

Abstract
Here evidence suggests that nitric oxide synthases (NOS) of tumor cells, in contrast to normal tissues, synthesize predominantly superoxide and peroxynitrite. Based on HPLC analysis, the underlying mechanism for this uncoupling is a reduced tetrahydrobiopterin: dihydrobiopterin ratio (BH4:BH2) found in breast, colorectal, epidermoid and head and neck tumors compared to normal tissues. Increasing BH4:BH2 and reconstitution of coupled NOS activity in breast cancer cells with the BH4 salvage pathway precursor, sepiapterin, causes significant shifts in downstream signaling including increased cGMP-dependent protein kinase (PKG) activity, decreased β-catenin expression and TCF4 promoter activity, and reduced NF-κB promoter activity. Sepiapterin inhibited breast tumor cell growth in vitro and in vivo as measured by clonogenic assay, Ki67 staining and 18F-deoxyglucose positron emission tomography (FDG-PET). In summary, using diverse tumor types, it is demonstrated that the BH4:BH2 ratio is lower in tumor tissues and as a consequence nitric oxide synthase activity generates more peroxynitrite and superoxide anion than nitric oxide resulting in important tumor growth promoting and anti-apoptotic signaling properties.
Implications
The synthetic BH4, Kuvan®, is used to elevate BH4:BH2 in some phenylketonuria patients and to treat diseases associated with endothelial dysfunction suggesting a novel, testable approach for correcting an abnormality of tumor metabolism to control tumor growth.
Keywords: Nitric Oxide, tetrahydrobiopterin, sepiapterin, cGMP, breast cancer
Introduction
Interest in nitric oxide (NO) as a signaling molecule in normal and cancer cells has developed because of links between chronic inflammatory diseases such as diabetes and reactive oxygen and reactive nitrogen species (ROS/RNS) with cancer (1). NO can have both pro- and anti-tumorigenic activities depending on concentration and levels of other ROS (2). For example, inhibition of endogenous NO synthases (NOS) blocks the growth of some but not all tumor cells by direct cytotoxic effects on the tumor cells or by inhibiting tumor endothelial cells (3, 4). In contrast, increased activity of inducible NOS (iNOS) in breast and colorectal cancer cells up-regulates tumor growth promoting Wnt/β-catenin signaling (5).
NO/RNS are essential for autocrine regulated oncogenic Ras-driven pancreatic tumor growth (6). Akt phosphorylation and activation of endothelial NOS (eNOS) stimulates RNS-dependent oxidation or S-nitrosylation of Cys118 of Ras resulting in cytoprotective signaling through Akt. Regulation of receptor Tyr kinase pathways, e.g. EGFR, can be achieved by inhibiting counteracting protein Tyr phosphatases by RNS-mediated oxidation of their active site Cys, e.g. SHP-2 (7, 8). PTEN also has an active site Cys sensitive to oxidation and its oxidation by RNS inhibits PTEN activating anti-apoptotic Akt signaling (9). RNS activate cytoprotective NF-κB transcriptional activity by nitrating Tyr181 of IκBα causing its dissociation from NF-κB (10). Simulating a chronic inflammatory environment by treating MCF-10A cells with low levels of RNS donors or co-culturing with activated macrophages stimulates Tyr nitration and activity of PP2A, leading to the down-regulation of BRCA1 expression and reduced homologous recombination DNA repair. The latter is compensated by enhanced non-homologous end-joining repair potentially promoting chromosomal instability, a hallmark of tumor progression (11).
Alternatively, NO can inhibit growth-promoting pathways of tumors. For example, S-nitrosylation of the p65 subunit of NF-κB or of IKKβ block NF-κB transcriptional activity (12). Increasing cellular cGMP with 5’-phosphodiesterase (PDE5) inhibitors or overexpression of cGMP dependent protein kinase G (PKG), a downstream effector of NO, inhibits colorectal and breast tumor growth (13, 14).
Given the technical difficulties of directly measuring NO in cells and tissues, most studies with tumors assume that NOS synthesize NO. However, as documented in the vasculature literature, NOS has two activities: NO or O2−/ONOO− synthesis (15). For all three isoforms of NOS, the catalytic cycle involves the cofactor BH4 donating electrons to the NOS Fe2+-O2 complex initiating Arg oxidation. The ratio of [BH4] to its oxidation product [BH2] is critical since both bind to the active site with equal affinity. When tissue BH4:BH2 is low as found in patients with chronic inflammatory diseases, more O2−/ONOO− and less NO are generated (16). This catalytic uncoupling also occurs at very low [Arg] or with increased levels of endogenous NOS inhibitors (17, 18).
We showed that sepiapterin (SP), a salvage pathway precursor for BH4, prophylactically blocked dextran sodium sulfate (DSS) induced colitis in mice and significantly reduced the incidence of colorectal tumors in an azoxymethane/DSS mouse model for colorectal cancer (19). HPLC analyses of total colon biopterins were inconclusive although there was a trend for lower BH4:BH2 in colons from mice treated with DSS that was reversed with SP. However, colonic cGMP levels were significantly reduced in colons of mice drinking DSS and this was reversed with SP by a mechanism sensitive to an inhibitor of the NO-activated soluble guanylate cyclase (sGC). Inhibiting sGC also blocked the protective effects of SP on colitis. Oral SP inhibited expression of inflammatory cytokines (IL-1β, IL-6, IL-17A), reduced infiltration of inflammatory cells (neutrophils and macrophages), and significantly inhibited the increased protein Tyr nitration in the diseased colons. These results are consistent with DSS/chronic inflammation induced uncoupling of NOS although we could not eliminate that BH4 or SP also protect sGC from oxidative inactivation (20).
To further explore the role of NOS uncoupling in tumor growth we measured BH4:BH2 in diverse tumors both in vitro and in vivo. Downstream signaling pathways relevant to breast cancer growth and sensitive to NO/RNS have been evaluated in terms of NOS coupling and tumor cell survival.
Materials and Methods
Biological Materials
Cell lines from American Tissue Culture Collection, authenticated through STR profiling, were frozen as early passage stocks after receipt from vendor. Cells were cultured for fewer than 6 months after thawing. All cell lines were grown in 10%FCS in RPMI1640 plus penicillin-streptomycin. Subcutaneous tumor xenografts were created in the hind legs of athymicNCr-nu/nu mice (3). MCF-7 xenograft growth was supported by 17β-estradiol pellets (1.5 mg, 60 day release, Innovative Research of America) inserted subcutaneously on the backs of mice. Breeding pairs of MMTVneu mice were from Jackson Laboratories. Drinking water was doped with 40 µg/ml SP or 0.64 mg/kg/day (19) significantly less than used in studies on vascular function (21). All animal experiments conform to protocols approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Human colorectal tumor samples and paired normal tissues were provided by the Tissue and Data Acquisition and Analysis Core of Massey Cancer Center as sections from OCT blocks prepared at the time of surgery. These samples were obtained through an Institutional IRB approved specimen anonymization agreement.
Reagents
Reagents included biopterins (Schircks Laboratories); KT5823 and the cGMP Elisa kit (Cayman Chemical Company). Antibodies were: Actin, IκBα,VASP (Santa Cruz); p65, pSer157 and pSer239 VASP (Cell Signaling Technology); PKG1β(Stressgen); Ki67 (NOVUS Biologicals); 3-nitrotyrosine (Life Technologies); β-catenin (BD Transduction); and fluorophore conjugated secondary antibodies (Rockland). Expression plasmids for VASP, iNOS, eNOS and GCH-1 were from Addgene.
Cell and Biochemical Analyses
Clonogenic assays and analytical methods for biopterins and cGMP have been described (3, 19). OCT blocks of human colon tissue were dissolved in PBS and the tissue collected by centrifugation prior to extracting in 0.1 N HCl for HPLC analysis. O2− generation was assayed by a fluorescence HPLC assay (22). Protein S-nitrosylation was detected by the biotin switch method (7).
For TCF/LEF promoter activity, cells were treated with PKGIβ siRNA premix (Qiagen) or SP. After 24 hours cells were transfected by lipofectamine/plus with a luciferase tagged TCF/LEF reporter construct (Addgene) in SP free medium. Three hours later medium was changed with fresh SP and luciferase activity measured 24 hr later. A luciferase reporter plasmid (Clontech) was used to measure NF-κB promoter activity (10).
PET/CT Imaging
Fasted and anesthetized (2% isoflurane) animals were tail vein injected with 300 µCi[18F]FDG (IBA Molecular, Sterling, VA). After 60 minutes of FDG uptake, PET/CT images were acquired in the Inveon Preclinical System (Siemens Healthcare, PA) without attenuation correction. Images were processed using manufacturer recommended calibration procedures and a phantom of known volume and activity acquired prior to the study. OSEM3D-MAP reconstructions used Inveon Acquisition Workplace 1.5 for region-of-interest (ROI) analysis in the Inveon Research Workplace 4.1. The percent injected dose/gram of tissue (%ID/g) was calculated after decay corrections with the formula, %ID/gm = Ct/ID × 100, where Ct is the concentration of radiotracer in the tissue obtained from the PET images after ROI analysis.
Immunohistochemistry
Tumors frozen in liquid N2 were embedded in OCT. Cryosections (6µm) fixed in ice-cold acetone were blocked with goat serum prior to staining with DAPI and antibodies. Images were captured using the Ariol Digital Pathology Platform. Quantitation was obtained with ImageJ software using DAPI staining for normalization.
Statistical Analysis
Except where indicated, a one way anova with Dunnett’s post hoc was used for statistical analysis with p≤0.05 considered significant.
Results
BH4:BH2 is low in tumor cells and increases with SP supplementation (Table 1)
Table 1. BH4:BH2 measurements in multiple cell types in vitro and in vivo.
| BH4:BH2 | BH4:BH2 + SP |
|
|---|---|---|
| Normal mouse tissuea | ||
| Colon | 7.1±0.6 | n/d |
| Lung | 11.1±2.4 | n/d |
| Liver | 8.3±0.8 | n/d |
| Kidney | 13.4±1.2 | n/d |
| In vitro tumor cell linesa | ||
| A431 | 0.3±0.5 | 2.1±0.6 |
| MCF7 | 0.6±0.2 | 10.1±0.4 |
| MDA231 | 1.0±0.1 | 5.8±0.2 |
| HT29 | 1.1±0.4 | 2.8±0.1 |
| MCF10A | 1.2±0.2 | 3.1±0.3 |
| MCF7-GCH1b | 3.6±0.2 | n/d |
| In vivo tumor xenograftsa | ||
| Fadu | 0.8±0.2 | 2.6±0.3 |
| MCF7 | 1.1±0.1 | 7.9±0.2 |
| MDA231 | 1.4±0.3 | 6.4±0.4 |
| Paired MMTVneu mouse tissuea | ||
| Mammary | 3.8±0.5 | n/d |
| Fat Pad | (n=4) | |
| Mammary | 1.0±0.2 | 2.8±0.5 |
| Tumors | (n=8) | (n=8) |
| Paired human colon tissuec | ||
| Normal | 4.5 | n/d |
| Colon | (3.5–6.1) | |
| Human | 2.3 | n/d |
| CRC | (1.5–3.5) | |
Mean±SEM. With in vitro samples, cells were harvested 16 hrs after incubation with 20 µM SP. With xenograft and spontaneous tumors, mice drank SP-loaded water for 2 days. For all comparisons between samples plus or minus SP, a p<0.005 was calculated by a 2-tailed student’s t-test.
MCF7 cells transiently transfected with GCH1 and BH4:BH2 levels measured 48 hrs later.
Paired samples of human colon tumor (CRC) and adjacent normal colon from the same patients (n=4) with average and range of values shown.
BH4:BH2 of tumor cells measured with a HPLC assay (Figure 1, Supplemental Data) is significantly lower relative to that of normal tissues examined. This is true regardless of their origin (breast, head and neck, epidermoid, colorectal) or whether grown in tissue culture, as xenografts, or in a spontaneous mouse breast cancer model (MMTVneu). Low BH4:BH2 is also observed in human colorectal carcinoma biopsies compared to that found in paired adjacent “normal” colon tissue (Table 1, paired T-test, n=4, p<0.001). That the paired “normal’ biopsy sample has a relatively lower BH4:BH2 from that expected for normal tissue is possibly a consequence of the inflammatory state of tissue adjacent to tumor. Insufficient amounts of normal human breast epithelial tissue for the HPLC analysis precluded a similar analysis for breast cancer tissues.
Figure 1. SP treatment in vitro and in vivo increases cGMP levels and PKG activity while reducing O2− formation.
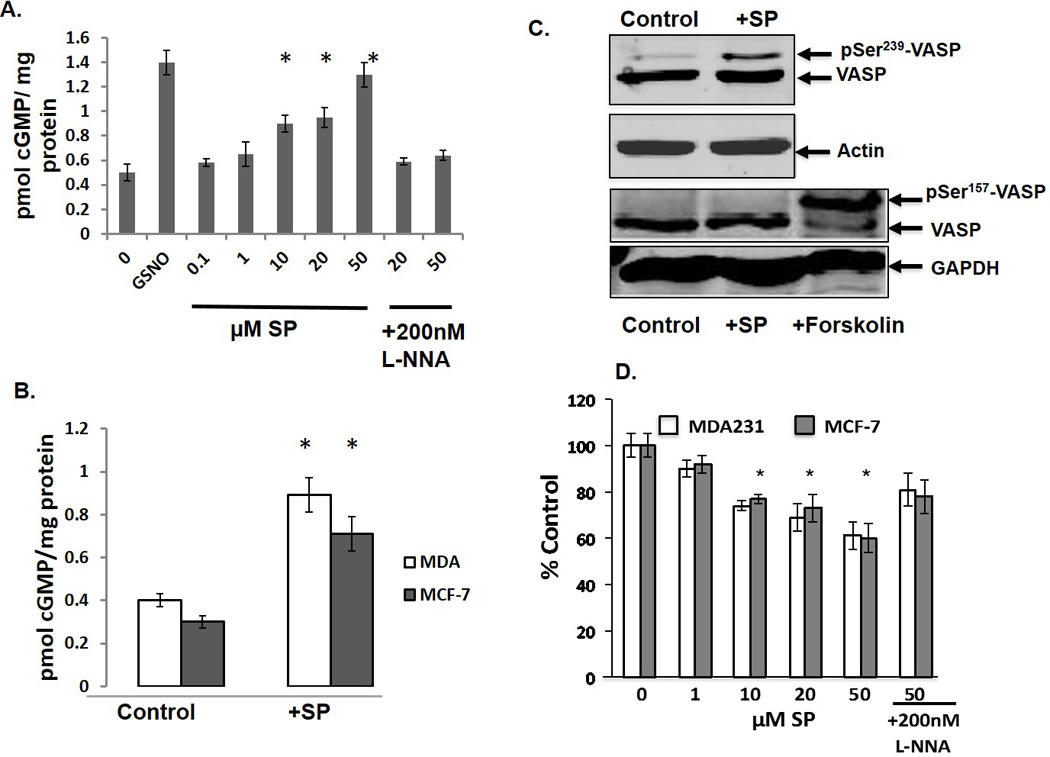
A. MCF-7 cells were incubated with SP for 6 hrs with or without 200 nM L-NNA. *p<0.05, n=3. B. MCF-7 and MDA-231 xenografts with or without 40 µg/ml SP in the drinking water. *p< 0.01, n =4. C. 24 hrs after transfection with VASP, MCF7 cells were treated for 6 hrs with 50 µM SP before harvesting and Western blot analysis for pSer239 (upper two panels) and pSer157 (lower two panels). As a positive control for PKA (pSer157-VASP, cells were incubated with 10µM forskolin. D. Cells were treated with SP for 6 hrs with or without 200 nM L-NNA. Cells were subsequently incubated with 10 µM dihydroethidium bromide for 30 mins prior to analysis by HPLC. *p<0.05, n=3.
BH4:BH2 is regulated in part by the rate limiting enzyme for BH4 synthesis, GTP-cyclohydrolase-1 (GCH1), or by a salvage pathway in which SP is converted to BH4 through the activities of SP reductase and dihydrofolate reductase (23). Targeting either pathway is effective in manipulating BH4:BH2. Transient transfection of MCF-7 cells at about 50% efficiency with an expression plasmid for GCH1 increases BH4:BH2 approximately 6-fold (Table 1). Incubating cells with SP increased BH4:BH2 maximally at 6 hrs and sustained for at least 16 hrs (Supplemental Data, Figure 2; Table 1). Depending on cell type, 20 µM SP for 16 hrs increases BH4:BH2 between 3 and 17 fold. For breast and squamous carcinoma xenografts and spontaneous MMTVneu mammary tumors, drinking water containing SP for 48 hrs elevates tumor BH4:BH2 from 3 to 7 fold.
To establish that SP promotes an increased BH4:BH2 via the salvage pathway, additional experiments used the SP reductase inhibitor, N-acetylserotonin (NAS), and the high affinity NOS inhibitor, Nω-nitro-L-arginine (L-NNA). Co-incubation of SP and NAS substantially blocks the SP-induced increase in BH4:BH2 with no effect on basal BH4:BH2 in MCF-7 or MDA-231 cells (Table 2). L-NNA modestly increases BH4:BH2 in cells not incubated with SP (p = 0.01, MCF-7; p =0.03 MDA-231). Co-incubation of L-NNA with SP also increases BH4:BH2 in MCF-7 cells relative to SP alone (p=0.002). This was not statistically significant with MDA-231 cells (p = 0.1). That NOS inhibition enhances BH4:BH2 is consistent with an uncoupling mechanism whereby NOS produced peroxynitrite directly oxidizes BH4 auto-propagating NOS uncoupling (17). We also demonstrated that NADPH oxidase activity contributes to BH4 oxidation in that incubation of cells with its peptide inhibitor, gp91 ds-tat, caused significant increases in the BH4:BH2 (Table 2).
Table 2. BH4:BH2 measurements in MCF7 and MDA-231 cells treated with without SP, L-NNA, NAS and a NADPH oxidase inhibitor.
Cells were incubated with 20 µM SP with or without 200 nM L-NNA,1 mM NAS or 5 µM gp91ds-tat for 6 hrs before harvesting. Mean ± SEM, n=3.
| MCF-7 BH4:BH2 |
MDA-231 BH4:BH2 |
|
|---|---|---|
| Control | 0.5±0.2 | 0.9±0.3 |
| +SP | 6.1±0.4 | 5.8±0.3 |
| +L-NNA | 1.1±0.1 | 1.6±0.2 |
| +L-NNA +SP | 8.1±0.3 | 6.4±0.4 |
| +NAS | 0.5±0.2 | 1.0±0.2 |
| +SP+NAS | 2.6±0.5 | 2.9±0.3 |
| +gp91ds-tat | 1.8±0.2 | 2.3±0.3 |
Recoupling NOS activity with SP
To test whether increased BH4:BH2 enhances NO production, cellular cGMP levels were measured. As a positive control, MCF-7 cells were incubated with the NO donor, S-Nitrosoglutathione (GSNO). There is a progressive increase in basal levels of cGMP with increasing SP concentration (Figure 1A). Co-incubation with L-NNA blocks the increase indicating that SP is acting through the NO-dependent sGC. SP also enhanced cGMP levels in MCF-7 and MDA-231 tumor xenografts (Figure 1B). Both MCF-7 and MDA-231 cells express cGMP-dependent PK G-1β (24, 25). To measure its enzymatic activity we monitored phosphorylation of Ser239 of the endogenous PKG substrate, VASP. SP treatment of MCF-7 cells transiently transfected with VASP enhances Ser239 VASP phosphorylation (Figure 1C, upper two panels). We also examined the cAMP-dependent protein kinase A (PKA) site, Ser157. Western blot analysis of VASP pSer157-VASP showed no change in phosphorylation with SP although there was a robust response to the PKA agonist, forskolin (Figure1C, lower two panels).
L-NNA inhibits both eNOS and iNOS with a selectivity of about 20-fold for eNOS (26). Since MCF-7 and MDA-231 cells express both eNOS and iNOS isoforms but iNOS catalytic activity is 100-fold greater than that of eNOS, we tested whether a highly selective inhibitor for iNOS, 1400W (5000-fold selectivity), also blocked SP-increased cGMP levels (27, 28). As shown in Supplemental Data Figure 3, 1400W effectively blocked SP-induced cGMP levels. That the catalytic activity of iNOS is 100-fold greater than eNOS argues that iNOS is the dominant but depending on physiological conditions perhaps not only NOS isoform stimulating sGC. Regardless of the isoform, exogenous SP increases BH4:BH2 thereby recoupling NOS and stimulating sGC.
Uncoupled NOS produces O2− and increasing cellular BH4 reduces cell production of O2− (15). To functionally assess NOS recoupling, O2− was measured using a HPLC-fluorescence assay that specifically measures its production from its oxidation of dihydroethidium to 2-hydroxy-ethidium (22). In both MCF-7 and MDA-231 cells, SP treatment partially decreased O2− generation by a mechanism inhibited by L-NNA (Figure 1D). These results combined with the cGMP measurements are consistent with SP recoupling of NOS activity.
Some investigations demonstrate that BH4 stabilizes a NOS dimer to SDS gel electrophoresis and is essential for a coupled catalysis (15, 29). We were unable to demonstrate this monomer-dimer transition with either iNOS or eNOS isoforms in SP-treated MCF-7 cells with or without transfection to overexpress the NOS isoforms. Other studies have also not shown this monomer-dimer transition with BH4 dependent recoupling of NOS activity (30, 31). It is possible that BH4 stabilizes a specific conformation of the dimer that is unstable in SDS but necessary for NO synthesis (30).
Recoupling NOS down regulates β-catenin expression
Reduced β-catenin expression is observed with PKG overexpression in cultured colorectal tumor cells (13, 32) and in both colorectal and breast tumor cells by incubating with a cGMP phosphodiesterase inhibitor (33, 34). MCF-7 cells were treated with 20 µM SP with or without the PKG inhibitor, KT5823 (a specific PKG inhibitor, IC50 20nM), for up to 3 days. Cells were harvested each day and analyzed for β-catenin expression. SP treatment in vitro for 72 hrs reduced β-catenin expression by more than 60% via a mechanism inhibited by KT5823. (Figures 2A,B). Almost complete ablation of β-catenin expression was observed in MCF-7 xenografts from mice drinking water supplemented with SP for 48 hrs (Figure 2C). In spontaneous tumors from MMTVneu mice, inclusion of SP in their drinking water reduced β-catenin expression by 50% (Figure 2D).
Figure 2. SP inhibits β-catenin expression and downstream signaling.
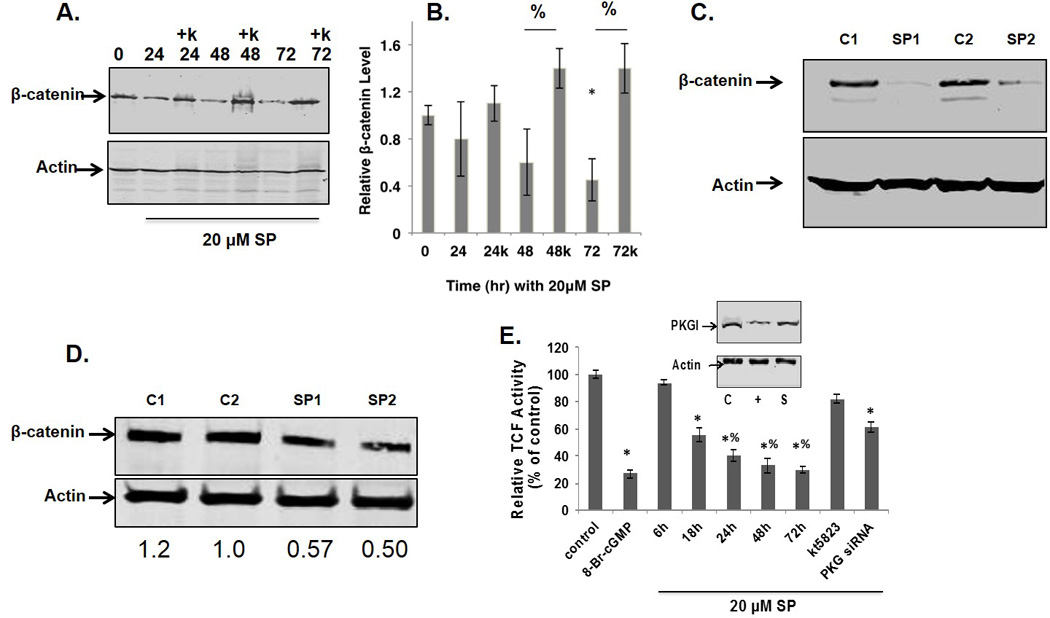
A. MCF7 cells incubated with SP with or without the PKG inhibitor, KT5823 (k), at 1 µM. β-catenin expression was analyzed by Western blot. B. Densitometry of 3 experiments with β-catenin expression normalized to actin. *relative to time zero, % comparing with or without KT5823, p<0.05 by two way anova. C. β-catenin levels relative to β-actin in MCF7 flank tumors with or without 40 µg/ml SP in their drinking water. C1 and C2, controls; SP1 and SP2 treated animals. D. β-catenin expression levels in tumors from MMTVneu mice with SP (mice SP1 and SP2) or without treatment (mice C1 and C2) for 4 days. E.TCF promoter activity measured with a luciferase reporter gene assay 24 hrs after transfection. Phosphodiesterase resistant 8-Br-cGMP (100 µM) was used as a positive control. Inset: PKG1β expression in control (C) cells transfected with scrambled siRNA (S) or siRNA targeting PKG (+).
β-catenin modulates tumor progression in part by interacting with the transcription factor TCF-4. We tested whether SP-induced down-regulation of β-catenin resulted in a decreased TCF-4 promoter activity using a luciferase reporter construct (Figure 2E). Luciferase activity was measured at 24 hrs post transfection with cells treated with SP with or without KT5823 for the indicated times or after siRNA knockdown of PKG. Significant inhibition by 20 µM SP was observed as early as 18 hrs with maximal inhibition of 80% at 72 hrs, the longest period tested. SP inhibition of TCF-4 promoter activity was mostly reversed with KT5823. Treatment of cells for 24 hrs with the cell permeant cGMP analog, 8-Br-cGMP, as a positive control reduced TCF-4 promoter activity by 80%. The pharmacological inhibition of PKG activity and its effect on TCF-4 activity by SP was confirmed by siRNA knockdown of PKG expression.
Recoupling NOS inhibits NF-κB Promoter Activity
A key kink between inflammation and oncogenesis is the transcription factor NF-κB. NF-κB is constitutively active in cancer cells due to autocrine and paracrine factors produced in the inflammatory microenvironment. NF-κB activity is both positively and negatively affected by ROS/RNS—nitration of Tyr181 of IκBα activates NF-κB (10) whereas S-nitrosylation of Cys38 of the p65 subunit inhibits (12). Treatment of MCF-7 cells with 20µM SP completely eliminated basal Tyr nitration of IκBα (Figure 3A). Figure 3B shows that increasing SP concentration augments levels of S-nitrosylated p65. A similar dose response was obtained when NF-κB promoter activity was measured in MCF-7 cells transiently transfected with a luciferase based reporter construct. Incubation of these cells with SP reduced basal NF-κB promoter activity by 40% (Figure 3C). A mutant Cys38 Ser p65, that renders NF-κB insensitive to S-nitrosylation (12), was co-transfected with the luciferase reporter and luciferase activity measured with and without pre-incubation with SP. The reduction in NF-κB reporter activity obtained with SP was completely blocked by the overexpression of the mutant protein (Figure 3C).
Figure 3. SP stimulates inhibits NF-κB transcriptional activity.

A. MCF-7 cells were incubated with or without 20µM SP for 6 hrs and lysates were immunopurified with anti-IκBα and probed for 3-nitrotyrosine. As a positive control cell lysates were treated with ONOO−. Results from 3 experiments were quantified by densitometry and shown in the bar graph (n=3, p<0.01). B. MCF7 cells were incubated with SP for 6 hrs before analysis of S-nitrosylation. GSNO (+) was added as a positive control. Total cell lysates were probed for actin as loading controls (bottom panel). (n =3, *p<0.05) C. The effect of 6 hrs SP on basal NF-κB promoter activity was determined with a luciferase-based reporter with tumor necrosis factor as a positive control. Some MCF-7 cells were co-transfected with the NF-κB reporter plasmid and the p65 Cys38 Ser mutant. Results represent the average of triplicate samples. (n =3, *p<0.05)
SP treatment is cytotoxic to MCF-7 and MDA-231 cells in tissue culture or as xenografts (Figure 4)
Figure 4. Sepiapterin is cytotoxic to mammary carcinoma cells in vitro and in tumor xenografts.

A. MCF-7 and MDA-231 breast carcinoma cells in vitro were incubated with SP for 16 hrs prior to plating for colony formation. (n=3, *p<0.05). B. For ex vivo clonogenic assay of tumor xenografts mice drank SP at 40 µg/ml for 48 hrs before isolating single tumor cells for clonogenic assay. (n =3, *p<0.05) Panel C. In vitro clonogenic assays comparing the effects of NHP4 and SP (at the indicated concentrations) and the PKG inhibitor KT5823 (1 µM) and NOS inhibitor L-NNA (200 nM) on SP cytotoxicity (*n=3, p<0.05). D. Animals bearing MDA-231 flank xenografts with and without drinking SP-doped water and with or without the SP reductase inhibitor NAS delivered ip at 10 mg/kg the day before starting SP, on the day starting SP and 24 hrs later. Animals were sacrificed at 48 hrs post-SP treatment. *n=3, p<0.05 comparing treated versus untreated and &n=3, p<0.05.
Clonogenic assay in vitro and with xenografts establish that SP treatment at the concentrations used is cytotoxic to both MCF-7 and MDA-231 breast carcinoma cells (Figure 4A,B). We tested whether the cytotoxic activity of SP could be inhibited by the NOS inhibitor, L-NNA, previously shown to inhibit NOS in MCF-7 but without cytotoxic activity in vitro by itself (19, 35). The present results confirmed these results but also demonstrated that L-NNA completely inhibited SP induced cell killing (Figure 4C). The PKG inhibitor, KT5823, was equally ineffective as a cytotoxic agent but abrogated SP cytotoxicity.
Because L-NNA is cytotoxic to tumors in vivo and because the SP reductase inhibitor NAS blocks the SP-induced increase in BH4:BH2 (Table 2), we tested whether inhibiting the BH4 salvage pathway with NAS, blocks the SP cytotoxicity in vivo. Treatment of mice with NAS blocked tumor cell killing by SP as measured by ex vivo clonogenic assay of MCF-7 xenograft tumors (Figure 4D).
SP is an antioxidant and shown to possibly protect sGC from oxidant-inactivation (20). To rule out this role in the cytotoxic activity of SP, cells were incubated with, tetrahydroneopterin (NHP4), a biopterin with equivalent free radical scavenging activity as SP but unable to participate as a cofactor for NOS (20). As shown in Figure 4C, NHP4 at concentrations equal to those used with SP is not cytotoxic.
The effects of prolonged SP treatment on tumor growth and animal survival was tested with MDA-231 xenografts. As shown in Supplemental Data Figure 3 continuous oral supplementation over weeks provides a significant survival advantage. Future studies are required to determine how much this is due to SP effects on tumor cells and how much on supporting stromal cells.
SP reduces tumor progression in the MMTVneu mouse assessed by FDG PET/CT and Ki67 staining
Anti-tumor activity of SP was also evaluated with a spontaneous cancer model, the MMTVneu mouse using [F18]- PET/CT and Ki67 staining of tumors after animal sacrifice (Figure 5). PET/CT scans before and after 5 days oral SP (Figure 5A) were quantified from 9 tumors (6 mice, 3 of which carried 2 tumors) and analyzed for changes in FDG uptake (Figure 5B). Percent decreases in FDG uptake by tumors varied significantly between animals and indeed between tumors from the same mouse (m001, m004, m005). In all mice except m005 SP reduced FDG uptake. When results from this mouse were censored in the analysis, SP reduced FDG uptake by 25% (p<0.019). When the data from this mouse were included in the analysis, FDG uptake was reduced by 21% (p<0.062).
Figure 5. PET-FDG and Ki67 IHC analyses of the effects of SP on MMTVneu tumor growth.

A. PET/CT scans for mouse m003, tumor #4 (see Panel B) before and 5 days after starting SP treatment by inclusion of SP in its drinking water. T=tumor, H=heart, K=kidney and B=bladder. B. Tabulated summary of results with 6 mice and 9 tumors (3 mice had 2 tumors each). The before and after results were analyzed by a matched 2-tail T-test for all 9 tumors and also with the results from animal m005 censored. C. % Change in FDG uptake plotted against the BH4:BH2 for those tumors for which biopterin measurements were made: Tumor #3, #4, #6, #7 and #8. The Pearson Product Moment Correlation calculation determined the linear correlation between BH4:BH2 and FDG uptake. D. All Images were tissue slices stained with DAPI and a fluorophore secondary antibody. Images A and B are from adjacent slices of a tumor from non-SP treated mouse. Images C and D are adjacent slices of a tumor from a mouse treated with SP for 5 days. Images B and D were incubated with a primary antibody against Ki67. The adjacent bar figure tabulates the results from tumors of 3 control animals and 3 SP treated animals. Five pairs of random slices from each tumor were analyzed and Ki67 image intensity normalized with respect to DAPI staining. The statistical analysis was an unpaired student’s t-test with a p < 0.01 for n =15 for control and n =15 for SP-treated animals.
Reasons for this variability in FDG uptake are unclear. However, there were some tumors with tissue extracts available for BH4:BH2 analysis including m005. A linear correlation (p = 0.01) was established between BH4:BH2 at the end of treatment and FDG uptake (Figure 5C). BH4:BH2 of the two tumors of SP-treated m005 (1.1 and 1.3) were not significantly different from the average ratio for untreated MMTVneu tumors (1.0 ± 0.2, Table 1). Since we did not monitor water consumption separately for each mouse, it was possible that m005 did not consume enough SP to enhance the BH4:BH2. Other mechanisms including poor tumor vascularization are also possible. For the tumor with greatest reduction in FDG uptake (−72%, m004, tumor 6), BH4:BH2 was 3.9, similar to that observed for the normal mammary fat pad of the MMTVneu mouse (3.8 ± 0.5, Table 1).
Ki67 staining, a marker for actively proliferating cells, was used to confirm that SP reduced tumor growth (Figure 5D). Representative immunofluorescence images of DAPI and Ki67 stained sections of tumors from mice with and without SP treatment are shown along with a statistical evaluation from 5 randomly chosen tumor slices from three control and three treated animals. SP treatment for 5 days reduced Ki67 staining by 60%.
Discussion
The major finding of this study is that regardless of tumor type and whether cells are cultured or derived from tumors, BH4:BH2 is significantly lower compared to normal tissues. Furthermore, BH4:BH2 is enhanced by supplementing the culture medium or animal drinking water with a salvage pathway precursor, SP. Therapeutically increasing BH4:BH2 caused tumor cell death in tissue culture or tumor xenografts as defined by clonogenic survival and in spontaneous tumors by FDG PET imaging and Ki67 staining.
Amino acid hydroxylases also use BH4 as a cofactor. Previous investigations suggest a growth promoting, anti-apoptotic role for serotonin, a product of tryptophan hydroxylase activity (36). This is contrary to the growth inhibitory properties observed with SP in vitro implying that any increase in tryptophan hydroxylase activity due to an increased BH4:BH2 is of lesser importance in effecting tumor growth. Future studies may need to consider both pro- and anti-angiogenic activities of serotonin (37, 38).
Low levels of cGMP due to enhanced expression of PDE5, a cGMP specific phosphodiesterase characterize some cancer cells (14). Increasing cGMP levels by inhibiting PDE5 blocks MDA-231 and ZR75-1 breast cancer cell growth but not growth of non-transformed human mammary epithelial cells that do not express PDE5 (14, 34). While not all downstream targets of cGMP/PKG are known, inhibition of PDE5 is associated with down-regulation of β-catenin expression and downstream TCF-4 promoter activity in these breast cancer cell lines (14, 34). Our findings demonstrate that increased cellular cGMP levels and PKG activity, reduced β-catenin expression and TCF-4 promoter activity, and enhanced tumor cell killing are also obtained by modulating BH4:BH2 with a natural endogenous molecule.
The effects of NOS recoupling on tumor cell viability cannot be completely explained by enhanced PKG activity. This is clearly seen in the experiments demonstrating that increasing BH4:BH2 results in decreased tyrosine nitration of IκBα but increased S-nitrosylation of the p65 subunit of NF-κB partially inhibiting the latter’s transcriptional activity (Figure 3). Other regulatory proteins potentially involved in tumor growth are also post-translationally modified by either Tyr nitration, e.g. PP2A, p53,(10, 11, 35) or Cys S-nitrosylation or oxidation, e.g. protein Tyr phosphatases, ras, (6, 7).
Several mechanisms potentially contribute to the decreased BH4:BH2 of cancer cells. Overexpression of the rate-limiting enzyme for BH4 synthesis, GCH1, significantly increases the BH4:BH2 in MCF-7 cells suggesting that either reduced GCH1 expression or activity may have a role. Some studies indicate that proteasomal degradation of GCH1 is a key regulatory factor in BH4 synthesis and it is thought to be one mechanism for the BH4 deficiency in diabetes mellitus (39). Proteasomal degradation of GCH1 is blocked by metformin-induced AMPK activation resulting in the recoupling of eNOS in endothelial cells (40, 41). On the other hand there is a recent report indicating that in some tumors GCH1 expression might actually be increased (42). This study did not, however, evaluate functional consequence such as BH4:BH2 or cGMP. GCH1 activity is also regulated by an inhibitory protein, GCH1 feedback regulatory protein (GFRP). GFRP inhibits GCH1 by a direct binding mechanism that requires BH4 at the interface between the two proteins (43).
Dihydrofolate reductase (DFHR) and dihydropteridine reductase (DHPR), acting together or individually can by salvage synthesize BH4. It is unlikely given the DNA synthesis requirements of cancer cells that reduced DHFR activity accounts for reduced BH4. DHPR reduces quinonoid dihydrobiopterin, an intermediate in the peroxynitrite oxidation of BH4 to BH2, back to BH4. There are reports that expression levels of DHPR are reduced in conditions where BH4:BH2 is low, e.g. diabetes (44). A genetic disorder of reduced DHPR is treated with BH4 (45). Early investigations also indicate that DHPR activities are lower in solid tumor tissue relative to their normal tissue counterpart (46).
Results in Table 2 demonstrate potential mechanisms by which NOS produced peroxynitrite oxidizes BH4 further propagating uncoupling (47). A related mechanism follows from the cellular proximity relationships between NOS and O2− generating activities. Both eNOS and iNOS, the major NOS isoforms in epithelial tumor cells, reside at least in part at the plasma membrane. eNOS has a complicated relationship to the plasma membrane being targeted to plasma membrane caveolae through a mechanism involving N-myristoylation and palmitoylation and forming a complex with caveolin-1 (48). Binding to caveolin-1 induces an inhibitory conformation of eNOS that is relieved upon Akt phosphorylation of eNOS. This positioning of eNOS at caveolae puts it in close proximity to the O2− generating NADPH oxidases potentially generating peroxynitrite that rapidly reacts with BH4 initiating uncoupling (49). GCH1 has also been localized to caveolae in some cells (50). Results in Table 2 demonstrate that NADPH oxidase is also a potential contributor in the oxidation of BH4.
NOS uncoupling can occur under other conditions, e.g. low [Arg], or elevated levels of endogenous NOS inhibitors (17, 18). Regardless of the mechanism, NOS uncoupling represents a critical switching mechanism for tumor cell growth. When the primary product of NOS is NO, downstream signaling is dominated by NO-dependent pathways (eg, sGC/PKG and S-nitrosylation). Uncoupled NOS, on the other hand, produces oxidants such as peroxynitrite and O2− initiating different downstream signaling that for tumor cells are pro-proliferative and anti-apoptotic, e.g. NF-κB. The focus of the present studies has been on the tumor cell. Given the chronic inflammatory conditions of solid tumors it is likely that stromal cells also exhibit uncoupled NOS activity and by this mechanism are subverted to support tumor growth. Our results also suggest a novel anti-cancer therapy that by its mode of action should have minimal normal tissue toxicity. Synthetic BH4 (Kuvan©) is presently being using to treat certain forms of phenylketonuria and is being tested in clinical trials to reduce endothelial dysfunction.
Supplementary Material
Acknowledgements
Dr. Catherine Dumur and Ms Pamela Grizzard of the Massey Cancer Center Tissue Acquisition Core provided the human tissue samples.
Financial Support: This work was supported by National Institutes of Health Grants 5R01CA090881 (RBM) and T32CA113277 (CSR and RJC). Additional funding for Tissue Acquisition Core and a pilot project came from the Massey Cancer Center Grant 5P30CA016059-33.
Footnotes
No real or potential conflicts of interest for the authors
References
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 2.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45(1):18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. PMCID: 2572721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardnell RJ, Mikkelsen RB. Nitric oxide synthase inhibition enhances the antitumor effect of radiation in the treatment of squamous carcinoma xenografts. PLoS One. 2011;6(5):e20147. doi: 10.1371/journal.pone.0020147. PMCID: 3102067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tozer GM, Prise VE, Chaplin DJ. Inhibition of nitric oxide synthase induces a selective reduction in tumor blood flow that is reversible with L-arginine. Cancer Res. 1997;57(5):948–955. [PubMed] [Google Scholar]
- 5.Du Q, Zhang X, Liu Q, Bartels CE, Geller DA. Nitric oxide production upregulates Wnt/beta-catenin signaling by inhibiting Dickkopf-1. Cancer Res. 2013;73(21):6526–6537. doi: 10.1158/0008-5472.CAN-13-1620. PMCID: 3818363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452(7187):646–649. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrett DM, Black SM, Todor H, Schmidt-Ullrich RK, Dawson KS, Mikkelsen RB. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005;280(15):14453–14461. doi: 10.1074/jbc.M411523200. [DOI] [PubMed] [Google Scholar]
- 8.Sturla LM, Amorino G, Alexander MS, Mikkelsen RB, Valerie K, Schmidt-Ullrichr RK. Requirement of Tyr-992 and Tyr-1173 in phosphorylation of the epidermal growth factor receptor by ionizing radiation and modulation by SHP2. J Biol Chem. 2005;280(15):14597–14604. doi: 10.1074/jbc.M413287200. [DOI] [PubMed] [Google Scholar]
- 9.Delgado-Esteban M, Martin-Zanca D, Andres-Martin L, Almeida A, Bolanos JP. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signaling pathway. J Neurochem. 2007;102(1):194–205. doi: 10.1111/j.1471-4159.2007.04450.x. [DOI] [PubMed] [Google Scholar]
- 10.Yakovlev VA, Barani IJ, Rabender CS, Black SM, Leach JK, Graves PR, et al. Tyrosine nitration of IkappaBalpha: a novel mechanism for NF-kappaB activation. Biochemistry. 2007;46(42):11671–11683. doi: 10.1021/bi701107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yakovlev VA. Nitric Oxide-Dependent Downregulation of BRCA1 Expression Promotes Genetic Instability. Cancer Res. 2013;73(2):706–715. doi: 10.1158/0008-5472.CAN-12-3270. PMCID: 3549017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marshall HE, Hess DT, Stamler JS. S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl Acad Sci U S A. 2004;101(24):8841–8842. doi: 10.1073/pnas.0403034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Browning DD, Kwon IK, Wang R. cGMP-dependent protein kinases as potential targets for colon cancer prevention and treatment. Future medicinal chemistry. 2010;2(1):65–80. doi: 10.4155/fmc.09.142. [DOI] [PubMed] [Google Scholar]
- 14.Windham PF, Tinsley HN. cGMP signaling as a target for the prevention and treatment of breast cancer. Semin Cancer Biol. 2014 doi: 10.1016/j.semcancer.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Cai S, Khoo J, Channon KM. Augmented BH4 by gene transfer restores nitric oxide synthase function in hyperglycemic human endothelial cells. Cardiovasc Res. 2005;65(4):823–831. doi: 10.1016/j.cardiores.2004.10.040. [DOI] [PubMed] [Google Scholar]
- 16.Luiking YC, Ten Have GA, Wolfe RR, Deutz NE. Arginine de novo and nitric oxide production in disease states. Am J Physiol Endocrinol Metab. 2012;303(10):E1177–E1189. doi: 10.1152/ajpendo.00284.2012. PMCID: 3517635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alkaitis MS, Crabtree MJ. Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep. 2012;9(3):200–210. doi: 10.1007/s11897-012-0097-5. PMCID: 3406312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zweier JL, Chen CA, Druhan LJ. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid Redox Signal. 2011;14(10):1769–1775. doi: 10.1089/ars.2011.3904. PMCID: 3078498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardnell RJ, Rabender CS, Ross GR, Guo C, Howlett EL, Alam A, et al. Sepiapterin ameliorates chemically induced murine colitis and azoxymethane-induced colon cancer. J Pharmacol Exp Ther. 2013;347(1):117–125. doi: 10.1124/jpet.113.203828. PMCID: 3781406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt K, Neubauer A, Kolesnik B, Stasch JP, Werner ER, Gorren AC, et al. Tetrahydrobiopterin protects soluble guanylate cyclase against oxidative inactivation. Mol Pharmacol. 2012;82(3):420–427. doi: 10.1124/mol.112.079855. [DOI] [PubMed] [Google Scholar]
- 21.Shimazu T, Otani H, Yoshioka K, Fujita M, Okazaki T, Iwasaka T. Sepiapterin enhances angiogenesis and functional recovery in mice after myocardial infarction. Am J Physiol Heart Circ Physiol. 2011;301(5):H2061–H2072. doi: 10.1152/ajpheart.00525.2011. [DOI] [PubMed] [Google Scholar]
- 22.Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc. 2008;3(1):8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- 23.Werner ER, Blau N, Thony B. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J. 2011;438(3):397–414. doi: 10.1042/BJ20110293. [DOI] [PubMed] [Google Scholar]
- 24.Fallahian F, Karami-Tehrani F, Salami S, Aghaei M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. Febs J. 2011;278(18):3360–3369. doi: 10.1111/j.1742-4658.2011.08260.x. [DOI] [PubMed] [Google Scholar]
- 25.Schwappacher R, Rangaswami H, Su-Yuo J, Hassad A, Spitler R, Casteel DE. cGMP-dependent protein kinase Ibeta regulates breast cancer cell migration and invasion via interaction with the actin/myosin-associated protein caldesmon. J Cell Sci. 2013;126(Pt 7):1626–1636. doi: 10.1242/jcs.118190. PMCID: 3647439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reif DW, McCreedy SA. N-nitro-L-arginine and N-monomethyl-L-arginine exhibit a different pattern of inactivation toward the three nitric oxide synthases. Arch Biochem Biophys. 1995;320(1):170–176. doi: 10.1006/abbi.1995.1356. [DOI] [PubMed] [Google Scholar]
- 27.Abdelmagid SA, Rickard JA, McDonald WJ, Thomas LN, Too CK. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J Cell Biochem. 2011;112(4):1084–1092. doi: 10.1002/jcb.23022. [DOI] [PubMed] [Google Scholar]
- 28.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, et al. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272(8):4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 29.Benson MA, Batchelor H, Chuaiphichai S, Bailey J, Zhu H, Stuehr DJ, et al. A pivotal role for tryptophan 447 in enzymatic coupling of human endothelial nitric oxide synthase (eNOS): effects on tetrahydrobiopterin-dependent catalysis and eNOS dimerization. J Biol Chem. 2013;288(41):29836–29845. doi: 10.1074/jbc.M113.493023. PMCID: 3795282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Crespo I, Gerber NC, Ortiz de Montellano PR. Endothelial nitric-oxide synthase. Expression in Escherichia coli, spectroscopic characterization, and role of tetrahydrobiopterin in dimer formation. J Biol Chem. 1996;271(19):11462–11467. doi: 10.1074/jbc.271.19.11462. [DOI] [PubMed] [Google Scholar]
- 31.Whitsett J, Martasek P, Zhao H, Schauer DW, Hatakeyama K, Kalyanaraman B, et al. Endothelial cell superoxide anion radical generation is not dependent on endothelial nitric oxide synthase-serine 1179 phosphorylation and endothelial nitric oxide synthase dimer/monomer distribution. Free Radic Biol Med. 2006;40(11):2056–2068. doi: 10.1016/j.freeradbiomed.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Kwon IK, Wang R, Thangaraju M, Shuang H, Liu K, Dashwood R, et al. PKG inhibits TCF signaling in colon cancer cells by blocking beta-catenin expression and activating FOXO4. Oncogene. 2010;29(23):3423–3434. doi: 10.1038/onc.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li N, Xi Y, Tinsley HN, Gurpinar E, Gary BD, Zhu B, et al. Sulindac Selectively Inhibits Colon Tumor Cell Growth by Activating the cGMP/PKG Pathway to Suppress Wnt/beta-Catenin Signaling. Mol Cancer Ther. 2013;12(9):1848–1859. doi: 10.1158/1535-7163.MCT-13-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tinsley HN, Gary BD, Keeton AB, Lu W, Li Y, Piazza GA. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/beta-catenin-mediated transcription in human breast tumor cells. Cancer Prev Res (Phila) 2011;4(8):1275–1284. doi: 10.1158/1940-6207.CAPR-11-0095. PMCID: 3151326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yakovlev VA, Bayden AS, Graves PR, Kellogg GE, Mikkelsen RB. Nitration of the tumor suppressor protein p53 at tyrosine 327 Promotes p53 oligomerization and activation. Biochemistry. 2010;49(25):5331–5339. doi: 10.1021/bi100564w. PMCID: 2892261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pai VP, Marshall AM, Hernandez LL, Buckley AR, Horseman ND. Altered serotonin physiology in human breast cancers favors paradoxical growth and cell survival. Breast Cancer Res. 2009;11(6):R81. doi: 10.1186/bcr2448. PMCID: 2815543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, et al. Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 2008;68(13):5152–5158. doi: 10.1158/0008-5472.CAN-08-0202. [DOI] [PubMed] [Google Scholar]
- 38.Qin L, Zhao D, Xu J, Ren X, Terwilliger EF, Parangi S, et al. The vascular permeabilizing factors histamine and serotonin induce angiogenesis through TR3/Nur77 and subsequently truncate it through thrombospondin-1. Blood. 2013;121(11):2154–2164. doi: 10.1182/blood-2012-07-443903. PMCID: 3596973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma S, Sun X, Kumar S, Rafikov R, Aramburo A, Kalkan G, et al. Preserving mitochondrial function prevents the proteasomal degradation of GTP cyclohydrolase I. Free Radic Biol Med. 2012;53(2):216–229. doi: 10.1016/j.freeradbiomed.2012.03.016. PMCID: 3527085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Xu J, Song P, Viollet B, Zou MH. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes. 2009;58(8):1893–1901. doi: 10.2337/db09-0267. PMCID: 2712774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim YH, Hwang JH, Kim KS, Noh JR, Gang GT, Oh WK, et al. Enhanced activation of NAD(P)H: quinone oxidoreductase 1 attenuates spontaneous hypertension by improvement of endothelial nitric oxide synthase coupling via tumor suppressor kinase liver kinase B1/adenosine 5'-monophosphate-activated protein kinase-mediated guanosine 5'-triphosphate cyclohydrolase 1 preservation. J Hypertens. 2014;32(2):306–317. doi: 10.1097/HJH.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 42.Pickert G, Lim HY, Weigert A, Haussler A, Myrczek T, Waldner M, et al. Inhibition of GTP cyclohydrolase attenuates tumor growth by reducing angiogenesis and M2-like polarization of tumor associated macrophages. Int J Cancer. 2013;132(3):591–604. doi: 10.1002/ijc.27706. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Rezvan A, Salerno JC, Husain A, Kwon K, Jo H, et al. GTP cyclohydrolase I phosphorylation and interaction with GTP cyclohydrolase feedback regulatory protein provide novel regulation of endothelial tetrahydrobiopterin and nitric oxide. Circ Res. 2010;106(2):328–336. doi: 10.1161/CIRCRESAHA.109.210658. PMCID: 2818799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee CK, Han JS, Won KJ, Jung SH, Park HJ, Lee HM, et al. Diminished expression of dihydropteridine reductase is a potent biomarker for hypertensive vessels. Proteomics. 2009;9(21):4851–4858. doi: 10.1002/pmic.200800973. [DOI] [PubMed] [Google Scholar]
- 45.Coughlin CR, 2nd, Hyland K, Randall R, Ficicioglu C. Dihydropteridine reductase deficiency and treatment with tetrahydrobiopterin: a case report. JIMD Rep. 2013;10:53–56. doi: 10.1007/8904_2012_202. PMCID: 3755580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez-Urretia L, Pierce CE, Greengard O. Dihydropteridine reductase activity of adult, fetal and neoplastic tissues. Enzyme. 1978;23(5):346–352. doi: 10.1159/000458599. [DOI] [PubMed] [Google Scholar]
- 47.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278(25):22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 48.Goligorsky MS, Li H, Brodsky S, Chen J. Relationships between caveolae and eNOS: everything in proximity and the proximity of everything. Am J Physiol Renal Physiol. 2002;283(1):F1–F10. doi: 10.1152/ajprenal.00377.2001. [DOI] [PubMed] [Google Scholar]
- 49.Reilly SN, Jayaram R, Nahar K, Antoniades C, Verheule S, Channon KM, et al. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation. 2011;124(10):1107–1117. doi: 10.1161/CIRCULATIONAHA.111.029223. [DOI] [PubMed] [Google Scholar]
- 50.Peterson TE, d'Uscio LV, Cao S, Wang XL, Katusic ZS. Guanosine triphosphate cyclohydrolase I expression and enzymatic activity are present in caveolae of endothelial cells. Hypertension. 2009;53(2):189–195. doi: 10.1161/HYPERTENSIONAHA.108.115709. PMCID: 2646898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.