

Highlights
-
•
RNA viruses passively evade host detection by masking viral PAMPs and replicating within vesicles.
-
•
Many emerging viruses harbor multiple strategies for innate immune evasion.
-
•
Viral antagonists have been found to target the pattern recognition receptor and interferon signaling pathways.
-
•
Knowledge of host–pathogen interactions is essential for vaccine/therapeutic development and understanding innate immunity.
Abstract
Recent outbreaks of Ebola, West Nile, Chikungunya, Middle Eastern Respiratory and other emerging/re-emerging RNA viruses continue to highlight the need to further understand the virus–host interactions that govern disease severity and infection outcome. As part of the early host antiviral defense, the innate immune system mediates pathogen recognition and initiation of potent antiviral programs that serve to limit virus replication, limit virus spread and activate adaptive immune responses. Concordantly, viral pathogens have evolved several strategies to counteract pathogen recognition and cell-intrinsic antiviral responses. In this review, we highlight the major mechanisms of innate immune evasion by emerging and re-emerging RNA viruses, focusing on pathogens that pose significant risk to public health.
Current Opinion in Virology 2015, 12:26–37
This review comes from a themed issue on Antiviral strategies
Edited by Michael Gale Jr. and Curt M Horvath
For a complete overview see the Issue and the Editorial
Available online 9th March 2015
http://dx.doi.org/10.1016/j.coviro.2015.02.005
1879-6257/© 2015 Elsevier B.V. All rights reserved.
Introduction
Several emerging and re-emerging RNA viruses have been the cause of several national and international epidemics within the past few years. These pathogens are no longer confined within national or even regional boundaries, and have become cause for global concern. In the summer of 2012, an outbreak of the flavivirus West Nile virus (WNV) in the United States, which since its discovery in 1937 has become endemic to all continents on earth except Antarctica, resulted in over 1800 cases in the state of Texas alone [1]. Moreover, recent epidemiological evidence has distinguished the emergence of novel Lineage 2 WNV isolates as the cause of several human outbreaks in Europe, which prior to 2011 was thought to be of low virulence to humans [2]. Later in 2012, the Middle East Respiratory Coronavirus (MERS-CoV) emerged in Saudi Arabia. In April 2014, an outbreak of over 300 cases of MERS occurred in Saudi Arabia (WHO, 2014), and has since spread to other countries in Europe. The US MERS-CoV shows similarity to the Severe Acute Respiratory-Coronavirus (SARS-CoV), which caused an epidemic of over 8000 cases in 2003 (World Health Organization, 2010). In March 2014, an outbreak of Ebolavirus (EBOV) began in West Africa, and has since caused over 9100 deaths (CDC, 2015) in Africa and now to other parts of the world. In December 2013, Chikungunya virus (CHIKV), a member of the Alphavirus family, was detected in the for the first time Western Hemisphere within Caribbean nations, and a few months later spread to both North and South America (CDC, 2014) [3]. Given the increase in global travel, international commerce and possible effects of climate change, we will continue to observe the emergence and re-emergence of viral strains that are of significant concern to human health worldwide.
The virulence of these human infections is undeniably linked to immune evasion mechanisms that can be found in nearly all RNA viruses. Activation of the host innate immune response begins with recognition of pathogen-associated molecular pattern (PAMP) by pattern recognition receptors (PRRs). Activation of host PRRs by non-self nucleic acids such as those found in RNA viruses trigger a signaling cascade resulting in the production of type I Interferon (IFN-α/β) and expression of hundreds of IFN-stimulated genes (ISGs) that target specific aspects of the viral life cycle, including virus binding to attachment receptors, virus entry, RNA synthesis, progeny virion assembly, and egress. In this review, we will discuss mechanisms by which emerging and re-emerging viruses utilize to evade PRR detection and inhibit innate immune signaling.
Masking viral PAMPs that trigger host PRRs
Hiding viral RNA replication from cytosolic PRRs
The mammalian immune system has two major classes of PRRs responsible for turning on type I IFN-mediated antiviral response to viral RNA: first, retinoic acid-inducible gene 1 (RIG-I)-like receptors, or RLRs and second, Toll-like receptors (TLRs). These intracellular ssRNA and dsRNA sensors have been reviewed in detail [4, 5] and recognize specific viral PAMPs such as 5′ diphosphate and triphosphate groups on non-self mRNA. The RLRs RIG-I, melanoma differentiation-associated gene 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2) are found in the cytosol and respond during RNA viral infection. In contrast, TLR3 and TLR 7/8 recognize dsRNA and ssRNA, respectively, and are localized within endosomes.
Many RNA viruses assemble replication complexes and drive viral RNA synthesis within a virally induced enclosed organelle inside the host cell. These membrane vesicles, also termed viroplasm-like structures (VLS), are thought to shield nascent viral RNAs from activating the RLRs during RNA synthesis [6] and have been found in various RNA viruses including CoV and flaviviruses (Table 1 ). Flavivirus-induced viroplasms contain RNA polymerase, nonstructural (NS) proteins NS2B, NS3, NS4A, NS4B and double-stranded RNA (dsRNA) [7•]. Immunoelectron microscopy revealed viral replication and virus budding occur within viroplasm and VLSs in Dengue virus (DENV)-infected mosquito cells [8], thus indicating these sites to be important for viral replication. Moreover, WNV, DENV and Japanese encephalitis virus (JEV) genomic RNA become sensitive to nuclease treatment after disruption of these membrane vesicles [9], which indicates these structures shield the viral genome from endogenous RNA degrading processes in the cytosol. It has also been suggested that membrane vesicle protection of viral RNA delays onset of IFN responses, but has not been definitively shown. A recent study utilizing flaviviruses JEV and DENV correlated high amounts of cytosolic JEV RNA with robust IFNβ production and low amounts of cytosolic DENV RNA with weak IFNβ production [10]. However, this work still does not directly address whether flavivirus viroplasms prevent IFN production. Interestingly, the Influenza virus (IAV), which causes annual seasonal epidemics worldwide, replicates within the nucleus of infected cells away from cytosolic RLRs and TLRs [11••]. Nonetheless, additional studies are warranted to elucidate the significance of viroplasm and VLSs to viral pathogenesis.
Table 1.
Summary of mechanisms of innate immune evasion
| Mechanism | Pathogen | Major protein(s) involved | Process | References |
|---|---|---|---|---|
| Membrane vesicle to hide replication, RNA PAMPs | CoV (MERS, SARS) | NSP3–NSP6, NSP13 | Double membrane vesicle (DMV) | [86, 87, 88] |
| Flavivirus | NS2B, NS3; NS4A, NS4B | Vesicle packet (VP); convoluted membrane (CM) | [7•, 8] | |
| ‘Cap binding’ | CoV (MERS, SARS) | Nsp13: RNA triphosphatase Nsp14: N7 Guanine-MTase Nsp16: 2′O-MTase Nsp10: Nsp16 cofactor |
Attachment of a 5′mRNA cap mimic similar to eukaryotic ‘Type-1 cap’ consisting of a GMP-bound diphosphate moiety bound to N-terminus containing methyl groups at the guanine at the N-7 and ribose 2′-O position (m7GpppNm-RNA) | [89, 90, 91, 92, 93] |
| Flavivirus | NS3: RNA triphosphatase NS5: RNA Polymerase, N7-Guanine MTase, 2′-O-MTase |
[94, 95, 96, 97, 98] | ||
| Mononegavirales (EBOV, Measles) | 2′O-MTase domain on C-terminus of viral genome | [99, 100] | ||
| CHIKV | nsP1: MTase nsP2: Nucleoside triphosphatase, MTase nsP3: protein recruitment nsP4: RNA Polymerase |
[101, 102, 103, 104] | ||
| ‘Cap Snatching’ | IAV | PB1 | Endonuclease excises cellular 5′ caps from host, attaches to viral RNA; also important for initiating viral transcription | [16, 105, 106] |
| Bunyaviruses | l-protein | [15] | ||
| Direct inhibition of PRRs | IAV | NS1: Directly complexes with RIG-I, ssRNA, MAVS PB1, PB2, PA, NP: Complexes with RIG-I |
Direct binding and inhibition of RLRs and RLR signaling components | [17, 18, 19] |
| Inhibitors of RIG-I Ubiquitination | IAV | NS1: interacts with TRIM25, Riplet | Inhibits ubiquitination of RIG-I | [21, 22] |
| Bunyavirus (SFTSV) | NS: Sequesters RIG-I, TRIM25, TBK1 into cytoplasmic structures | [23, 24, 25] | ||
| Inhibition of mitochondria-associated molecules | Flavivirus (DENV, HCV, YFV?) | NS2B/NS3 protease (DENV) NS4B (HCV/YFV) NS3/4A (HCV) |
NS2B/NS3 cleaves human STING; NS4B inhibition of STING, MAVS, TBK1 by interaction; NS3/4A cleaves MAVS |
[26••, 27, 28, 29, 30, 31, 38] |
| CoV (human CoV, SARS) | PLP domains in nsp3 | Proteolytic cleavage inhibits STING dimerization | [32, 33] | |
| IAV | PB1-F2, PB2 | Interacts with and inhibits MAVS | [34, 35] | |
| Suppressors of IKK Kinases | DENV | NS2B/3 | Inhibits IKKɛ | [39] |
| IAV | NS1 | Interacts with IKK α/β, inhibits IκBα phosphorylation | [41] | |
| EBOV | VP35 | Inhibits IKKɛ | [40] | |
| SARS-CoV | M | Interacts with IKKβ, TRAF6, TBK1, IKKɛ, inhibits NFκB | [42, 43] | |
| Inhibition of IRF3, NFκB | SARS-CoV | ORF3b, ORF6, N | Inhibits activation and nuclear translocation of IRF3 (and NFκB, MERS ORF4a) by unknown mechanism | [44] |
| MERS-CoV | ORF4a, 4b, 5, M | [45] | ||
| Interactions with PACT, PKR | IAV | NS1: Binds PACT, PKR | Disrupts PACT:PKR and PACT:RIG-I interactions to inhibit IFN production | [46, 48] |
| EBOV | VP35: Binds PACT | [50] | ||
| Dicer and RNAi inhibition | EBOV | VP30: Interacts with TRBP, Dicer VP35: Interacts with Dicer, TRBP, PACT; RNA binding partners in RISC? VP40:? |
Inhibition of host RNAi mechanisms that target foreign RNA for degradation | [52] |
| IAV | NS1: Binds PACT | [46, 47] | ||
| DENV | NS4B: Inhibits conversion of dsRNA → siRNA by Dicer | [55] | ||
| Inhibition of JAK1, TYK2 activation | Flavivirus JEV | NS5 | Inhibits phosphorylation of JAK1, TYK2 | [61] |
| Flavivirus WNV | NS4B | [60] | ||
| Inducer of SOCS1, SOCS3 | EBOV | VP40, glycoprotein | Upregulates expression of SOCS1, SOCS3 | [65] |
| Flavivirus JEV | [62] | |||
| Flavivirus WNV | [63] | |||
| IAV | NS1 | [66, 67] | ||
| CHIKV | [64•] | |||
| Inhibitor of STAT1 activation | CHIKV | nsP2 | Inhibits STAT1 and/or STAT2 phosphorylation; DENV NS4B binds and promotes degradation of STAT2 | [76] |
| Flavivirus JEV | NS4A, NS5 | [61, 72] | ||
| Flavivirus WNV | Core, E, NS1-5 | [68, 75•] | ||
| Flavivirus DENV | NS4B, NS5 | [69, 73, 74] | ||
| IAV | NS1 | [66] | ||
| EBOV | VP24 | Binds STAT1 directly; inhibits Karyopherin α proteins | [79, 80] | |
| SARS-CoV | nsp1; ORF6; PLP | (1) Inhibits STAT1 phosphorylation (2) Sequesters Karyopherin α2 to prevent STAT1 nuclear translocation (3) Increases E2-25k Ub ligase to promote Ub-dependent proteosomal degradation of Erk1 to inhibit STAT1 phosphorylation |
[44, 81, 82, 84] | |
| Inhibitor of STAT2 activation | DENV | NS5 | Increases STAT2 degradation | [69, 73, 74] |
| IAV | NS1 | Inhibits STAT2 phosphorylation | [66] | |
5′ cap that masks viral PAMPs
The RLRs RIG-I and MDA5 are activated upon recognizing non-self dsRNAs consisting of unmethylated 5′ tri-phosphate or di-phosphate ends [12]. Multiple RNA viruses have evolved enzymatic processes that catalyze post-translational modification of nascent mRNAs that effectively mask viral PAMPs by mimicking eukaryotic RNA structures. These processes include the addition of 5′ cap structures similar to eukaryotic ‘Type 1 caps’ composed of a GMP-bound diphosphate moiety bound to the N-terminus of the transcribed mRNA molecule (GpppN-RNA) with methyl groups at guanine residues at the N-7 and ribose 2′-O position (m7GpppNm-RNA). These post-translational modifications are catalyzed by phosphatases and methyltransferases (MTases). Studies examining various viral pathogens have demonstrated that loss of these post-translational modifications via mutating virally encoded phosphatases and MTases leads to decreased viral replication and enhancement of host antiviral responses. Notably, a recent study using SARS-CoV lacking Nsp16, which acts as a 2′ O-MTase, showed that loss of 2′ O-methylation of the SARS-CoV genome enhanced host type I IFN production [13]. Moreover, mutant JEV strains lacking the 2′-O MTase are attenuated compared to wildtype JEV strains [14•]. Therefore, post-translational modification of viral mRNA is an effective immunoevasion strategy that prevents induction of host antiviral processes. Table 1 summarizes other known cap-binding enzymes found in various RNA viruses.
Some RNA viruses lack the catalytic enzymes to generate their own RNA cap. These viruses hijack the host's cellular mRNA by expressing viral endonucleases and utilize these caps to mask viral RNA PAMPs. In particular, IAV and other negative-strand segmented viruses such as Bunyaviruses have been shown to utilize this mechanism of ‘cap snatching’ [15]. IAV RNA polymerase complexes with PB1, an endonuclease with cap binding activity that captures a N7 methylguanosine 5′ cap from host cellular pre-mRNA transcribed by host RNA polymerase II as a means to generate primers required for initiation of viral RNA transcription [16]. A similar endonuclease is also found in Bunyaviruses encoded by the l-protein [15]. The presence of this 5′ cap is important for initiation of viral transcription during replication, maintaining RNA stability, and cap-dependent translation as well as evading detection by cytosolic PRRs.
Viral blockade of PRR signaling cascades and type I IFN signaling
RLRs preferentially bind and detect dsRNA, leading to downstream production of type I IFN and activation of other antiviral genes. Within the three RLRs, RIG-I has been studied in the best detail in the context of host–pathogen interactions and signaling. Specifically, RIG-I (and MDA5) signal through mitochondrial antiviral signaling (MAVS) protein to activate Tank Binding Kinase 1 (TBK1) and I-kappa-B kinase ɛ (IKKɛ), leading to activation of transcription factors Interferon Regulatory Factor (IRF) 3 and 7 as well as NFκB. In contrast, recognition of RNA PAMPs by endosomal TLRs signals through adaptors TRIF in TLR3 and MyD88 in TLR7/8. Similar to RLRs, triggering endosomal TLR3/TRIF results in both NFκB and IRF3/7 activation, the latter leading to production of type I IFN and induction of other antiviral genes, yet few viral proteins that directly antagonize TLR signaling adaptor proteins have been identified. In this section, we highlight common antiviral signaling molecules targeted by various emerging and re-emerging RNA viral pathogens (Figure 1 ).
Figure 1.
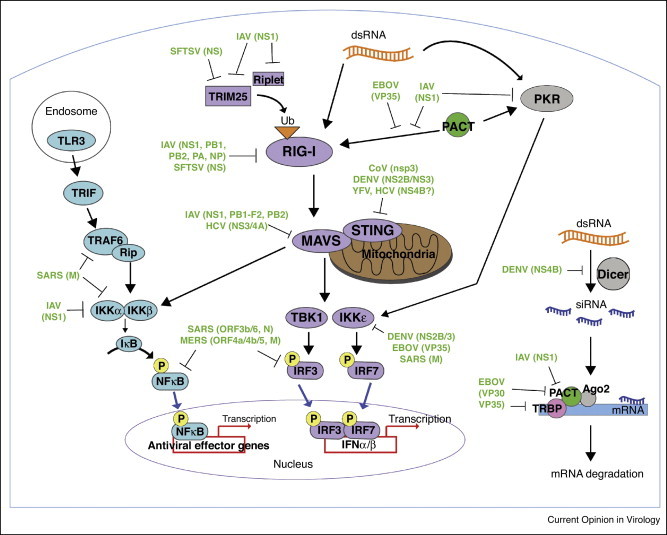
Summary of RNA virus inhibition of cytosolic innate immune signaling pathways. Viral protein antagonists of three cytosolic signaling pathways are represented here: (1) TLR3 activation leads to signaling through TRIF, TRAF6/Rip, and IKKα/β to turn on NFκB. (2) RIG-I activation requires binding to dsRNA and subsequent ubiquitination by E3 ubiquitin ligases, TRIM25 and Riplet. RIG-I is also independently activated by interactions with PACT. This signals through mitochondrial-bound MAVS, leading to TBK1/IKKɛ activation to initiate transcription factors NFκB, IRF3/IRF7. Additionally, PKR is activated by PACT or dsRNA binding, which also turns on TBK1/IKKɛ independently of RIG-I activation. (3) The RNAi pathway involves Dicer conversion of dsRNA to siRNA. The siRNA recognizes its complementary sequence in the target mRNA molecule, which recruits the RISC comprised of Argonaute 2, PACT and TRBP. The resulting mRNA is proteolytically degraded. Viral proteins that inhibit host signaling molecules are indicated. TRIF, TIR-domain-containing adapter-inducing interferon-β; TRAF6, TNF receptor associated factor; RIP, receptor-interacting protein.
Direct inhibition of RIG-I activation
IAV NS1 complexes with 5′-phosphorylated ssRNA and RIG-I [17], but also interacts with RIG-I and MAVS [18]. Mutant NS1 that is unable to bind RNA does not co-precipitate with RIG-I [17], supporting the notion that NS1 may require RNA intermediates to associate with RIG-I. More recently, subunits of the IAV RNA polymerase complex, basic polymerase 1 and 2 (PB1, PB2), the acidic polymerase (PA) and nucleoprotein (NP) were found to directly complex with RIG-I, yet the physiological effects of these interactions have not been identified [19]. Interestingly, interactions between RIG-I and the IAV RNA Polymerase complex occurred in the absence of RNA and were not competitively inhibited by IAV NS1, illustrating that these interactions do not require RNA intermediates and that IAV may indeed directly inhibit RIG-I.
After recognizing viral dsRNA, RIG-I undergoes conformational changes and post-translational modification that includes K63-linked polyubiquitinated on lysine residues of their CARD and C-terminal domains. This reaction is catalyzed by the E3 ubiquitin ligase TRIM25 [20]. The multifunctional NS1 protein of IAV and NS protein of Bunyavirus Severe Fever with Thrombocytopenia Syndrome virus (SFTSV) both target TRIM25. IAV NS1 interacts directly with TRIM25, thus reducing RIG-I polyubiquitination [21, 22]. Studies by Rajsbaum et al. also implicated NS1 in interacting with and inhibiting Riplet, an E3 ligase that polyubiquitinates the C-terminal region of RIG-I [22]. Thus IAV NS1 exerts multiple mechanisms in suppressing RIG-I ubiquitination. In contrast, SFTSV exerts a different mechanism to antagonize RIG-I. SFTSV encodes NS protein that sequesters RIG-I, TRIM25 and TBK1 into cytoplasmic structures similar to the aforementioned viroplasms to inhibit RIG-I triggering of type I IFN [23, 24, 25]. SFTSV NS proteins can inhibit IFNβ promoter activation, and was shown to induce re-localization of RIG-I, TRIM25 and TBK1 into cytoplasmic structures that resemble inclusion bodies. Therefore RNA viruses are able to directly suppress PRR activation as a mode of immune evasion.
Inhibition of mitochondrial-associated signaling molecules: MAVS and STING/MITA
ER protein stimulator of IFN genes, or STING (also known as MITA) is responsible for activation of TBK1-dependent phosphorylation of cytosolic IRF3 and turning on production of type I IFN. Flaviviruses DENV and YFV as well as SARS-CoV can directly inhibit STING and its downstream constituents, albeit through distinct mechanisms. The DENV NS2B/NS3 protease complex can directly cleave human STING, but not its mouse homologue MPYS [26••, 27, 28]. YFV NS4B, like DENV NS4B, displays similar homology to the catalytic domain found on the N-terminus of STING that is required for its function, and by association it was speculated that YFV NS4B also inhibits STING [29]. Indeed, YFV NS4B was found to co-localize with human STING via confocal microscopy, but functional inhibition of STING by YFV NS4B has still yet to be confirmed. In contrast, NS4B of another flavivirus, Hepatitis C virus (HCV) was experimentally demonstrated to inhibit STING [30, 31]. It is unclear whether HCV NS4B also cleaves STING, yet it is likely that STING inhibition via NS4B is conserved among all flaviviruses, including YFV.
Human CoV and SARS-CoV also encode an inhibitor of STING through expression of papain-like protease (PLP) domains within nonstructural protein 3 (nsp3) that function as both proteases and deubiquitinating enzymes [32, 33]. Inhibition of STING through PLPs in SARS-CoV and PLP2 in human CoV occurs via protease-dependent and protease-independent mechanisms. CoV PLPs can inhibit the dimerization of STING, which is required for its activation, as well as inhibit its ability to form complexes with MAVS and IKKɛ [33]. CoV PLPs can also decrease the ubiquitination of STING as well as RIG-I, TBK1 and IRF3, but interestingly does not require ubiquitin catalytic activity to reduce ubiquitinylation on STING [32, 33]. Likewise inhibition of proteolytic activity does not affect CoV PLPs from inhibiting type I IFN production. Thus the exact mechanism of how PLP domains within CoV nsp3 can antagonize cytosolic STING and inhibit IFN production requires further investigation.
Although no known inhibitors of STING have been characterized in IAV, PB1-F2 and PB2 proteins have been shown to interact with and inhibit MAVS [34, 35]. Both PB2 and PB1-F2 are characterized as inhibitors of IFNβ production via direct interaction with MAVS. A recent study illustrated that a single Threonine to Isoleucine mutation at position 588 (T588I) increased IAV PB2's ability to bind MAVS and inhibit type I IFN production [36]. These mutations were originally isolated in swine IAV variants, but also found in the H1N1 IAV that caused the 2009 pandemic. This implicates PB2 as a potent antagonist of MAVS that may have broader functions in determining the virulence of IAV strains. MAVS is also inhibited by the actions of HCV protease NS3 and its required cofactor NS4A [37, 38]. HCV NS3/NS4A was shown to cleave MAVS at Cysteine-508 and is a potent inhibitor of IFNβ. Therefore inhibition of MAVS and STING are effective viral mechanisms of immune evasion.
Viral suppressors of IκB kinases: effects on TLR and RLR IFN signaling pathways
Activation of dsRNA via TLRs and RLRs involve distinct players that converge downstream of PRR activation. Recognition of dsRNA through TLR3, RLRs RIG-I/MDA5 or the broad-spectrum dsRNA responder protein kinase RNA (PKR) converges upon activation of TBK1 and IKKɛ, which phosphorylates IRF3 and IRF7, respectively, and allows IRF3/7 translocation into the nucleus to mediate IFN gene transcription. In addition, TLR3 and RLR pathways both activate IKKα/β kinases that activate NFκB the former via TRIF recruitment of TRAF6/RIP1 and the latter via MAVS. Several inhibitors of IKKα/β and IKKɛ have been identified among RNA viruses. DENV NS2B/3, which we have discussed as a human STING protease, and the multifunctional EBOV VP35 can inhibit IKKɛ and production of type I IFN by preventing translocation of transcription factors IRF3 and IRF7. Both DENV NS2B/3 and EBOV VP35 bind directly to IKKɛ to disrupt complex formation with IRF3 [39, 40].
In contrast, suppression of IKKα/β will lead to inhibition of NFκB expression of NFκB-dependent genes that regulate a myriad of biological processes, spanning survival to surface molecule expression. IAV and CoV have been shown to target IKKα/β resulting in NFκB inhibition. IAV and CoV have been shown to target IKKα/β resulting in NFκB inhibition. IAV NS1 interacts with the N-terminal kinase domains of IKKα and IKKβ [41] and also inhibits phosphorylation of IκBα, the kinase responsible for phosphorylating NFκB and allowing its translocation to the nucleus. SARS-CoV membrane glycoprotein (M) physically interacts specifically with IKKβ but not with IKKα [42]. Interaction with IKKβ correlates with SARS-CoV inhibition IκBα phosphorylation required for its degradation resulting in sustained inhibition of NFκB nuclear translocation. Moreover, SARS-CoV M glycoprotein directly complexes with TRAF6, TBK1, and IKKɛ but does not inhibit IRF3 activation [43].
The genome of CoV contains several open reading frames (ORFs) that encode accessory proteins. These accessory proteins play no role in replication or release of infectious viruses, but may have immunoevasion functions. The SARS-CoV genome contains ORFs 3b and 6, and along with nucleocapsid (N) protein, was shown to be potent inhibitors of IRF3 phosphorylation and nuclear translocation; N protein was also able to inhibit NFκB activity [44]. MERS-CoV Membrane (M), ORF 4a, ORF 4b, and ORF 5 are also potent antagonists of IFN production [45]. ORF 4a, ORF 4b, ORF 5, and M inhibit the activation and translocation of IRF3, while ORF 4a also inhibits the activity of NFκB. The exact mechanism by which these CoV accessory proteins abrogate IRF3/NFκB activity remains to be determined.
Interactions with PACT and its effect on PKR and RIG-I
EBOV VP35 and IAV NS1 both interact with PKR activating protein (PACT), a dsRNA binding partner involved with eukaryotic RNAi as well as activating RIG-I and PKR (Protein Kinase R). PKR is a nonspecific dsRNA sensor that can turn on TBK1/IKKɛ and downstream IFN production, among other functions. IAV NS1 can inhibit PKR-mediated type I IFN production in two manners: first, binding to PACT and preventing its interaction with PKR [46, 47] or second, directly binding to PKR [46, 48]. In addition, PACT directly activates and sustains RIG-I activation independent of Dicer and PKR via binding to the C-terminal domain of RIG-I and stimulating its ATPase activity [49]. Both IAV NS1 and EBOV VP35 have also been shown to block RIG-I activation through interaction with PACT. IAV NS1 binds PACT directly through its RNA binding domain, and is able to interfere with PACT interactions with RIG-I, thus reducing IFNβ production [47]. Similarly, EBOV VP35 binds to PACT via its C-terminal IFN inhibitory domain, which is sufficient to disrupt PACT:RIG-I interactions and subsequent IFN production [50]. Therefore, IAV NS1 and EBOV VP35 interactions with PACT interfere with type I IFN production by suppressing dsRNA-binding proteins PKR and RIG-I. Thus viral inhibition of type I IFN production via PACT can occur through PKR-dependent or PKR-independent means.
Inhibition of Dicer and eukaryotic RNAi processes
RNAi is an important gene regulation pathway that negatively regulates gene expression via short nucleotide RNA molecules. The generation of small RNAs is mediated by an RNase called Dicer that digests dsRNA into siRNA, which then become a part of the RNA-induced silencing complex (RISC). The RISC includes first, the endonuclease Argonaute 2 (Ago2); second, dsRNA binding partners Transactivation response RNA binding protein (TRBP) and PACT; and third, the unwound strand of siRNA serving as the ‘guide strand’ that identifies ssRNA transcripts with complementary sequence as a target for degradation [51•] (Figure 1). Several RNA viruses encode proteins that antagonize the mammalian RNAi pathway. EBOV nonstructural proteins VP30, VP40 and VP35 have been identified as mammalian RNAi suppressors, with VP35 exhibiting the most potent antagonistic function [52, 53]. VP35 readily immunoprecipitates with Dicer, TRBP and PACT, while VP30 only interacts with Dicer and TRBP. The ability of VP35 and VP30 to interact with TRBP and PACT also implicated their participation in inhibiting RIG-I signaling and the IFN production pathway, as both TRBP and PACT regulate PKR [54]. IAV NS1 has also been identified as a suppressor of eukaryotic RNAi. Similar to EBOV VP35, IAV NS1 can also bind PACT within the RNAi pathway, but also affects type I IFN production by disrupting PACT:RIG-I interactions [46, 47]. Recently, DENV NS4B has also been implicated in suppressing Dicer-dependent RNAi activation [55]. Specifically, NS4B could inhibit in vitro conversion of dsRNA to siRNA by Dicer. Transient knockdown of the central components of endogenous RNAi (e.g. Dicer, Argonaute) enhanced DENV replication in Huh7 cells, which supports that mammalian RNAi limits viral pathogenesis of DENV.
Although there have been various siRNAs directly involved in antiviral functions in plants and invertebrates, a similar function in the mammalian immune system has been difficult to discern. Few mammalian siRNA/microRNAs (miRNAs) specific to viral mRNA have been identified, and there is conflicting evidence as to whether RNAi components are directly required to limit viral pathogenesis. A previous study by Schopman et al. conducted deep-sequencing of HIV-infected T cells to identify various HIV-specific siRNAs that inhibited HIV replication, representing the first convincing report of mammalian RNAi as an antiviral mechanism [56]. Two studies identified mammalian RNAi mechanisms from cells infected with mosquito-borne Nodamura virus (NoV) [57, 58]. Specifically, NoV encodes a suppressor of mammalian RNAi, B2, and functional B2 was required for NoV replication in hamster and mice [57]. However, a study by Seo and colleagues observed that mammalian RISC formation was inhibited by poly I:C and other viruses that triggered TLRs and RLRs, which argues that at least in some contexts, mammalian RNAi may not function as an antiviral process [59]. Thus to date, it is still unknown whether mammalian RNAi is a primary antiviral pathway to curtail virus replication.
Inhibition of JAK1/TYK2
Type I IFN signaling is a crucial component in control of viral replication, and is initiated upon IFNα/β binding of interferon α/β receptor (IFNAR). This leads to recruitment of janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), which phosphorylate transcription factors signal transducers and activators of transcription (STAT) 1 and 2. STAT1 and STAT2 form a complex with IRF9, which translocates to the nucleus to initiate transcription of interferon stimulated genes (ISGs) (Figure 2 ). IFN antagonists have been well-characterized in a number of RNA viruses, and target multiple components of the IFNAR signaling pathway. Flaviviruses JEV and WNV inhibit phosphorylation and activation of JAK1 via NS5 and NS4B, respectively [60, 61]. WNV NS4B also inhibits phosphorylation and activation of TYK2 [60]. Viral IFN antagonism is also mediated through induction of the suppressors of cytokine signaling (SOCS) 1 and 3 that negatively regulate JAK1. Flaviviruses WNV and JEV both induce transcriptional expression of SOCS1/3 early after infection [62, 63] in addition to other mechanisms. Likewise, CHIKV and EBOV both induce increased expression of SOCS1 [64•, 65]. Finally, IAV NS1 also induces upregulation of SOCS1/3 [66, 67].
Figure 2.
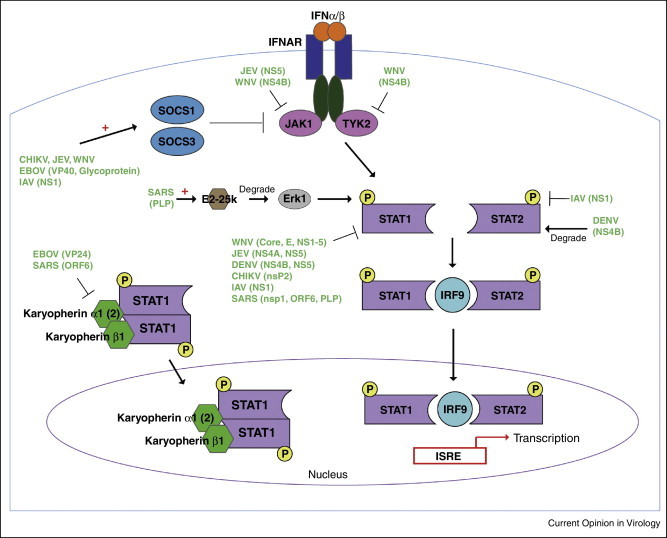
RNA virus inhibition of type I IFN signaling. Binding of IFNAR by type I IFN (IFNα/β) initiates downstream kinases, JAK1 and TYK2. JAK1 and TYK2 phosphorylates STAT1 and STAT2, which leads to the formation of the STAT1:IRF9:STAT2 complex. This complex translocates to the nucleus and initiates transcription of IFN-stimulated response element (ISRE). SOCS1 and/or SOCS3 negatively regulate JAK-STAT signaling by interacting with JAK1/TYK2. Nuclear accumulation of phosphorylated STAT1 also depends on Karyopherins. Karyopherin α1 (or α2) complexes with phosphorylated STAT1, which then recruits Karyopherin β1. The complex of phospho-STAT1:Karyopherin α:Karyopherin β1 is able to cross the nuclear membrane and regulate gene expression. Viral proteins that either inhibit or degrade host signaling factors are indicated. ‘+’ denotes upregulation of expression.
Inhibition of STAT1/STAT2
Numerous RNA viral pathogens antagonize STAT1 and/or STAT2. The nonstructural proteins of multiple flaviviruses have been shown to suppress STAT1/2 activity. Specifically, NS5 (JEV, WNV, DENV; [61, 68, 69]) NS4B (WNV, DENV; [70, 71]) and NS4A (JEV; [72]) can inhibit STAT phosphorylation and their subsequent activation. DENV NS5 has also been shown to promote the proteosomal degradation of STAT2 [69, 73, 74]. Specifically, DENV NS5 directly binds STAT2 to UBR4, an E3 ubiquitin ligase, to initiate the proteolytic degradation of STAT2 [74]. This suggests that other NS proteins may contribute to NS5-mediated targeting of STAT2 for proteolytic cleavage. Additionally, the structural (core and envelope, E) and nonstructural NS1-NS5 proteins of a pathogenic WNV strain (Tx-HC 2002) inhibits STAT1/2 phosphorylation and IFN production, and introduction of these proteins into a non-pathogenic strain of WNV (Madagascar 78) enhances IFN inhibition and virus-induced cytopathology [75•]. This indicates that STAT1/2 suppressor function is a major determinant of virulence within WNV strains, which is likely to be the case for other RNA viruses that target STAT signalisng.
Several RNA viruses suppress STAT1/2 function by inhibiting phosphorylation in addition to nuclear translocation. CHIKV nsP2 inhibits phosphorylation of STAT1/2 and their nuclear translocation [76], which was similar for another alphavirus Sindbis virus nsP2 [77]. CHIKV nsP2 does not induce degradation of STAT1/2 as observed in other viruses like DENV, and the mechanism for how CHIKV nsP2 inhibits STAT nuclear accumulation is unclear. IAV NS1 is also capable of downregulating STAT1/2 phosphorylation and inhibiting the nuclear translocation of phosphorylated STAT1/2 [66, 78]. NS1 may hinder nuclear accumulation by disrupting phospho-STAT:DNA complexes that form and migrate to the nucleus to initiate gene transcription [66]. Interestingly, H5N1 and H1N1 IAV NS1 proteins can also downregulate expression of IFNAR 1 and 2 to inhibit type I IFN signaling [66]. Thus IAV NS1 mediates three modes of IFN signaling antagonism: first, upregulation of SOCS1/3 to diminish activation of JAK1/TYK2; second, inhibition of STAT1/2 phosphorylation and nuclear translocation; and third, downregulation of IFNAR1/2 expression.
EBOV VP24 prevents the nuclear accumulation of STAT1 by binding to Karyopherin α1 protein (importin α1). Karyopherin α1 belongs to a group of nuclear transport carriers that upon recognition of the nuclear localization signal (NLS) of a nascent protein recruits Karyopherin β1 (importin-1) to the complex. Ultimately the complex containing the nascent protein, Karyopherin α1 and Karyopherin β1 can cross the nuclear membrane. VP24 specifically binds to Karyopherin α1 and other members of the Karyopherin α subgroup and inhibits its interaction with phosphorylated STAT1 [79, 80]. This function was not observed for EBOV VP35, nor did VP24 affect upregulation of STAT1 expression. Therefore VP24 interactions with Karyopherin α proteins limit STAT1 accumulation in the nucleus.
SARS-CoV is capable of suppressing IFN signaling via three distinct mechanisms that all target STAT1. First, SARS-CoV nsp1 inhibits STAT1 phosphorylation, but has little or no effect on STAT2, JAK1 or TYK2 phosphorylation [81]. This study did not examine whether nsp1 affects STAT1 nuclear transport, but two independent studies showed that SARS-CoV open reading frame 6 (ORF6) encodes accessory proteins that inhibit STAT1 nuclear translocation [44, 82]. Similar to EBOV VP24, SARS-CoV ORF6 also inhibited nuclear accumulation of STAT1 by sequestering Karyopherin α2 at the ER/Golgi and preventing its interaction with cytosolic phospho-STAT1 [82]. ORF6 also relocalizes Karyopherin β1 at the ER/Golgi membrane, thus inhibiting the formation of the STAT1:Karyopherin complex required for nuclear transport. Importantly, infection with MERS-CoV, which does not encode ORF6, does not inhibit nuclear translocation of phospho-STAT1 [83]. This illustrates a clear distinction between SARS and MERS-CoV, which may reflect differences in virulence and pathogenesis between these viruses.
Finally, SARS-CoV also mediates a third mechanism of IFN signaling inhibition via PLPs. SARS PLP upregulates the expression of cellular E3 ubiquitin ligase E2-25k, which is upregulated by IFNα signaling [84]. PLP transfection into human pro-monocytes significantly induced expression of cellular E2-25k. Increased E2-25k correlated with significantly decreased extracellular signal-regulated kinase (Erk) 1, but not Erk2, protein in the cell, which was mediated through ubiquitin-dependent proteolytic degradation of Erk1. The degradation of Erk1 was concomitant with decreased IFNα-dependent STAT1 and c-Jun phosphorylation. Thus SARS-CoV antagonizes STAT1 activity through three distinct mechanisms: first, nsp1-dependent inhibition of STAT1 phosphorylation; second, ORF6-dependent sequestration of Karyopherin α1 and STAT1 translocation; third, PLP-dependent induction of E2-25k to promote Erk1 degradation and decreased STAT1 phosphorylation.
Conclusions
The emergence and re-emergence of RNA viruses continues to pose a significant public health threat worldwide. However, few vaccines for use in humans or specific antiviral therapeutics exist for several emerging/re-emerging viruses, including SARS, MERS-CoV, WNV, or BUNV. Thus, it is crucially important to understand the virus–host interactions that govern immunity, disease severity, and infection outcome. Recent findings have found that several viruses utilize multifaceted strategies to evade multiple arms of the innate immune response. Functionally pleiotropic suppressors such as IAV NS1 and EBOV VP35 reveal the complexity and level of sophistication by which these viral pathogens have developed as immune escape artists, but their nature as multifunctional antagonists also makes them attractive candidates in vaccines and therapeutics. For example, interventions that target IAV NS1 would suppress multiple IAV immunoevasion mechanisms, increasing the potential effectiveness of a candidate vaccine or therapy. Indeed, deletion mutants of IAV have already been tested in live attenuated vaccines in Phase I/II clinical trial, resulting in enhanced IAV-specific antibody responses [85••]. With the ongoing Ebola epidemic in West Africa, it is tempting to imagine that targeting the multifunctional EBOV VP35 may lead to the development of effective therapeutics and even a possible vaccine. Most importantly, studies aimed at expanding our understanding of viral evasion strategies undoubtedly reveal new insights into innate immune signaling and host antiviral defenses. Indeed, discovery of the RIG-I signaling pathway led to the identification of viral factors that antagonize RLR signaling. As viral pathogens continue to cause threat of global epidemics, improved understanding of host defenses will be crucial to limiting the burden of human disease.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
The Suthar laboratory is supported by start-up funds provided by the Children's Healthcare of Atlanta, Emory Vaccine Center, the Georgia Research Alliance, the Emory University Research Council and NIH Grants R03AI109194, U19AI083019, R56AI110516 and 5U19AI057266.
References
- 1.Murray K.O., Ruktanonchai D., Hesalroad D., Fonken E., Nolan M.S. West Nile virus, Texas, USA, 2012. Emerg Infect Dis. 2013;19:1836–1838. doi: 10.3201/eid1911.130768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papa A., Xanthopoulou K., Gewehr S., Mourelatos S. Detection of West Nile virus lineage 2 in mosquitoes during a human outbreak in Greece. Clin Microbiol Infect. 2011;17:1176–1180. doi: 10.1111/j.1469-0691.2010.03438.x. [DOI] [PubMed] [Google Scholar]
- 3.Kuehn B.M. Chikungunya virus transmission found in the United States: US health authorities brace for wider spread. JAMA. 2014;312:776–777. doi: 10.1001/jama.2014.9916. [DOI] [PubMed] [Google Scholar]
- 4.Quicke K.M., Suthar M.S. The innate immune playbook for restricting West Nile virus infection. Viruses. 2013;5:2643–2658. doi: 10.3390/v5112643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkins C., Gale M., Jr. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.den Boon J.A., Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol. 2010;64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 7•.Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P., Fuller S.D., Antony C., Krijnse-Locker J., Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dengue virus replication occurs in membrane-bound organelles. This study was one of the initial studies that visualized Dengue viroplasms and demonstrated these structures as sites of viral replication.
- 8.Junjhon J., Pennington J.G., Edwards T.J., Perera R., Lanman J., Kuhn R.J. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J Virol. 2014;88:4687–4697. doi: 10.1128/JVI.00118-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uchil P.D., Satchidanandam V. Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J Biol Chem. 2003;278:24388–24398. doi: 10.1074/jbc.M301717200. [DOI] [PubMed] [Google Scholar]
- 10.Uchida L., Espada-Murao L.A., Takamatsu Y., Okamoto K., Hayasaka D., Yu F., Nabeshima T., Buerano C.C., Morita K. The dengue virus conceals double-stranded RNA in the intracellular membrane to escape from an interferon response. Sci Rep. 2014;4:7395. doi: 10.1038/srep07395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11••.Jackson D.A., Caton A.J., McCready S.J., Cook P.R. Influenza virus RNA is synthesized at fixed sites in the nucleus. Nature. 1982;296:366–368. doi: 10.1038/296366a0. [DOI] [PubMed] [Google Scholar]; Influenza viral replication occurs within the nucleus. This was one of the first studies to illustrate that Influenza replicates within the host nucleus.
- 12.Loo Y.M., Gale M., Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menachery V.D., Yount B.L., Jr., Josset L., Gralinski L.E., Scobey T., Agnihothram S., Katze M.G., Baric R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-o-methyltransferase activity. J Virol. 2014;88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.Li S.H., Dong H., Li X.F., Xie X., Zhao H., Deng Y.Q., Wang X.Y., Ye Q., Zhu S.Y., Wang H.J. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J Virol. 2013;87:5812–5819. doi: 10.1128/JVI.02806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Attenuation of JEV can be attained by abrogating 2′-O methylase activity. This study illustrates that targeting the 2′-O methylase of JEV results in an attenuated mutant that elicits robust immune responses, which may be useful for vaccine design.
- 15.Reguera J., Weber F., Cusack S. Bunyaviridae RNA polymerases (l-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010;6:e1001101. doi: 10.1371/journal.ppat.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih S.R., Krug R.M. Surprising function of the three influenza viral polymerase proteins: selective protection of viral mRNAs against the cap-snatching reaction catalyzed by the same polymerase proteins. Virology. 1996;226:430–435. doi: 10.1006/viro.1996.0673. [DOI] [PubMed] [Google Scholar]
- 17.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 18.Mibayashi M., Martinez-Sobrido L., Loo Y.M., Cardenas W.B., Gale M., Jr., Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W., Chen H., Sutton T., Obadan A., Perez D.R. Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J Virol. 2014;88:10432–10447. doi: 10.1128/JVI.01383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maelfait J., Beyaert R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol Mol Biol Rev. 2012;76:33–45. doi: 10.1128/MMBR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gack M.U., Albrecht R.A., Urano T., Inn K.S., Huang I.C., Carnero E., Farzan M., Inoue S., Jung J.U., Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajsbaum R., Albrecht R.A., Wang M.K., Maharaj N.P., Versteeg G.A., Nistal-Villan E., Garcia-Sastre A., Gack M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012;8:e1003059. doi: 10.1371/journal.ppat.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ning Y.J., Wang M., Deng M., Shen S., Liu W., Cao W.C., Deng F., Wang Y.Y., Hu Z., Wang H. Viral suppression of innate immunity via spatial isolation of TBK1/IKKepsilon from mitochondrial antiviral platform. J Mol Cell Biol. 2014;6:324–337. doi: 10.1093/jmcb/mju015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qu B., Qi X., Wu X., Liang M., Li C., Cardona C.J., Xu W., Tang F., Li Z., Wu B. Suppression of the interferon and NF-kappaB responses by severe fever with thrombocytopenia syndrome virus. J Virol. 2012;86:8388–8401. doi: 10.1128/JVI.00612-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santiago F.W., Covaleda L.M., Sanchez-Aparicio M.T., Silvas J.A., Diaz-Vizarreta A.C., Patel J.R., Popov V., Yu X.J., Garcia-Sastre A., Aguilar P.V. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J Virol. 2014;88:4572–4585. doi: 10.1128/JVI.03021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26••.Aguirre S., Maestre A.M., Pagni S., Patel J.R., Savage T., Gutman D., Maringer K., Bernal-Rubio D., Shabman R.S., Simon V. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012;8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study illustrates that Dengue nonstructural proteins NS2B/NS3 proteolytically cleaves human STING.
- 27.Rodriguez-Madoz J.R., Belicha-Villanueva A., Bernal-Rubio D., Ashour J., Ayllon J., Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J Virol. 2010;84:9760–9774. doi: 10.1128/JVI.01051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu C.Y., Chang T.H., Liang J.J., Chiang R.L., Lee Y.L., Liao C.L., Lin Y.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012;8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding Q., Cao X., Lu J., Huang B., Liu Y.J., Kato N., Shu H.B., Zhong J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol. 2013;59:52–58. doi: 10.1016/j.jhep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 31.Nitta S., Sakamoto N., Nakagawa M., Kakinuma S., Mishima K., Kusano-Kitazume A., Kiyohashi K., Murakawa M., Nishimura-Sakurai Y., Azuma S. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology. 2013;57:46–58. doi: 10.1002/hep.26017. [DOI] [PubMed] [Google Scholar]
- 32.Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Clementz M.A., Banach B.S., Li K., Baker S.C. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graef K.M., Vreede F.T., Lau Y.F., McCall A.W., Carr S.M., Subbarao K., Fodor E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol. 2010;84:8433–8445. doi: 10.1128/JVI.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varga Z.T., Grant A., Manicassamy B., Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol. 2012;86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Z., Yi C., Zhao L., Wang S., Zhou L., Hu Y., Zou W., Chen H., Jin M. PB2-588I enhances 2009 H1N1 pandemic influenza virus virulence by increasing viral replication and exacerbating PB2 inhibition of beta interferon expression. J Virol. 2014;88:2260–2267. doi: 10.1128/JVI.03024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loo Y.M., Owen D.M., Li K., Erickson A.K., Johnson C.L., Fish P.M., Carney D.S., Wang T., Ishida H., Yoneyama M. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 39.Anglero-Rodriguez Y.I., Pantoja P., Sariol C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IkappaB kinase epsilon interaction. Clin Vaccine Immunol. 2014;21:29–38. doi: 10.1128/CVI.00500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prins K.C., Cardenas W.B., Basler C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao S., Song L., Li J., Zhang Z., Peng H., Jiang W., Wang Q., Kang T., Chen S., Huang W. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol. 2012;14:1849–1866. doi: 10.1111/cmi.12005. [DOI] [PubMed] [Google Scholar]
- 42.Fang X., Gao J., Zheng H., Li B., Kong L., Zhang Y., Wang W., Zeng Y., Ye L. The membrane protein of SARS-CoV suppresses NF-kappaB activation. J Med Virol. 2007;79:1431–1439. doi: 10.1002/jmv.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siu K.L., Kok K.H., Ng M.H., Poon V.K., Yuen K.Y., Zheng B.J., Jin D.Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y., Zhang L., Geng H., Deng Y., Huang B., Guo Y., Zhao Z., Tan W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S., Min J.Y., Krug R.M., Sen G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology. 2006;349:13–21. doi: 10.1016/j.virol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Tawaratsumida K., Phan V., Hrincius E.R., High A.A., Webby R., Redecke V., Hacker H. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J Virol. 2014;88:9038–9048. doi: 10.1128/JVI.00830-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Min J.Y., Li S., Sen G.C., Krug R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007;363:236–243. doi: 10.1016/j.virol.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 49.Kok K.H., Lui P.Y., Ng M.H., Siu K.L., Au S.W., Jin D.Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9:299–309. doi: 10.1016/j.chom.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Luthra P., Ramanan P., Mire C.E., Weisend C., Tsuda Y., Yen B., Liu G., Leung D.W., Geisbert T.W., Ebihara H. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Tanguy M., Miska E.A. Antiviral RNA interference in animals: piecing together the evidence. Nat Struct Mol Biol. 2013;20:1239–1241. doi: 10.1038/nsmb.2708. [DOI] [PubMed] [Google Scholar]; This review provides an informative perspective of the mammalian RNAi pathway and its possible role as a natural antiviral response.
- 52.Fabozzi G., Nabel C.S., Dolan M.A., Sullivan N.J. Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J Virol. 2011;85:2512–2523. doi: 10.1128/JVI.01160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haasnoot J., de Vries W., Geutjes E.J., Prins M., de Haan P., Berkhout B. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007;3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perry A.K., Chen G., Zheng D., Tang H., Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005;15:407–422. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 55.Kakumani P.K., Ponia S.S., S R.K., Sood V., Chinnappan M., Banerjea A.C., Medigeshi G.R., Malhotra P., Mukherjee S.K., Bhatnagar R.K. Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J Virol. 2013;87:8870–8883. doi: 10.1128/JVI.02774-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schopman N.C., Willemsen M., Liu Y.P., Bradley T., van Kampen A., Baas F., Berkhout B., Haasnoot J. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012;40:414–427. doi: 10.1093/nar/gkr719. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mammalian siRNA to HIV may function as a natural antiviral response. This was the first convincing report that demonstrated that mammalian siRNAs can inhibit HIV replication.
- 57.Li Y., Lu J., Han Y., Fan X., Ding S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science. 2013;342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maillard P.V., Ciaudo C., Marchais A., Li Y., Jay F., Ding S.W., Voinnet O. Antiviral RNA interference in mammalian cells. Science. 2013;342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seo G.J., Kincaid R.P., Phanaksri T., Burke J.M., Pare J.M., Cox J.E., Hsiang T.Y., Krug R.M., Sullivan C.S. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe. 2013;14:435–445. doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo J.T., Hayashi J., Seeger C. West Nile virus inhibits the signal transduction pathway of alpha interferon. J Virol. 2005;79:1343–1350. doi: 10.1128/JVI.79.3.1343-1350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin R.J., Chang B.L., Yu H.P., Liao C.L., Lin Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J Virol. 2006;80:5908–5918. doi: 10.1128/JVI.02714-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kundu K., Dutta K., Nazmi A., Basu A. Japanese encephalitis virus infection modulates the expression of suppressors of cytokine signaling (SOCS) in macrophages: implications for the hosts’ innate immune response. Cell Immunol. 2013;285:100–110. doi: 10.1016/j.cellimm.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 63.Mansfield K.L., Johnson N., Cosby S.L., Solomon T., Fooks A.R. Transcriptional upregulation of SOCS 1 and suppressors of cytokine signaling 3 mRNA in the absence of suppressors of cytokine signaling 2 mRNA after infection with West Nile virus or tick-borne encephalitis virus. Vector Borne Zoonotic Dis. 2010;10:649–653. doi: 10.1089/vbz.2009.0259. [DOI] [PubMed] [Google Scholar]
- 64•.Patil D.R., Hundekar S.L., Arankalle V.A. Expression profile of immune response genes during acute myopathy induced by chikungunya virus in a mouse model. Microbes Infect. 2012;14:457–469. doi: 10.1016/j.micinf.2011.12.008. [DOI] [PubMed] [Google Scholar]; This study identifies gene targets upregulated during Chikunguna virus infection in the mouse.
- 65.Okumura A., Pitha P.M., Yoshimura A., Harty R.N. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J Virol. 2010;84:27–33. doi: 10.1128/JVI.01462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jia D., Rahbar R., Chan R.W., Lee S.M., Chan M.C., Wang B.X., Baker D.P., Sun B., Peiris J.S., Nicholls J.M. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS One. 2010;5:e13927. doi: 10.1371/journal.pone.0013927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pauli E.K., Schmolke M., Wolff T., Viemann D., Roth J., Bode J.G., Ludwig S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4:e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laurent-Rolle M., Boer E.F., Lubick K.J., Wolfinbarger J.B., Carmody A.B., Rockx B., Liu W., Ashour J., Shupert W.L., Holbrook M.R. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol. 2010;84:3503–3515. doi: 10.1128/JVI.01161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mazzon M., Jones M., Davidson A., Chain B., Jacobs M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J Infect Dis. 2009;200:1261–1270. doi: 10.1086/605847. [DOI] [PubMed] [Google Scholar]
- 70.Evans J.D., Seeger C. Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J Virol. 2007;81:11809–11816. doi: 10.1128/JVI.00791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Munoz-Jordan J.L., Laurent-Rolle M., Ashour J., Martinez-Sobrido L., Ashok M., Lipkin W.I., Garcia-Sastre A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol. 2005;79:8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin C.W., Cheng C.W., Yang T.C., Li S.W., Cheng M.H., Wan L., Lin Y.J., Lai C.H., Lin W.Y., Kao M.C. Interferon antagonist function of Japanese encephalitis virus NS4A and its interaction with DEAD-box RNA helicase DDX42. Virus Res. 2008;137:49–55. doi: 10.1016/j.virusres.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 73.Ashour J., Laurent-Rolle M., Shi P.Y., Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol. 2009;83:5408–5418. doi: 10.1128/JVI.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morrison J., Laurent-Rolle M., Maestre A.M., Rajsbaum R., Pisanelli G., Simon V., Mulder L.C., Fernandez-Sesma A., Garcia-Sastre A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013;9:e1003265. doi: 10.1371/journal.ppat.1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75•.Suthar M.S., Brassil M.M., Blahnik G., Gale M., Jr. Infectious clones of novel lineage 1 and lineage 2 West Nile virus strains WNV-TX02 and WNV-Madagascar. J Virol. 2012;86:7704–7709. doi: 10.1128/JVI.00401-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Inserting structural and nonstructural proteins from the infectious clone of a pathogenic strain of WNV to a non-pathogenic strain confers increased inhibition of host interferon responses. This study identifies virulence factors that determines pathogenicity within different lineages of WNV.
- 76.Fros J.J., Liu W.J., Prow N.A., Geertsema C., Ligtenberg M., Vanlandingham D.L., Schnettler E., Vlak J.M., Suhrbier A., Khromykh A.A. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J Virol. 2010;84:10877–10887. doi: 10.1128/JVI.00949-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frolov I., Akhrymuk M., Akhrymuk I., Atasheva S., Frolova E.I. Early events in alphavirus replication determine the outcome of infection. J Virol. 2012;86:5055–5066. doi: 10.1128/JVI.07223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uetani K., Hiroi M., Meguro T., Ogawa H., Kamisako T., Ohmori Y., Erzurum S.C. Influenza A virus abrogates IFN-gamma response in respiratory epithelial cells by disruption of the Jak/Stat pathway. Eur J Immunol. 2008;38:1559–1573. doi: 10.1002/eji.200737045. [DOI] [PubMed] [Google Scholar]
- 79.Reid S.P., Leung L.W., Hartman A.L., Martinez O., Shaw M.L., Carbonnelle C., Volchkov V.E., Nichol S.T., Basler C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. 2006;80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reid S.P., Valmas C., Martinez O., Sanchez F.M., Basler C.F. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol. 2007;81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Wilde A.H., Raj V.S., Oudshoorn D., Bestebroer T.M., van Nieuwkoop S., Limpens R.W., Posthuma C.C., van der Meer Y., Barcena M., Haagmans B.L. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J Gen Virol. 2013;94:1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li S.W., Lai C.C., Ping J.F., Tsai F.J., Wan L., Lin Y.J., Kung S.H., Lin C.W. Severe acute respiratory syndrome coronavirus papain-like protease suppressed alpha interferon-induced responses through downregulation of extracellular signal-regulated kinase 1-mediated signalling pathways. J Gen Virol. 2011;92:1127–1140. doi: 10.1099/vir.0.028936-0. [DOI] [PubMed] [Google Scholar]
- 85••.Mossler C., Groiss F., Wolzt M., Wolschek M., Seipelt J., Muster T. Phase I/II trial of a replication-deficient trivalent influenza virus vaccine lacking NS1. Vaccine. 2013;31:6194–6200. doi: 10.1016/j.vaccine.2013.10.061. [DOI] [PubMed] [Google Scholar]; An intriguing report on a clinical trial of an attenuated influenza vaccine that targets the NS1 protein.
- 86.Angelini M.M., Akhlaghpour M., Neuman B.W., Buchmeier M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio. 2013;4 doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prentice E., McAuliffe J., Lu X., Subbarao K., Denison M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J Virol. 2004;78:9977–9986. doi: 10.1128/JVI.78.18.9977-9986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stertz S., Reichelt M., Spiegel M., Kuri T., Martinez-Sobrido L., Garcia-Sastre A., Weber F., Kochs G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. 2007;361:304–315. doi: 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bouvet M., Lugari A., Posthuma C.C., Zevenhoven J.C., Bernard S., Betzi S., Imbert I., Canard B., Guillemot J.C., Lecine P. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J Biol Chem. 2014;289:25783–25796. doi: 10.1074/jbc.M114.577353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y., Cai H., Pan J., Xiang N., Tien P., Ahola T., Guo D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci U S A. 2009;106:3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K., Gorbalenya A.E., Snijder E.J., Canard B. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J Virol. 2008;82:8071–8084. doi: 10.1128/JVI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jin X., Chen Y., Sun Y., Zeng C., Wang Y., Tao J., Wu A., Yu X., Zhang Z., Tian J. Characterization of the guanine-N7 methyltransferase activity of coronavirus nsp14 on nucleotide GTP. Virus Res. 2013;176:45–52. doi: 10.1016/j.virusres.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.von Grotthuss M., Wyrwicz L.S., Rychlewski L. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li X.D., Shan C., Deng C.L., Ye H.Q., Shi P.Y., Yuan Z.M., Gong P., Zhang B. The interface between methyltransferase and polymerase of NS5 is essential for flavivirus replication. PLoS Negl Trop Dis. 2014;8:e2891. doi: 10.1371/journal.pntd.0002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ray D., Shah A., Tilgner M., Guo Y., Zhao Y., Dong H., Deas T.S., Zhou Y., Li H., Shi P.Y. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J Virol. 2006;80:8362–8370. doi: 10.1128/JVI.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Szretter K.J., Daniels B.P., Cho H., Gainey M.D., Yokoyama W.M., Gale M., Jr., Virgin H.W., Klein R.S., Sen G.C., Diamond M.S. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wengler G., Wengler G. The NS 3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology. 1993;197:265–273. doi: 10.1006/viro.1993.1587. [DOI] [PubMed] [Google Scholar]
- 98.Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K.A., Shi P.Y., Li H. Structure and function of flavivirus NS5 methyltransferase. J Virol. 2007;81:3891–3903. doi: 10.1128/JVI.02704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferron F., Longhi S., Henrissat B., Canard B. Viral RNA-polymerases — a predicted 2′-O-ribose methyltransferase domain shared by all Mononegavirales. Trends Biochem Sci. 2002;27:222–224. doi: 10.1016/s0968-0004(02)02091-1. [DOI] [PubMed] [Google Scholar]
- 100.Yoshikawa Y., Mizumoto K., Yamanouchi K. Characterization of messenger RNAs of measles virus. J Gen Virol. 1986;67(Pt 12):2807–2812. doi: 10.1099/0022-1317-67-12-2807. [DOI] [PubMed] [Google Scholar]
- 101.Karpe Y.A., Aher P.P., Lole K.S. NTPase and 5′-RNA triphosphatase activities of Chikungunya virus nsP2 protein. PLoS One. 2011;6:e22336. doi: 10.1371/journal.pone.0022336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rana J., Rajasekharan S., Gulati S., Dudha N., Gupta A., Chaudhary V.K., Gupta S. Network mapping among the functional domains of Chikungunya virus nonstructural proteins. Proteins. 2014;82:2403–2411. doi: 10.1002/prot.24602. [DOI] [PubMed] [Google Scholar]
- 103.Sreejith R., Rana J., Dudha N., Kumar K., Gabrani R., Sharma S.K., Gupta A., Vrati S., Chaudhary V.K., Gupta S. Mapping interactions of Chikungunya virus nonstructural proteins. Virus Res. 2012;169:231–236. doi: 10.1016/j.virusres.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 104.Vasiljeva L., Merits A., Auvinen P., Kaariainen L. Identification of a novel function of the alphavirus capping apparatus. RNA 5′-triphosphatase activity of Nsp2. J Biol Chem. 2000;275:17281–17287. doi: 10.1074/jbc.M910340199. [DOI] [PubMed] [Google Scholar]
- 105.Boivin S., Cusack S., Ruigrok R.W., Hart D.J. Influenza A virus polymerase: structural insights into replication and host adaptation mechanisms. J Biol Chem. 2010;285:28411–28417. doi: 10.1074/jbc.R110.117531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Olson A.C., Rosenblum E., Kuchta R.D. Regulation of influenza RNA polymerase activity and the switch between replication and transcription by the concentrations of the vRNA 5′ end, the cap source, and the polymerase. Biochemistry. 2010;49:10208–10215. doi: 10.1021/bi101011j. [DOI] [PMC free article] [PubMed] [Google Scholar]