 to Ala (G93A) (Gurney et al., 1994). The G93A mutant transgenic mouse model of ALS is the most widely studied and well-characterized pre-clinical model of ALS. These mice develop severe paralysis and die at approximately 6 months of age. We tested the hypothesis that mild-TBI will affect the progression of disease in an animal model of ALS (G93A). Mild-TBI was given at 84 days of age, prior to disease onset, to investigate the ability of brain injury to potentiate disease course. Further, as part of this process, we have also generated the first comprehensive report on the pathological effects of mild-TBI on the lumbar spinal cord in a closed skull mouse model of mild-TBI in wild type C57Bl6 mice.
EXPERIMENTAL PROCEDURES
Animals
Mice used in the current study were housed in the Veterinary Medical Unit of the Audie L. Murphy VA Hospital in San Antonio Texas. Mice carrying the G93A mutation in SOD1 were originally generated by Gurney et. al. Founder breeder pairs that carried approximately 20 copies of the G93ASOD1 transgene and express mutant SOD1 approximately 10-fold as compared to the levels of endogenous SOD1, the G93AGur line [B6-Tg(SOD1-G93A)1Gur], were purchased from Jackson Laboratories (Jackson Laboratories, Bar Harbor, Maine). Mice were bred on a C57Bl6 background within our laboratory and a colony was established for use in this study. Female littermate G93A transgenic mice were subjected to traumatic brain injury or sham surgery and sacrificed at multiple time points for histological and biochemical analysis. Additional animals were monitored past onset of disease in order to collect behavioral and EMG data throughout the study. All of the procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio and the Audie L. Murphy Veterans Hospital.
Assessment of age of disease onset and life span
G93A mice were weighed once per week starting at the pre-symptomatic stage of the disease (84 days) in order to monitor disease course. The G93A mice develop complete paralysis at the end-stage of disease. Mice were examined daily after the onset of symptoms and end-stage was determined by the inability of the animal to right itself within 20 seconds when placed on its side. Mice that were unable to right themselves were euthanized immediately and recorded as dead for the purpose of life span studies. Onset of disease was considered the first loss of body weight that was not recovered by the next weighing for each individual animal. Within our G93A colony, disease onset is at approximately 100 days of age (wild type, n=13; G93A, n=12). Body weight was plotted as percent of the initial body weight at 84 days of age. In this way, body weight graphs represent the change in body weight as compared to body weight at day 84. Further, survival curves were plotted to determine changes in onset, utilizing the day of initial body weight loss as onset for each individual mouse.
Western Blot
Western Blot analysis was performed on cytosolic fractions from the ipsilateral (impacted) hemisphere for wild type/Sham, wild type/TBI, G93A/Sham and G93A/TBI groups at 24 and 72 hours following injury, n=4. Equivalent amounts of protein from each sample were analyzed by sodium dodecyl sulfate-polyacrylamide electrophoresis using 4–12% Bis–Tris precast gels (Invitrogen, California, USA) under reducing conditions (1 hour, room temperature (RT)) and electro-blotted onto a nitrocellulose membrane (18 hours, overnight). After a blocking step (0.1% Tween-20/5% nonfat milk in PBS, 1 hour, RT), membranes were incubated with primary antibodies overnight (12–14 hours, 4°C) with gentle agitation. The following primary antibodies were used (1:1000): rabbit polyclonal to GFAP (Abcam, ab7260), rabbit polyclonal to Iba1 (WAKO), mouse polyclonal to beta Actin (Cell Signaling, 4967). Membranes were washed, incubated with secondary antibody (RT, 1 hour, Cell Signaling) and developed with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). Densitometry results were collected using ImageJ (NIH) and analyzed by multiple Students t-tests where p < 0.05 was considered statistically significant.
Closed Skull Impact Model
Mild traumatic brain injury (TBI) was induced at 84 days of age with the TBI 0310 impact device (Precision Systems LLC) as we have previously described and validated using MRI and histology.(Evans et al., 2014) TBI was administered as a closed cortical injury using pneumatic force. Prior to surgery mice were anesthetized in a chamber using 2–4% isoflurane in 100% oxygen and anesthesia was maintained at 1% for the remaining procedures. The mice were fixed to a pad in the prone position under a heating lamp to maintain body temperature and a midline incision in the scalp was made and the skin and periosteum retracted. A stainless steel disc (7mm in diameter and 3mm thick) was glued to the skull between the coronal and lambdoid sutures over the somatosensory cortex using super glue. TBI was induced using the TBI 0310 impact device calibrated to deliver a blow at 4.5 m/s, 100ms dwell time and a depth of 2mm directly to the disc. Following injury, the disc and glue were removed and the incision sutured. Antibiotic ointment was applied to wounds. Animals were allowed to wake in a warm/dry cage with a sterile liner and monitored for at least 1 hour. Sham animals were subjected to all procedures except that the impact device was calibrated to a level just above the disk resulting in no impact. All animals were observed and weighed weekly until completion of experimentation.
Rotarod
Neuromuscular function was tested using Rotamex 4/8 (Columbus Instruments, Columbus, OH). Each mouse was trained prior to sham or TBI treatment for five consecutive days (six trials/day) where the speed of the rotor was accelerated from 4 to 40rpm with an acceleration of 0.2 rpm/sec. Twenty-four hours after the last training session, the mice were tested in a probe trial consisting of six trials as previously described. The average latency to fall was then recorded. This trial was used as the baseline trial and was performed the day before brain injury or sham surgery. Mice were tested following sham or TBI treatment at 1, 7, and 30 days.
Grip Strength
Hindlimb muscle strength was determined by measuring peak force (in pounds) using the Digital Grip Strength meter equipped with a Hind Limb Pull Bar Assembly (Columbus Instruments, Columbus, OH). Mice were allowed to grip the metal grids with their forepaws, and gently pulled backwards by the tail until they could no longer hold the grids. The grip force was observed over 10 trials and the maximum force was recorded starting at 3 days post injury to avoid confounding other behavioral tests (Martin et al., 2009).
Electromyography (EMG)
Bilateral hind limb muscle groups were examined with a Nicolet Viking Quest portable EMG apparatus (CareFusion San Diego, CA. USA). A monopolar electrode (active) was inserted into the muscle of interest. An identical electrode (reference) was inserted subcutaneously into the lateral and distal-most tendinous portion of the gastroc-soleus complex, ipsilateral to the muscle studied. A subdermal needle (ground) was inserted subcutaneously into tendinous tissue posterior to and near the reference electrode. Abnormal spontaneous activity in the form of denervation potentials (positive sharp waves and fibrillations) were recorded as a score of denervation potentials per muscle group per limb in each animal at 1, 7, and 30 days post injury, as described previously (Evans et al., 2014). Notably, EMG was performed after behavioral testing at each time point in order to avoid complications.
Real-Time PCR
Total RNA was isolated from gastrocnemius muscle using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA yield and purity were determined by measuring the absorbance at 260 and 280 nm. First-strand cDNA was synthesized from 1 μg of total RNA using iScript™ cDNA synthesis kit (BioRad, CA, USA). 2.5 nanogram of cDNA samples was PCR amplified using primers for AChR alpha, Runx1, MyoD, GADD45a or 18S rRNA and iQ™ SYBR green supermix (BioRad, CA, USA). Real-time PCR was performed using the LightCycler Real–Time PCR instrument. Relative mRNA expression was calculated by the ΔΔCt method.
Nissl Staining
Standard procedures were used for detection of Nissl bodies found within neurons as has been previously described (Talley Watts et al., 2013). Briefly, brains were harvested, sectioned at 20um and placed on plus slides. The slides were dried at 37°C overnight. Slides were hydrated with distilled water, 0.1% cresyl violet was applied for 7 minutes, followed by a wash in distilled water. The slides were then hydrated, cleared in xylene and cover slipped. Images were acquired on a Nikon Eclipse TE2000-U microscope (Nikon Inc.). Cell area was quantified using the ImageJ analyze particle function. This function allows for the exclusion of particles below 100 pixels, resulting in the collection of data from only those particles from cell somas. Soma size was determined in the ipsilateral cortex of 5 cryosections taken from the area of impact. The spinal cord sections were taken from the lumbar spinal cord 24 hours post injury or sham surgery and the soma size was determined in the contralateral lumbar spinal cord to assess the changes in the motor track associated with the ipsilateral brain trauma.
Isoprostane Measurements
Briefly, oxidative damage to lipids was measured as the level of F2-isoprostanes in the intact spinal cord 24 hours post injury or sham surgery using the gas chromatography–mass spectrometry (GC/MS) method of Morrow and Roberts (Morrow and Roberts, 1999) as previously described (Ward et al., 2005).
Statistics
One-way ANOVA was used to compare the differences among three or more groups followed by multiple t-tests corrected for multiple comparisons using the Holm-Sidak method. Student’s t-test was used to compare the difference between two groups. The significance level was set at p<0.05. Data are presented as mean ± standard error of the mean (SEM). The Log-rank Mantel Cox test was used to compare differences in survival and rotarod curves. GraphPad Prism software (GraphPad Software Inc.) was used to perform statistical analyses.
RESULTS
Disease course in G93A mice following mild TBI
The interaction between injury and time was significant in the wild type mice [F(7,161) = 2.881, p=0.0073, Figure 1A]. TBI causes a significant decrease in body weight in wild type mice at 87 days, which was recovered one week post injury [87 days *p=0.0038, Multiple t tests, Figure 1A]. The interaction between injury and time was also significant in G93A [F(13,233) = 8.206, p<0.0001], Figure 1B. Loss of body weight after disease onset is an established marker of ALS disease progression in the G93A mouse model. The loss of body weight in G93A following mild-TBI was not significantly different from Sham/G93A mice, although there appeared to be a trend toward reduced body weight following injury [87 days p=0.0001, 94, 107, 161 days p<0.05 Multiple t-tests, Figure 1B]. Body weight was significantly lower in TBI/G93A mice at the end stage of disease (after 160 days). Changes in body weight were used to determine disease onset in the G93A mice. Onset was determined by recording the age of the first sign of body weight loss that was not recovered. When the percent of the population at onset across time was compared between groups there were no significant differences in onset as a result of brain injury (Figure 1C). Further, there was no significant difference in median or maximum lifespan between TBI and Sham G93A groups (Figure 1D). These data indicate no major effects of mild-TBI on the initiation or duration of the ALS disease course.
Figure 1. Effect of TBI on body weight, onset and survival in wild type mice and in a mouse model of ALS (G93A).
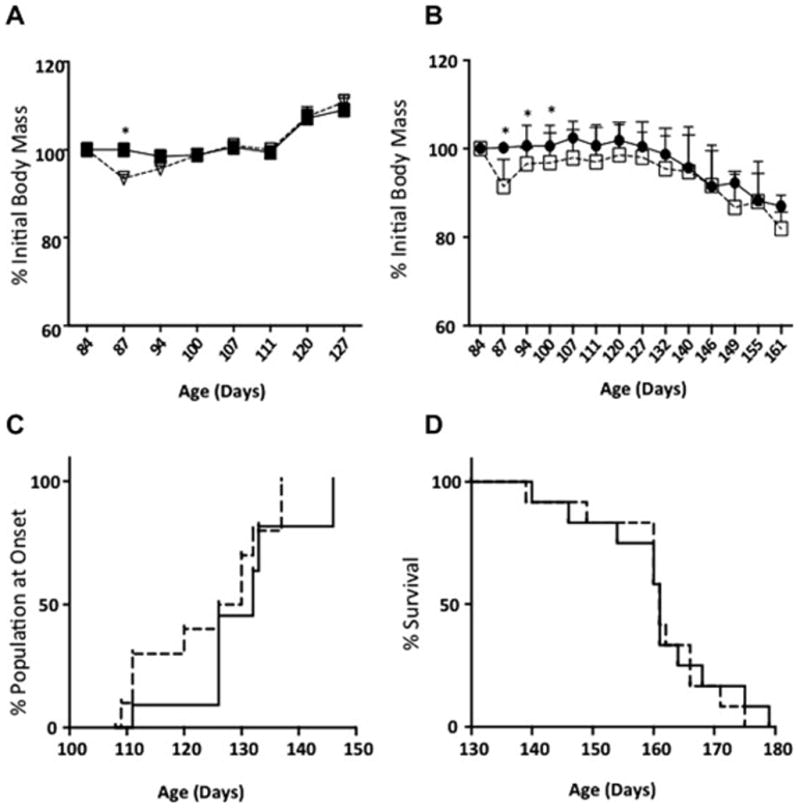
A. Body weight changes after TBI in wild-type mice. Sham group indicated by solid squares TBI group indicated by open triangles. B. Body weight changes in G93A transgenic mice after TBI. Sham group indicated by solid circles and TBI group indicated by open squares. C. Percent of G93A population at onset of disease as determined by the day of the first sign of body weight loss. There was no significant difference in the percent of the population at onset between sham (solid line) and TBI (dashed line) G93A mice. D. Survival analysis shows no effect of mild-TBI on median or maximum lifespan in G93A mice. Sham (solid line) and TBI (dashed line). Significance was measured using one way ANOVA followed by multiple t-tests corrected for multiple comparisons using the Holm-Sidak method. * denotes a statistical difference between groups, p<0.05.
Motor function deficits are found in G93A and wild type mice following mild-TBI
In order to assess the effect of mild-TBI on the progression of motor deficits in the G93A mouse model of ALS, we utilized three well-established methods: rotarod, grip strength, and electromyography (EMG). Our results demonstrate that rotarod performance, an assay of motor coordination in rodents (Hamm et al., 1994), was reduced 1 day post mild-TBI in wild type mice compared to sham mice [Log-Rank Mantel Cox Test preformed; 1 day wild-type/Sham vs. wild-type/TBI, p=0.0292, G93A/Sham vs. G93A/TBI, p=0.6964, Figure 2A]. We determined this reduction by representing the data on survival curves as the percent of the population still remaining on the rotating rod (y-axis) across time in seconds (x-axis). In this way, we could determine the relative difference between groups by comparing the survival curves. The latency to fall was reduced for the wild type population significantly at 7 days following injury [Log-Rank Mantel Cox test, wild type/Sham vs. wild type/TBI p=0.0302, G93A/Sham vs. G93A/TBI p=0.7, Figure 2B]. Further, at this time point we found a significant reduction in performance in G93A/sham mice as compared to wild-type/Sham mice [Log-Rank Mantel Cox test, wild-type/Sham vs. G93A/Sham p=0.0049, Figure 2B]. At 30 days post mild-TBI G93A mice still tend to show a trend toward impairment in rotarod performance when compared to G93A/sham mice [Log-Rank Mantel Cox test, wild-type/Sham vs. wild-type/TBI p=0.4490, G93A/Sham vs. G93A/TBI p=0.05, wild type/Sham vs. G93A/Sham p=0.05, Figure 2C]. Rotarod deficits develop later in G93A mice, presenting at 30 days the last time point tested.
Figure 2. Motor function deficits are evident in G93A and wild-type mice following mild-TBI.

Time points represent days post TBI (1 day, 7 days and 30 days) in G93A and wild type-mice (n=wild-type/Sham (11), wild-type/TBI (12), G93A/Sham (7), G93A/TBI (7)). Data are expressed as the percent of each group to remain on the rotating rod (y-axis) at a given time point in seconds (x-axis). Results were analyzed using the Log-Rank Mantel Cox Test. A. 1 day after mild-TBI, the average latency to fall was significantly reduced as a result of TBI in wild type mice, p=0.0292. G93A mice did not show significantly reduced performance compared to wild type mice at this time point, p>0.05. B. At 7 days, the amount of time spent on the rotating rod (latency) remained lower in wild-type mice after TBI as compared to sham (p=0.0302). Additionally, G93A/Sham mice performed for a shorter time as compared to wild-type/Sham mice (p=0.0049). C. At 30 days post injury the latency to fall was reduced in G93A/Sham versus G93A/TBI mice (p=0.0049). This deficit is not seen in wild-type Sham as compared to TBI mice.
To further assess the effects of mild-TBI on motor function, grip strength was tested at four time-points (3, 7, 14, and 30 days) post mild-TBI. Grip strength is a measure of muscle strength and is commonly used to assess G93A disease course. As expected, there were consistent and significant reductions in grip strength in G93A mice compared to wild type mice at 3, 7, 14, and 30 days. There was no effect of mild-TBI on grip strength in wild type mice. However, at 14 and 30 days post injury there were significant reductions in the G93A/TBI group compared to the G93A/Sham group [Two Way ANOVA, Time Variation, F(3, 148) = 5.406, p<0.01, Multiple t-tests G93A/Sham vs. G93A/TBI, t=5.661, +p<0.05; wild type/Sham vs. G93A/Sham, t=0.0088, *p<0.01; n=13–17, Figure 3].
Figure 3. The effect of TBI on grip strength in wild-type and G93A mice.
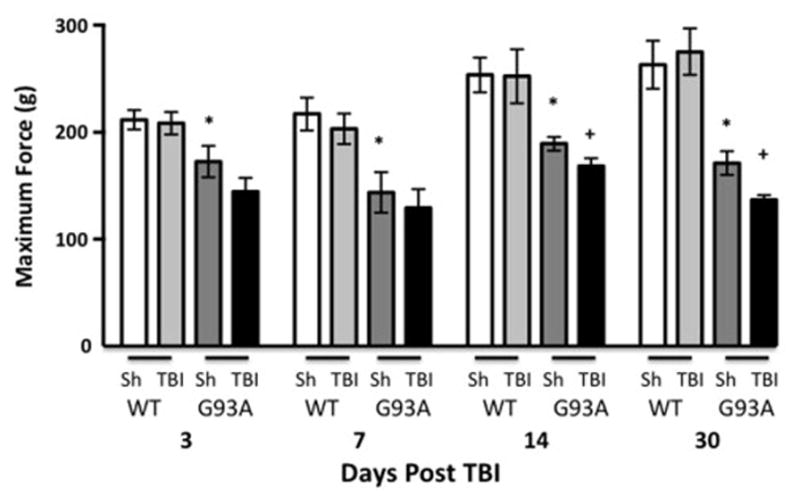
Grip strength was measured at four time points following TBI in wild-type and G93A mice (n=13 and 17). Results were analyzed using Two Way repeated measures ANOVA followed by multiple t-tests corrected for multiple comparisons using the Holm-Sidak method and revealed a difference among groups as a variation of time, p=0.0015. *denotes a statistical difference between wild-type/Sham and G93A/Sham, p<0.05), +denotes a statistical difference between G93A/Sham and G93A/TBI mice, (p<0.05). G93A/TBI mice have a significantly reduced grip strength at 1 month following injury as compared to G93A/Sham. Sh=Sham, WT=wild-type
Electromyography (EMG) was utilized to investigate the role of mild-TBI on muscle innervation in our animal model. EMG is used clinically to diagnose peripheral nerve injury and motor neuron diseases, such as ALS. Abnormal EMG findings were compiled as a score of denervation potentials per muscle group per limb in each animal as described previously (Evans et al., 2014). Linear regression showed a significant difference between the curves of EMG abnormalities for each treatment group [Linear Regression, p<0.001, F87.08 (6,69) n=5–7, Figure 4]. In wild-type mice, EMG abnormalities were evident as early as 1 day after injury and remained significantly increased 30 days post mild-TBI. In G93A mice, the EMG abnormalities recorded at 1 and 7 days post injury were increased compared to wild-type mice and higher in G93A/TBI mice than in G93A/sham mice. At 30 days post injury G93A/Sham and G93A/TBI have equal levels of EMG abnormality present [One-way ANOVA for Sham vs. TBI; wild-type p<0.0001, Bonferroni’s Post Hoc Test Sham vs. TBI; wild-type: 7 days p=0.06, 30 days p=0.007, One-way ANOVA for G93A/Sham vs. G93A/TBI p<0.0001, Bonferroni’s Post Hoc Test Sham vs. TBI; G93A: 1 day p=0.006, 7 days p=0.001. n=5–7, Figure 4].
Figure 4. Electromyography changes as a result of TBI.
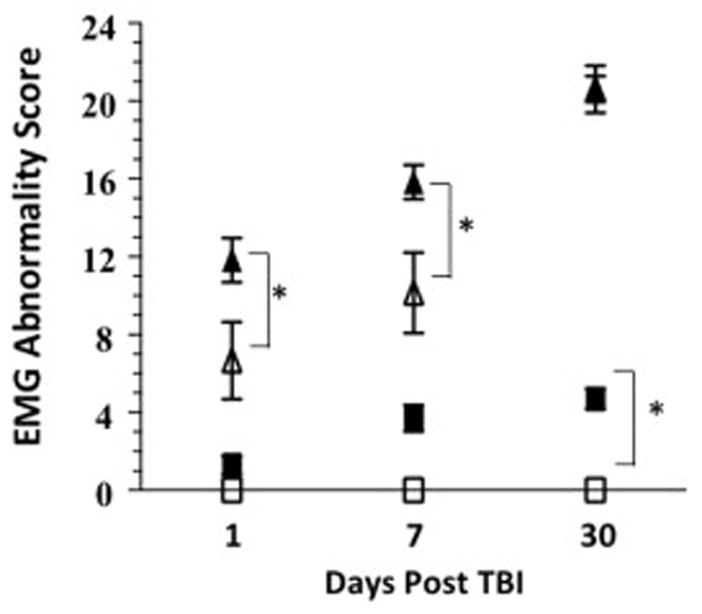
EMG abnormality score is significantly increased following TBI in wild-type and G93A mice. When compared to the wild-type cohort, G93A mice have a significantly higher EMG abnormality score at each time point tested. n=5 (wild-type) and 7 (G93A) closed triangle=G93A/TBI, open triangle=G93A/Sham, closed square=wild-type/TBI, open square=wild-type/Sham
Denervation increases mRNA levels of several genes as an adaptive response. We investigated, in the gastrocnemius muscle, AChR mRNA (Jang and Van Remmen, 2011), as well as the levels of other denervation markers Runx1 (Wang et al., 2005), MyoD (Ishido et al., 2004) and Gadd45a (Bongers et al., 2013). One month after TBI compared with WT sham mice, G93A sham mice showed a several fold increase in AChR alpha, Runx1, MyoD and GADD45a mRNA levels (Figure 5 A, B, C, D) indicating denervation in the gastrocnemius muscle of these mice. Mice from the G93A/TBI group showed further increases in these denervation markers which was significant with AChR and Runx1 mRNA levels. Although MyoD and GADD45a mRNA levels were increased, it did not reach significance. MyHC remained unchanged (Figure 5 E).
Figure 5. TBI increased markers of denervation in G93A mice.
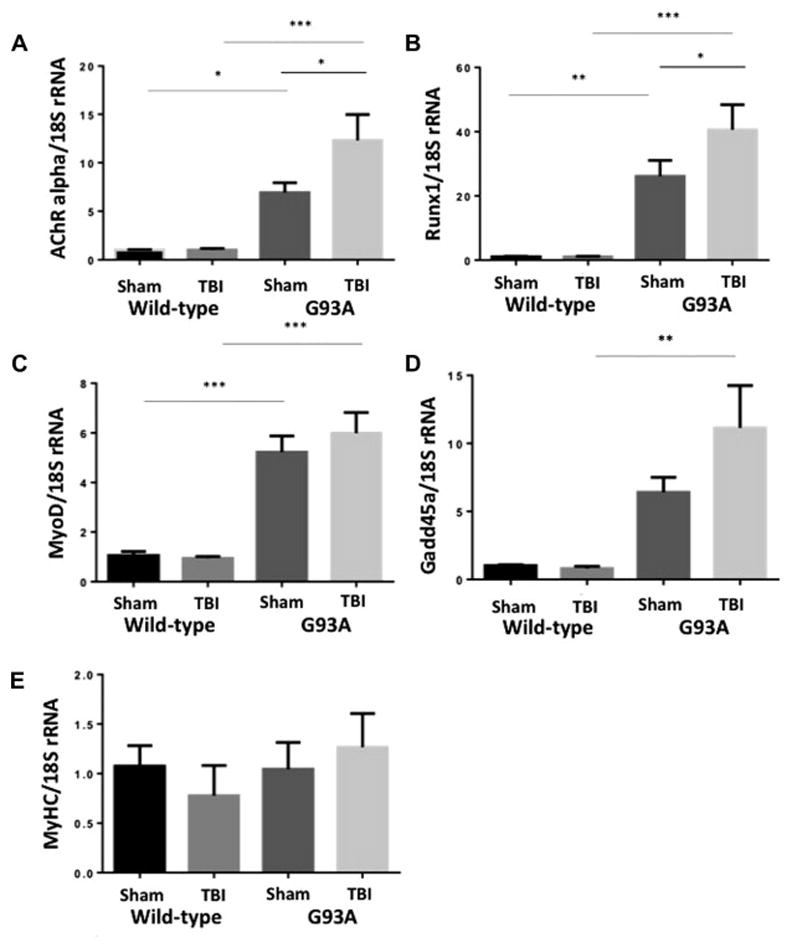
Total RNA was prepared and used for real time RT-PCR to detect A. AChR alpha, B. Runx1, C. MyoD D. GADD45a and E. MyHC mRNA as described in the methods section. Mean ± SEM of n=5 mice/group is shown. *p<0.05, **p<0.01, ***p<0.001 vs WT sham, Tg sham, WT TBI, Tg TBI.
Increased edema, astrogliosis, and microgliosis in brain after mild-TBI in wild type and G93A mice
In order to assess the effect of mild-TBI in the G93A mouse model of ALS, brain sections were prepared 24 hours following injury to measure histological changes. An increase in cellular edema was seen in the ipsilateral cortex of both wild type and G93A mice following TBI, as indicated by increases in cell soma size (Figure 6 B). There were no significant differences in cell soma size in the cortex of G93A mice compared to wild type mice (Figure 6 B).
Figure 6. Increased edema in cortex 24 hours after mild-TBI in wild-type and G93A mice.
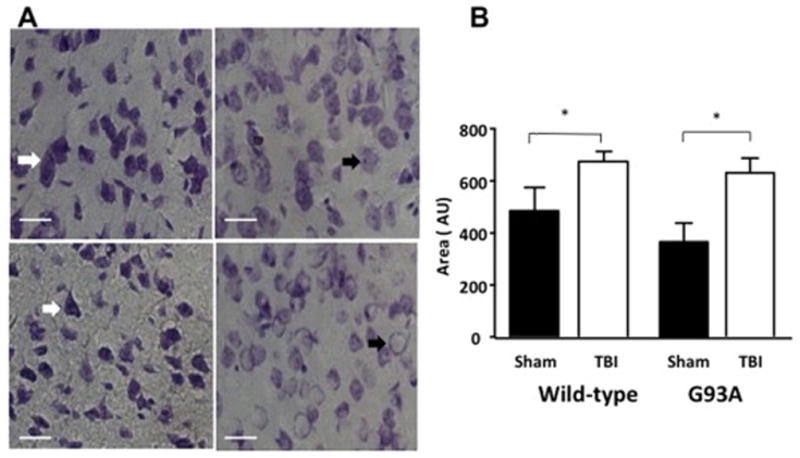
A. Images of Nissl stained wild-type and G93A cortex 24 hours following sham or TBI treatment. Images (40X) show an increase in the presence of neuronal edema in G93A/TBI and wild-type/TBI 24 hours following injury as compared to Sham. The white arrow indicates non-injured neurons (nucleolus intact with regular distribution of Nissl bodies throughout cytoplasm) the black arrow indicates edema. Graph of cell area indicating an increase in cell soma size 24 hours after TBI in B. wild type mice and C. G93A mice. n=3, Student’s t-test was performed *p<0.05, **p<0.001.
To determine the level of astrogliosis in the cortex, glial fibrillary acidic protein (GFAP) was used as a marker for activated astrocytes (Brahmachari et al., 2006). Western blot analysis showed no significant increase in ipsilateral hemisphere total GFAP levels at 24 hours as a result of mild-TBI but there was an increase in GFAP as a result of G93A mouse pathology (Figure 7 A, TTest, p<0.05). At 72 hours following injury a significant increase in GFAP in the ipsilateral hemisphere was seen in only in wild type mice following mild-TBI (Figure 7 B, TTest, p<0.05).
Figure 7. Astrogliosis in the injured cortex after mild-TBI.
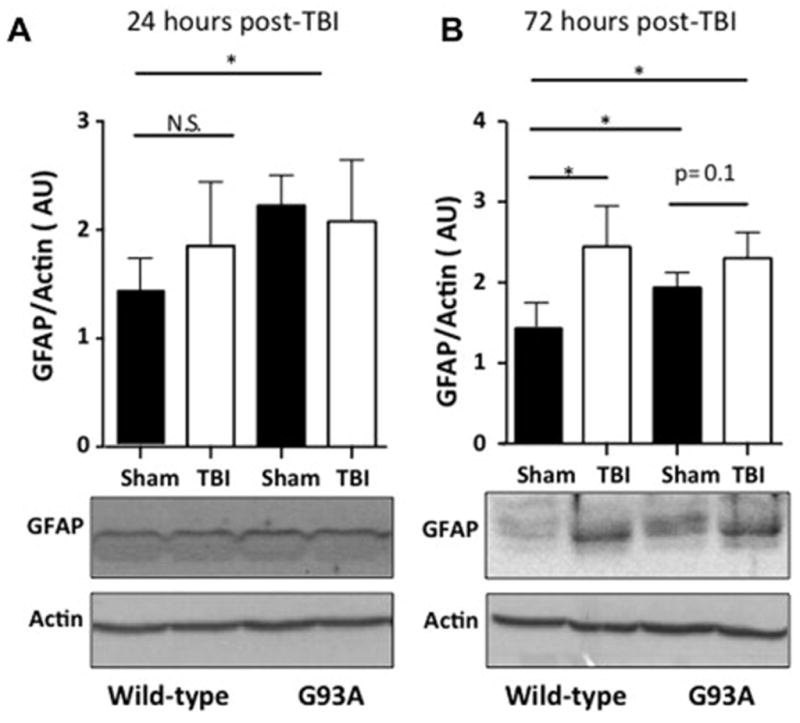
A. Western blot analysis reveals no significant increase in total GFAP levels at 24 hours as a result of mild-TBI but a clear increase in GFAP as a result of G93A mouse pathology. B. A significant increase as a result of mild-TBI is seen by western blot analysis at 72 hours post injury in wild-type mice but not in G93A mice. Western blot analysis N=4, Student’s t-test was performed *p<0.05.
To measure the activation of microgliosis, in the ipsilateral cortex we utilized a common marker for microglia, Iba1. Western blot analysis showed a significant increase in total ipsilateral hemisphere Iba1 levels at 24 hours as a result of mild-TBI in wild type mice with a trend toward an increase in G93A/TBI mice was not significant (Figure 8 A, TTest, p<0.05). By 72 hours post injury, Iba1 increased to the same extent in both wild type and G93A mice (Figure 8 B, TTest, p<0.05).
Figure 8. Microgliosis in the injured cortex following TBI.
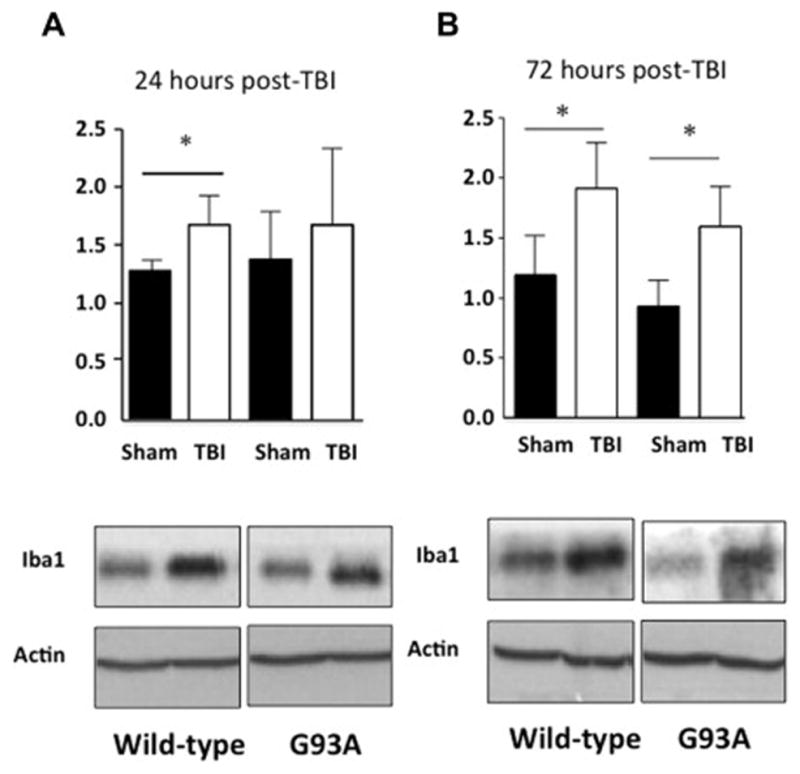
A. Western blot analysis a significant increase in total Iba1 levels at 24 hours as a result of mild-TBI in wild-type mice B. This increase becomes significant by western blot analysis in G93A mice as well as wild-type mice at 72 hours post injury. Western blot analysis n=4, p values represent result of Students t-test where *p<0.05
Increased lumbar spinal cord edema, astrogliosis, and microgliosis after mild-TBI
To determine if mild-TBI also resulted in changes in the spinal cord we examined lumbar spinal cord at 24 hours post injury. Our results show a significant increase in cell soma size 24 hours after mild-TBI in wild type mice as compared to sham (Figure 9 B, TTest, p<0.05). Cell soma size was also increased in the lumbar spinal cord of G93A mice following brain injury as compared to sham (Figure 9 B, TTest, p<0.05). Interestingly, we found a reduction in cell soma size of sham G93A mice when compared to sham wild type mice.
Figure 9. Increased neuronal edema in lumbar spinal cord 24 hours after mild-TBI.
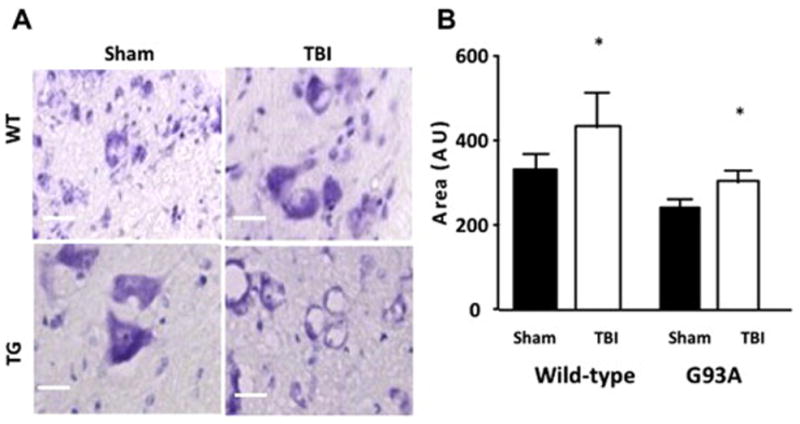
A. Images of Nissl stained wild-type and G93A lumbar spinal cord ventral horn 24 hours following sham or TBI treatment. 20X magnification shows an increase in the presence of neuronal edema in G93A/Sham, G93A/TBI and wild-type/TBI 24 hours following injury or sham surgery. B. Graph of cell area indicating an increase in cellular soma size at 24 hours after TBI in wild-type mice and in G93A mice 24 hours following TBI. n=3, Student’s t-test was performed and p-value reported. A comparison between wild-type/Sham and G93A/Sham reveals a significant difference p<0.01.
To investigate the effect of mild-TBI on inflammation in the lumbar spinal cord we investigated microgliosis and astrogliosis utilizing Iba1 as a marker for microglia and GFAP as a marker for reactive astrocytes, respectively. Western blot analysis showed a significant increase of GFAP in wild-type mice after mild-TBI at 24 and 72 hours. Levels of GFAP in spinal cord from G93A mice are elevated even in sham G93A and show no further as a result of mild-TBI (Figure 10 A). The pattern of GFAP expression is similar at 72 hours post injury (Figure 10 B). Western blot analysis of total Iba1 levels revealed an increase at 24 hours as a result of mild-TBI in wild type mice, but no further increase in G93A mice above that resulting from ALS disease progression (Figure 11 A). At 72 hours post injury, both wild type and G93A mice show elevated levels of Iba1 in spinal cord (Figure 11 B).
Figure 10. Elevated levels of GFAP indicate increased astrogliosis in the spinal cord following mild-TBI.
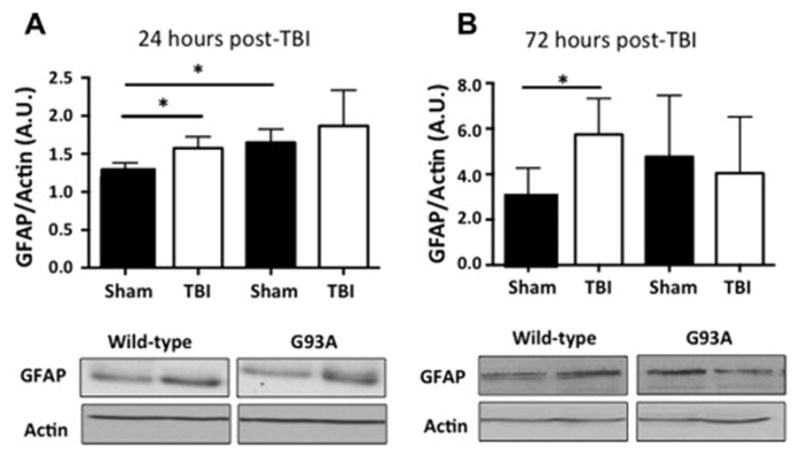
A. Western blot analysis of GFAP indicates a significant increase in wild type mice as a result of mTBI and an increase in G93A mice as a result of disease progression. B. This increase is still present in wild-type mice at 72 hours post injury.
Figure 11. Elevated levels of Iba1 indicate increased microgliosis in the spinal cord following mild-TBI.

A. Western blot analysis reveals a significant increase in total Iba1 levels at 24 hours as a result of mild-TBI in wild-type mice. B. Significantly increased Iba1 in G93A mice by western blot analysis at 72 hours post injury. n=4, Significance determined by Student’s t-test *p<0.05
Increased oxidative stress in the spinal cord after brain injury
To assess increases in oxidative stress total F2-isoprostane levels were measured in whole spinal cords 24 hours after mild-TBI. As shown in Figure 10, we found a significant increase in F2-isoprostanes in spinal cord from wild type/TBI mice as compared to wild type/Sham mice. Further, there was an increase in F2-isoprostanes in spinal cord from G93A/Sham mice compared to wild type/Sham (Figure 12).
Figure 12. Elevated oxidative damage in the spinal cord following mild-TBI.

Total F2-Isoprostanes were measured in whole spinal cords 24 hours after mild-TBI and a significant increase was seen in wild-type/TBI mice as compared to wild-type/Sham. Further, there is an increase in G93A/Sham mice as compared to wild-type/Sham. n=4, Significance determined by Student’s t-test *p<0.05
DISCUSSION
Our study investigated the effects of mild-TBI on the motor pathway. We sought to determine whether mild-TBI could induce spinal cord and peripheral nerve pathology as well as motor deficits in wild-type mice and potentiate the neurodegenerative phenotype associated with ALS in the G93A mutant mouse model. Following mild-TBI, disease course, as indicated by the progressive loss of motor function, was exacerbated in G93A mice. Lifespan was not affected by mild-TBI. We also report increases in astrogliosis, microgliosis and oxidative stress in the lumbar spinal cord within days of mild-TBI in wild-type mice, suggesting that oxidative stress and inflammation could be potential mechanisms resulting in the initiation of motor deficits. This is the first report of peripheral motor neuron pathology in response to mild-TBI.
We found that mild-TBI results in a moderate loss of body weight in the initial week following injury in wild-type mice, but has minimal effects on the rate of body weight loss resulting from disease progression in the G93A mouse model. The initial loss of body weight after TBI in wild type mice has been reported previously (Evans et al., 2014) and could be caused by decreased food and water intake as a result of nausea, and/or dizziness experienced after mild TBI (O’Neil et al., 2013). G93A mice characteristically lose body weight after disease onset as a result of reduced food intake and wasting of muscle tissue in the later stages of disease progression (Gurney et al., 1994). The slight exacerbation of disease related weight loss following mild-TBI in the G93A mice could be a result of the short-term effects of the brain injury, as seen in wild type mice. However, additional findings of our study support the presence of long-term repercussions of TBI in this model, therefore we are unable to rule-out the possibility of a potentiation of muscle wasting in G93A mice following mild-TBI. Long-term studies are warranted to determine if this trend in weight loss across the lifespan is valid, which tissues might be affected, and the underlying mechanisms.
Problems with motor coordination occur in the acute phase after mild-TBI in humans and animal models (Guskiewicz, 2003, Slobounov et al., 2008, Yang et al., 2013b). In the closed skull weight drop murine models of mild TBI, changes in motor function and behavior last from 72 hours to one week post injury depending on the study (Yang et al., 2013b) (Flierl et al., 2009). In our closed skull model of mild-TBI there are induced deficits in the rotarod task for balance and motor coordination in wild type mice at these early time points, as well as further reduced performance in G93A mice as far as 30 days after injury. At 1 and 7 days post mild-TBI, wild-type/TBI and G93A/Sham mice performed similarly on the rotarod task, which suggests that the detrimental effects on motor function, due to the G93ASOD1 mutation, are comparable to that of mildTBI in a wild type mouse. By 30 days post injury this deficit is no longer detected in wild-type mice.
To investigate the ability of our mild-TBI model to cause muscle weakness we tested the effect of mild-TBI on overt muscle strength using grip strength. Mild-TBI caused reductions in grip strength in G93A mice beginning 14 days after injury, which were not seen at any time point tested in wild-type mice after mild-TBI. The lack of an effect of mild-TBI on grip strength in wild-type mice can be explained by the mild nature of the injury. G93A mice experience muscle wasting as a symptom of disease progression. Therefore, reduced grip strength in G93A mice after brain injury suggests that the trend toward reduced body weight seen in this cohort could be a result, at least in part, of a potentiated loss of muscle mass and integrity. In a retrospective study of patients with persistent muscle weakness following severe TBI, injury to the cortical spinal tract (CST), most commonly in the form of diffuse axonal injury as identified by diffusion tensor imaging, was suggested as a potential cause of prolonged muscle weakness. (Katz et al., 1998, Choi et al., 2012) This suggests that reduced muscle strength in the G93A mouse model could be due to an increased susceptibility to injury of the CST and that mild-TBI is not sufficient to induce comparable CST injuries in wild-type mice. Future pre-clinical studies utilizing imaging technologies to investigate the effects of mild-TBI on the CST in this model could shed light on the temporal progression of injury along the CST.
To investigate the effect of mild-TBI on motor unit integrity we utilized electromyography (EMG), a commonly used clinical tool for ALS diagnosis. We found that mild-TBI results in acute and prolonged presence of EMG abnormalities (positive sharp waves and fibrillations) in wild type/TBI mice from 1 to 30 days post injury. Additionally, mild-TBI resulted in increased EMG abnormalities in G93A mice at 1 and 7 days post injury indicating a possible exacerbated loss of motor neuron innervation at the muscle. This is the only study, to report the presence and persistence of EMG abnormalities, as a result of mild-TBI in a mouse. This phenomenon was described in humans in a study by Spaans et al., that examined 10 subjects with severe acquired brain injuries and reported the presence of fibrillation potentials and positive sharp waves, in upper and lower extremity muscles (Spaans and Wilts, 1982). This study speculated that EMG abnormalities found in the muscle after brain injury were due to a lesion to the central nervous system, which resulted in a dying back process of lower motor neurons (trans-synaptic degeneration), stating that the spontaneous activity appeared within 2–3 weeks of injury and was diminished considerably by 6 months (Spaans and Wilts, 1982). However, findings of this study were confounded by the heterogeneity of TBI in their participants and the possibility that peripheral (nerve) injuries had also occurred. This pattern is comparable to what was found in the wild type mice subjected to mild-TBI in this study. In contrast, our model allows us to reproduce the intensity and location of the TBI and control for peripheral nerve/extremity injuries that might co-occur during traumatic incidences in humans. We also investigated the effects of our mild-TBI on nerve conduction velocity (NCV, data not shown) and found that there were no effects of mild-TBI on motor NCV in either wild type or G93A mice. Indicating that the identified deficits were not due to myelin pathology.
We suggest that mild-TBI results in lesions to the central nervous system that result in the dysfunction or loss of motor neurons within the anterior horn of the spinal cord. This may lead to axonal degeneration, possibly via the dying back process. One limitation of our EMG approach is the inability to estimate motor unit number and function. Furthermore, EMG grossly examines the peripheral neuromuscular junction and although the more distal portion of the nerve innervates more distal muscles, EMG does not indicate the precise location where peripheral nerve changes or pathology may be occurring. This question would be better addressed using axonal tracing techniques.
To further investigate the effect of mild TBI on neuromuscular junction integrity we examined mRNA levels of several genes that have been shown to have an adaptive response to denervation, one month after mild-TBI in the gastrocnemius muscle. We have shown previously, that mice lacking SOD1 showed significant increase in AChR alpha mRNA levels indicating the denervation process (Jang and Van Remmen, 2011). Along with AChR mRNA, we investigated the levels of Runx1 (Wang et al., 2005), MyoD (Ishido et al., 2004) and Gadd45a (Bongers et al., 2013) and found that compared with wild-type/Sham mice, G93A/Sham mice showed several fold increases in AChR alpha, Runx1, MyoD and GADD45a mRNA levels (Figure 5 A, B, C, D) as would be expected. G93A mice who experienced a mild TBI showed further increases in these denervation markers, which were significant with AChR and Runx1 mRNA levels. Although MyoD and GADD45a mRNA levels were increased, they did not reach significance. These data clearly indicate that 1.) Mild-TBI in wild type mice does not result in sustained denervation at 30 days and 2.) G93A mice have sustained denervation as seen in elevation of AChR alpha and Runx1 mRNA and mild-TBI further aggravates this denervation by potentiating increases in AChR alpha and Runx1 mRNA (Fig 5). Taken together these data support that as far as 1 month following mild TBI, brain injury is sufficient to potentiate detrimental phenotypes in the neuromuscular junction integrity in G93A mice.
In line with previous work, we have shown that there is a consistent increase in cellular edema and a subsequent up-regulation of astrogliosis and microgliosis in the brain of wild type and G93A mice as a result of our closed skull model of mild-TBI (Davis, 2000). The magnitude of this increase was not significantly different in the wild type and G93A mice after injury. These results indicate that the severity of primary and secondary injury within the cortex in G93A mice is similar to that of wild-type mice.
We therefore decided to investigate the lumbar spinal cord to determine if the differential effects of mild-TBI on motor coordination and integrity could be the result of downstream injury. We found a significant reduction in cellular soma size within the contralateral lumbar spinal cord of sham G93A mice as compared to wild type mice. One explanation of these results may be that early disruptions in the blood-spinal cord barrier in rodent models of ALS carrying the G93A SOD1 mutation lead to early motor-neuron dysfunction and loss (Dal Canto and Gurney, 1994, Winkler et al., 2014). Increases in cellular area in TBI groups as compared to sham groups for both wild-type and G93A mice were found to be of similar magnitude. The results of these studies indicate a rapid induction of astrogliosis and microgliosis indicated by increased GFAP and Iba1 expression in the lumbar spinal cord. An increase in astrogliosis was seen following mild-TBI in wild type mice as compared to Sham, 72 hours following injury. We also showed that G93A mice have increased microgliosis when compared to wild type mice and that mild-TBI increased these levels in both groups. These results indicate that secondary injury mechanisms following mild-TBI (astrogliosis and microgliosis) are increased in the lumbar spinal cord as early as 24 hours post mild brain injury and we hypothesize that this increase is in part responsible for the motor dysfunction phenotypes we have shown in our model. Our data show that mild-TBI is capable of causing a disruption in cellular homeostasis in the lumbar spinal cord as a potential mechanism for the acute motor coordination symptoms experienced by mild-TBI patients. It is possible that the force generated by the mTBI could be propagated across the body axis, resulting in a propagation of inflammatory and excitotoxic signals along the motor axis. However, in our model the mouse is fixed in a prone position that limits movement making a secondary spinal injury from a mild impact less likely
A key mechanism of ALS disease progression is increased oxidative damage in the lumbar spinal cord (Gurney et al., 1996, Hall et al., 1998). F2-isoprostanes are a well-established marker of oxidative stress and ALS disease progression. They are often used to assess the level of disease pathology in the G93A murine models of ALS (Milatovic et al., 2011). Furthermore, levels of isoprostanes are elevated in the CSF of human pediatric and adult TBI patients as well as in the brain of severe adult TBI patients, as determined by cerebral microdialysis (Bayir et al., 2002, Bayir et al., 2004, Clausen et al., 2012). We found that F2-isoprostanes are significantly increased at the level of the spinal cord following TBI in wild type mice to a level comparable to that of sham G93A mice. This indicates that mild-TBI results in a rapid increase in oxidative stress in the lumbar spinal cord of wild type mice to levels similar to that of G93A transgenic mice at 85 days of age. The role of secondary injury mechanisms in the spinal cord in the progression of motor pathology following mild-TBI warrants further investigation. No further increase in F2-isoprostanes within the spinal cord of G93A mice were seen following mild-TBI possibly due to a plateau affect, which suggests that by 80 days of age the levels of F2-Isoprostanes are at their peak in the spinal cord of this mouse model. Additional studies are needed to confirm this finding.
CONCLUSIONS
Taken together, our results suggest increased oxidative stress may be a potential mechanism for the long-term effects of mild-TBI on motor function. The effects on motor function were indicated in part by the observed elevated denervation potentials and in part by increased markers of denervation in the gastrocnemius muscle 30 days after injury. We also demonstrated that mild-TBI does not affect the lifespan of G93A mice, indicating that the mechanisms associated with G93A disease progression and TBI pathology are not additive and therefore are potentially exclusive from one another. The results of this study signify that future investigations using animal models of TBI should examine how injury affects the entire motor system pathway and that sensitive clinical testing approaches may identify resulting motor pathologies after brain injury.
Highlights for review.
Mild-TBI did not alter lifespan of G93A mice or the age of onset.
TBI resulted in impaired rotarod performance, grip strength was reduced in TG mice
Electromyography indicates that TBI causes peripheral effects increased in G93A mice
Inflammation was detected in brain & spinal cord by 72 hours in WT & G93A mice
Isoprostanes increase in WT spinal cord post TBI to levels comparable to G93A mice
Acknowledgments
This research was funded in part by an independent National Research Service Award, National Institute for Neurological Diseases and Stroke (1F31NS080508-01; Evans TM) and the Hartford Foundation/American Federation for Aging Research Scholars in Geriatric Medicine Program (Jaramillo CA) and a VA Merit Review grant to Dr. Van Remmen. We also acknowledge the assistance of Vivian Diaz and the Nathan Shock Center in animal care and maintenance and collection of body weight and body composition data. We would also like to acknowledge the help and guidance of Shane Sprague and the Neurosurgery Department Brain Injury Core for their support in establishing our TBI procedures.
ABBEVIATIONS
- TBI
traumatic brain injury
- ALS
Amyotrophic lateral sclerosis
- EMG
electromyography
- WT
wildtype
- CCI
closed cortical injury
- GFAP
glial fibrillary acidic protein
Footnotes
AUTHOR’S DISCLOSURE STATEMENT
| Author: | Conflicts of interest: |
|---|---|
| Teresa Marie Evans, PhD | Dr. Evans reports no conflicts of interest. |
| Carlos Jaramillo, MD, PhD | Dr. Jaramillo reports no conflicts of interest. |
| Kavithalakshmi Sataranatarajan, PhD | Dr. Sataranatarajan reports no conflicts of interest |
| Lora Watts, PhD | Dr. Watts reports no conflicts of interest. |
| Marian Sabia | Ms. Sabia reports no conflicts of interest. |
| Wenbo Qi, PhD | Dr. Qi reports no conflicts of interest. |
| Holly Van Remmen, PhD | Dr. Van Remmen reports no conflicts of interest. |
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Teresa Marie Evans, Email: EvansTM@uthscsa.edu.
Carlos A. Jaramillo, Email: jaramilloc3@uthscsa.edu.
Kavithalakshmi Sataranatarajan, Email: Kavithalakshmi-Sataranatarajan@omrf.org.
Lora Watts, Email: wattsl@uthscsa.edu.
Marian Sabia, Email: sabia@uthscsa.edu.
Wenbo Qi, Email: Qi@uthscsa.edu.
Holly Van Remmen, Email: Holly-VanRemmen@omrf.org.
References
- Armon C, Nelson LM. Is head trauma a risk factor for amyotrophic lateral sclerosis? An evidence based review. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2012;13:351–356. doi: 10.3109/17482968.2012.660954. [DOI] [PubMed] [Google Scholar]
- Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochimica et biophysica acta. 2012;1822:675–684. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatric research. 2002;51:571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Bayir H, Marion DW, Puccio AM, Wisniewski SR, Janesko KL, Clark RS, Kochanek PM. Marked gender effect on lipid peroxidation after severe traumatic brain injury in adult patients. Journal of neurotrauma. 2004;21:1–8. doi: 10.1089/089771504772695896. [DOI] [PubMed] [Google Scholar]
- Bongers KS, Fox DK, Ebert SM, Kunkel SD, Dyle MC, Bullard SA, Dierdorff JM, Adams CM. Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. American journal of physiology Endocrinology and metabolism. 2013;305:E907–915. doi: 10.1152/ajpendo.00380.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:4930–4939. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broglio SP, Sosnoff JJ, Shin S, He X, Alcaraz C, Zimmerman J. Head impacts during high school football: a biomechanical assessment. Journal of athletic training. 2009;44:342–349. doi: 10.4085/1062-6050-44.4.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. American journal of epidemiology. 2007;166:810–816. doi: 10.1093/aje/kwm153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A, Meineri P, Tribolo A, Schiffer D. Risk factors in motor neuron disease: a casecontrol study. Neuroepidemiology. 1991;10:174–184. doi: 10.1159/000110267. [DOI] [PubMed] [Google Scholar]
- Choi GS, Kim OL, Kim SH, Ahn SH, Cho YW, Son SM, Jang SH. Classification of cause of motor weakness in traumatic brain injury using diffusion tensor imaging. Archives of neurology. 2012;69:363–367. doi: 10.1001/archneurol.2011.1930. [DOI] [PubMed] [Google Scholar]
- Clausen F, Marklund N, Lewen A, Enblad P, Basu S, Hillered L. Interstitial F(2)- isoprostane 8-iso-PGF(2alpha) as a biomarker of oxidative stress after severe human traumatic brain injury. Journal of neurotrauma. 2012;29:766–775. doi: 10.1089/neu.2011.1754. [DOI] [PubMed] [Google Scholar]
- Czeiter E, Pal J, Kovesdi E, Bukovics P, Luckl J, Doczi T, Buki A. Traumatic axonal injury in the spinal cord evoked by traumatic brain injury. Journal of neurotrauma. 2008;25:205–213. doi: 10.1089/neu.2007.0331. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. The American journal of pathology. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Davis AE. Mechanisms of traumatic brain injury: biomechanical, structural and cellular considerations. Critical care nursing quarterly. 2000;23:1–13. doi: 10.1097/00002727-200011000-00002. [DOI] [PubMed] [Google Scholar]
- Evans TM, Remmen HV, Purkar A, Mahesula S, Gelfond JAL, Sabia M, Qi W, Lin A-L, Jaramillo CA, Haskins WE. Microwave and magnetic (M2) proteomics of a mouse model of mild traumatic brain injury. Translational Proteomics. 2014;3:10–21. doi: 10.1016/j.trprot.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nature protocols. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- Funk JR, Rowson S, Daniel RW, Duma SM. Validation of concussion risk curves for collegiate football players derived from HITS data. Annals of biomedical engineering. 2012;40:79–89. doi: 10.1007/s10439-011-0400-8. [DOI] [PubMed] [Google Scholar]
- Gallagher JP, Sanders M. Trauma and amyotrophic lateral sclerosis: a report of 78 patients. Acta neurologica Scandinavica. 1987;75:145–150. doi: 10.1111/j.1600-0404.1987.tb07909.x. [DOI] [PubMed] [Google Scholar]
- Gessel LM, Fields SK, Collins CL, Dick RW, Comstock RD. Concussions among United States high school and collegiate athletes. Journal of athletic training. 2007;42:495–503. [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Andrus PK, Hall ED. Pathogenic mechanisms in familial amyotrophic lateral sclerosis due to mutation of Cu, Zn superoxide dismutase. Pathologie-biologie. 1996;44:51–56. [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Guskiewicz KM. Assessment of postural stability following sport-related concussion. Current sports medicine reports. 2003;2:24–30. doi: 10.1249/00149619-200302000-00006. [DOI] [PubMed] [Google Scholar]
- Hall ED. Antioxidant therapies for acute spinal cord injury. Neurotherapeutics. 2011;8:152–167. doi: 10.1007/s13311-011-0026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J Neurosci Res. 1998;53:66–77. doi: 10.1002/(SICI)1097-4547(19980701)53:1<66::AID-JNR7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. Journal of neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- Ishido M, Kami K, Masuhara M. Localization of MyoD, myogenin and cell cycle regulatory factors in hypertrophying rat skeletal muscles. Acta physiologica Scandinavica. 2004;180:281–289. doi: 10.1046/j.0001-6772.2003.01238.x. [DOI] [PubMed] [Google Scholar]
- Jang YC, Van Remmen H. Age-associated alterations of the neuromuscular junction. Experimental gerontology. 2011;46:193–198. doi: 10.1016/j.exger.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamper JE, Pop V, Fukuda AM, Ajao DO, Hartman RE, Badaut J. Juvenile traumatic brain injury evolves into a chronic brain disorder: behavioral and histological changes over 6months. Experimental neurology. 2013;250:8–19. doi: 10.1016/j.expneurol.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DI, Alexander MP, Klein RB. Recovery of arm function in patients with paresis after traumatic brain injury. Archives of physical medicine and rehabilitation. 1998;79:488–493. doi: 10.1016/s0003-9993(98)90060-0. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Povlishock JT. Oxygen radicals in brain injury. Central nervous system trauma: journal of the American Paralysis Association. 1986;3:257–263. doi: 10.1089/cns.1986.3.257. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF, Beebe GW. Epidemiology of amyotrophic lateral sclerosis: 1. A casecontrol comparison based on ALS deaths. Neurology. 1980;30:453–462. doi: 10.1212/wnl.30.5.453. [DOI] [PubMed] [Google Scholar]
- Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurodegenerative causes of death among retired National Football League players. Neurology. 2012;79:1970–1974. doi: 10.1212/WNL.0b013e31826daf50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Xu R, Rodino-Klapac LR, Oglesbay E, Camboni M, Montgomery CL, Shontz K, Chicoine LG, Clark KR, Sahenk Z, Mendell JR, Janssen PM. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am J Physiol Cell Physiol. 2009;296:C476–488. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadyen BJ, Cantin JF, Swaine B, Duchesneau G, Doyon J, Dumas D, Fait P. Modalityspecific, multitask locomotor deficits persist despite good recovery after a traumatic brain injury. Archives of physical medicine and rehabilitation. 2009;90:1596–1606. doi: 10.1016/j.apmr.2009.03.010. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. Journal of neuropathology and experimental neurology. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M, Budson AE. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. Journal of neuropathology and experimental neurology. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, Montine TJ, Aschner M. Measurement of isoprostanes as markers of oxidative stress. Methods Mol Biol. 2011;758:195–204. doi: 10.1007/978-1-61779-170-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., 2nd Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods in enzymology. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K, McNeal DW, Morecraft RJ, Combs CK. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. Journal of neurotrauma. 2007;24:1719–1742. doi: 10.1089/neu.2007.0377. [DOI] [PubMed] [Google Scholar]
- O’Neil ME, Carlson K, Storzbach D, Brenner L, Freeman M, Quinones A, Motu’apuaka M, Ensley M, Kansagara D. Complications of Mild Traumatic Brain Injury in Veterans and Military Personnel: A Systematic Review. Washington (DC): 2013. [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Allen KD, Loiacono VT, Norman B, Stanwyck CL, Nord KM, Williams CD, Kasarskis EJ, Kamel F, McGuire V, Nelson LM, Oddone EZ. Genes and Environmental Exposures in Veterans with Amyotrophic Lateral Sclerosis: the GENEVA study. Rationale, study design and demographic characteristics. Neuroepidemiology. 2008;30:191–204. doi: 10.1159/000126911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Kwee LC, Allen KD, Oddone EZ. Association of ALS with head injury, cigarette smoking and APOE genotypes. Journal of the neurological sciences. 2010;291:22–29. doi: 10.1016/j.jns.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobounov S, Cao C, Sebastianelli W, Slobounov E, Newell K. Residual deficits from concussion as revealed by virtual time-to-contact measures of postural stability. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology. 2008;119:281–289. doi: 10.1016/j.clinph.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Spaans F, Wilts G. Denervation due to lesions of the central nervous system. An EMG study in cases of cerebral contusion and cerebrovascular accidents. Journal of the neurological sciences. 1982;57:291–305. doi: 10.1016/0022-510x(82)90036-3. [DOI] [PubMed] [Google Scholar]
- Talley Watts L, Sprague S, Zheng W, Garling RJ, Jimenez D, Digicaylioglu M, Lechleiter J. Purinergic 2Y1 receptor stimulation decreases cerebral edema and reactive gliosis in a traumatic brain injury model. Journal of neurotrauma. 2013;30:55–66. doi: 10.1089/neu.2012.2488. [DOI] [PubMed] [Google Scholar]
- Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes & development. 2005;19:1715–1722. doi: 10.1101/gad.1318305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WF, Qi W, Van Remmen H, Zackert WE, Roberts LJ, 2nd, Richardson A. Effects of age and caloric restriction on lipid peroxidation: measurement of oxidative stress by F2-isoprostane levels. The journals of gerontology Series A, Biological sciences and medical sciences. 2005;60:847–851. doi: 10.1093/gerona/60.7.847. [DOI] [PubMed] [Google Scholar]
- Williams DB, Annegers JF, Kokmen E, O’Brien PC, Kurland LT. Brain injury and neurologic sequelae: a cohort study of dementia, parkinsonism, and amyotrophic lateral sclerosis. Neurology. 1991;41:1554–1557. doi: 10.1212/wnl.41.10.1554. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, Cleveland DW, Zlokovic BV. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1401595111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Ying Z, Schubert D, Gomez-Pinilla F. Brain and spinal cord interaction: a dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabilitation and neural repair. 2011;25:332–342. doi: 10.1177/1545968310397706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Gangidine M, Pritts TA, Goodman MD, Lentsch AB. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock. 2013a;40:471–475. doi: 10.1097/SHK.0000000000000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Gustafson J, Gangidine M, Stepien D, Schuster R, Pritts TA, Goodman MD, Remick DG, Lentsch AB. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. The Journal of surgical research. 2013b;184:981–988. doi: 10.1016/j.jss.2013.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Wang Z, Tanaka M, Chiu CT, Leeds P, Zhang Y, Chuang DM. Posttrauma cotreatment with lithium and valproate: reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. Journal of neurosurgery. 2013;119:766–773. doi: 10.3171/2013.6.JNS13135. [DOI] [PMC free article] [PubMed] [Google Scholar]