

With cardiovascular disease remaining a major cause of morbidity and mortality in spite of recent therapeutic advances, the molecular basis of heart failure continues to be an active field of investigation. In particular, the renin-angiotensin system (RAS), which plays a major role in blood pressure control and cardiac hypertrophy, has been the subject of countless studies. The RAS comprises a complex set of proteolytic processing steps and interconnected signaling pathways, acting both systemically and locally, and involving multiple angiotensin (Ang) peptides and receptors. Of these, the most extensively studied are AngII, considered to be the major effector molecule of the RAS, and one of its receptors, the AngII type 1 receptor (AT1R).
Two decades ago, AngII treatment and mechanical stretch were shown to have similar effects upon cardiomyocyte signal transduction pathways and gene expression, resulting in hypertrophic growth that was found to be primarily mediated by the AT1R1–4. Later studies showed that mechanical stress can activate the AT1R even in the absence of AngII5, with ligand binding and mechanical stress causing distinct conformational changes in the receptor6. The AT1R is a G protein-coupled receptor (GPCR) that mediates the hypertrophic effects of AngII by triggering downstream signaling through coupled heterotrimeric G proteins. Binding of AngII also causes recruitment of β-arrestin, which, in turn, leads to receptor desensitization and internalization. Ligand-independent or β-arrestin biased activation of the receptor, on the other hand, is independent of G protein signaling and induces distinct conformational changes in the receptor and in β-arrestin that trigger an alternative, cardioprotective downstream signaling pathway.7,8
What happens to the AT1R after internalization? In the current model, sorting of endosomal AT1R leads to one of three fates: interaction with other molecules to participate in intracellular signaling, resensitization and recycling of the receptor back to the plasma membrane or lysosomal degradation.9 In this issue of Circulation, Pironti et al provide evidence of a fourth possibility, with potentially far-reaching consequences. Cardiac cells are capable of secreting multiple autocrine/paracrine factors, including AngII, thereby exerting effects beyond the individual cell. In addition, stress may promote release of exosomes, extracellular membrane vesicles capable of carrying a variety of cargoes, including proteins and nucleic acids, for transfer to other cells in the local environment or farther off via the systemic circulation. Exosomes, which are secreted by a variety of cell types and exist at high concentrations in the blood, originate from endosomal formation of intraluminal vesicles within multivesicular bodies, and are released from the cell by fusion of the multivesicular bodies with the cell membrane. In this study, using rigorous methods of purification and quantification, the authors show that exosome secretion increases significantly following either osmotic stretch or AngII treatment of HEK293T cells (with or without stable expression of AT1R) in vitro or seven days of cardiac pressure overload in wild type (WT) mice in vivo. Furthermore, some of the exosomes were found to contain AT1R, with the receptor density in the cell media and serum increasing substantially following stimulation with osmotic stretch or AngII in vitro or pressure overload in vivo. Transfer of these exosomes to WT HEK293T cells (which do not express AT1R) or AT1R knockout mice caused the recipients to become responsive to AngII treatment, demonstrating that the exosomal AT1Rs remain functional.10 Thus, it appears that not only is internalization not necessarily the end of the line for AT1Rs but it may actually be part of a process that allows cardiac cells to extend the reach of their signaling to more distant tissues (Figure 1).
Figure 1.
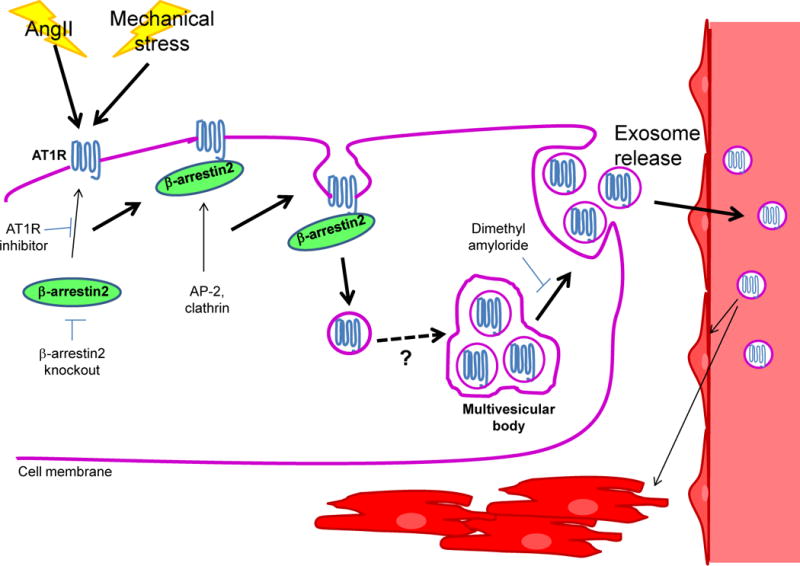
Schematic representation of the formation of AT1R-containing exosomes. Stimulation by AngII or mechanical stress activates the AT1R, triggering recruitment of β-arrestin2. β-arrestin2, in turn, recruits components of the endocytic machinery, leading to internalization of the AT1R. Endosomal AT1R is packaged into intraluminal vesicles within multivesicular bodies via a currently unknown mechanism, which are released as exosomes upon fusion of the multivesicular bodies with the cell membrane. Circulating AT1R-exosomes target the cardiomyocytes and mesenteric vessels in the heart, as well as skeletal muscle.
Packaging of AT1R into exosomes was found to be dependent upon the presence of β-arrestin, and β-arrestin2 in particular.10 This is not surprising given that β-arrestin is known to play an essential role in AT1R internalization by disrupting receptor/G protein interaction and recruiting components of the endocytic machinery.9 One question that has not yet been addressed, however, is precisely what role, if any, β-arrestin2 plays in directing AT1R to exosomes rather than one of the alternate possible fates. Although global β-arrestin2 knockout prevented AT1R packaging, exosomal release was unaffected.10 Nor does the increase in exosome release appear to be dependent upon the AT1R itself, since WT HEK293T cells also released an increased number of exosomes in response to osmotic stretch,10 although it might be helpful to confirm this observation in vivo by examining exosomal release in response to pressure overload in AT1R knockout mice. Both AngII and mechanical stress were used to stimulate exosome release, but these two stimuli have been previously shown to induce different β-arrestin conformations and trigger different downstream signaling pathways, with the latter being independent of ligand binding, at least in the short term.7 In the long term, however, some of the effects of stretch and pressure overload are also mediated by AngII/AT1R signaling. Hypotonic stress-induced c-fos upregulation in cardiomyocytes was unaffected by treatment with an AT1R antagonist,11 but mechanical stretch and pressure overload have been shown to induce secretion of AngII.12,13 What, then, is the common mechanism by which the different types of stimulus increase AT1R-containing exosome release, and how is the AT1R, rather than other receptors, specifically targeted for exocytosis? If AngII ligand binding is required, testing the effects of treatment with AT1R blockers or angiotensinogen knockout on AT1R packaging could be informative. Mechanical stretch induces internalization of the AT1R, which forms a more stable class B type interaction with β-arrestin, but not of the β1-adrenergic receptor, which forms a less stable class A type interaction,7 and this may perhaps account for the specific inclusion of AT1Rs. However, even under stimulated conditions, the AT1R density was less than 10 receptors/100 exosomes,10 suggesting that something else may be packaged into the exosomes under these conditions as well. If so, what else are the exosomes carrying and why?
As mentioned above, global knockout of β-arrestin2 prevented transfer of AT1Rs to the exosomes during pressure overload in vivo. Similarly, cardiomyocyte-specific β-arrestin2 knockout markedly reduced serum AT1R levels of tamoxifen-treated β-arrestin2flox/flox/αMHCMerCreMer mice after seven days of pressure overload. Based on this observation, the authors concluded that cardiomyocytes are the major source of AT1R-containing exosomes during pressure overload in vivo.10 Cardiomyocytes have been shown to secrete a number of soluble factors that modulate the functions of surrounding cells, and it was recently demonstrated that regulation of Mediator complex subunit 13 (MED13) signaling in cardiomyocytes controls lipid utilization and oxygen consumption in white adipose tissue and the liver, presumably through circulating factors released by the heart.14 Nevertheless, exocytosis of functional receptors would represent a significant extension of the mechanistic network by which cardiomyocytes can exert control over other cells and tissues at a distance. However, a possibility exists that this conclusion may prove to be premature. No direct evidence is presented to confirm that cardiomyocytes are the source of the exosomes, nor is there any data on the cardiac function of the cardiac-specific β-arrestin2 knockout mice. It is, therefore, possible that the AT1R-containing exosomes are released from other cardiac cells as a secondary effect of changes in cardiac function following pressure overload. AT1R expression is relatively low in cardiomyocytes, whereas it is substantially higher in cardiac fibroblasts.15 Furthermore, cardiac fibroblasts have been shown to secrete cytokines and exosomes that induce hypertrophy in cardiomyocytes,16,17 raising the question of whether they could be involved in the observed AT1R-containg exosome release during pressure overload as well. The authors do not address a possible secondary effect of the cardiac-specific β-arrestin2 knockout that makes interpretation of the data difficult, namely that, while cardiomyocyte levels of β-arrestin2 were approximately 80% lower in the cardiac-specific β-arrestin2 knockout mice than in WT mice, there was an approximately 4-fold increase in β-arrestin2 levels in cardiac fibroblasts.10 It is unclear how this might affect circulating AT1R levels. However, it may be that the exosomes are actually derived from cardiac fibroblasts rather than cardiomyocytes, and that it is the excess β-arrestin2 in the fibroblasts of the knockout mice that is responsible for the decrease in circulating AT1R, possibly by allowing AT1R internalization but somehow preventing its exocytosis, rather than the decrease in cardiomyocyte β-arrestin2. Further investigation of AT1R internalization and exosome tracking in the different cell types is required to address this issue.
This study clearly represents a significant advance in understanding how the local cardiac RAS can exert its effects systemically. Although AngII is secreted into the circulation, the responsiveness of target cells was until now thought to be dependent upon their intrinsic AT1R densities. It now appears that cardiac cells take part in determining not just how much of the effector molecule is released but also how capable its targets are of responding to it. What remains unclear at this point is the functional significance of this mechanism. Pironti et al used AT1R knockout mice to demonstrate that the AT1Rs in exosomes remain functional, causing increases in blood pressure and phosphorylation of extracellular signal-regulated kinase (ERK) in response to AngII treatment in vivo.10 However, they did not inject WT mice, so that it is unknown whether the introduction of exogenous AT1R-containing exosomes would have a significant additive effect over that of the endogenous receptors. Examination of receptor density in the tissues of WT and AT1R knockout mice revealed that the exosomes specifically target the heart and skeletal muscle but not the kidneys or lungs, and that, within the heart, they traffic to cardiomyocytes and the endothelial and smooth muscle cells of mesenteric vessels but not to cardiac fibroblasts.10 Exosomal markers reflect their parent cells, and it is thought that uptake depends upon binding of targeting peptides to receptors on recipient cells. Indeed, increasing targeting peptide stability was recently shown to significantly increase exosomal uptake.18 Furthermore, there is evidence that the endocytic pathway by which exosomes are internalized varies and may be cell type-specific.19,20 Thus, how the specific targeting of AT1R-exosomes is achieved and how they are taken up by the target cells remain to be elucidated. Likewise, the purpose of the receptor transfer has yet to be determined. The authors speculate that the transfer of AT1Rs may represent an attempt to offset receptor downregulation in the presence of increased AngII levels and that this may aggravate cardiac dysfunction and remodeling during pressure overload.10 If AT1R-containing exosome release is involved in the progression of heart failure, selectively inhibiting this process could represent a novel therapeutic modality. GPCR kinases (GRKs) are required for GPCR internalization, and paroxetine, which specifically inhibits GRK2, was recently demonstrated to be of greater benefit in protecting the heart after myocardial infarction in mice than currently used β-blocker therapy.21 However, AT1R/β-arrestin2-dependent ERK activation in response to mechanical stretch was shown to require GRK5 and GRK6 rather than GRK2.7 GRK5-specific small molecule inhibitors are currently under investigation22 and may prove to be of use in targeting pressure overload-induced AT1R exocytosis. On the other hand, knockout of either AT1R or β-arrestin2 has been shown to decrease Akt phosphorylation and increase apoptosis in the mouse heart following mechanical stress, indicating that activation of the AT1R by mechanical stress triggers prosurvival signaling.7 This raises the possibility that the AT1R-exosome release, which is also triggered by mechanical stress in a β-arrestin2-dependent manner, may also represent a protective mechanism. A recent study found that plasma exosomes collected from normal rats or humans were highly protective against cardiac ischemia/reperfusion injury in rats, supporting the idea that, at least under some conditions, exosomes may have a cardioprotective role.23 If so, it remains to be determined whether further upregulation of the process would be of therapeutic value for combating the progression of heart failure.
Acknowledgments
We sincerely thank Dr. Susmita Sahoo for her helpful suggestions.
Funding Sources: This work was supported in part by U.S. Public Health Service Grants HL102738, HL67724, HL91469, AG23039 and HL112330. This work was also supported by the Fondation Leducq Transatlantic Networks of Excellence.
Footnotes
Disclosures: None.
References
- 1.Sadoshima J, Jahn L, Takahashi T, Kulik TJ, Izumo S. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. An in vitro model of load-induced cardiac hypertrophy. J Biol Chem. 1992;267:10551–10560. [PubMed] [Google Scholar]
- 2.Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 1993;12:1681–1692. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadoshima J, Izumo S. Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 4.Sadoshima J, Izumo S. Signal transduction pathways of angiotensin II-induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers. Circ Res. 1993;73:424–438. doi: 10.1161/01.res.73.3.424. [DOI] [PubMed] [Google Scholar]
- 5.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda N, Miura S, Akazawa H, Tanaka T, Qin Y, Kiya Y, Imaizumi S, Fujino M, Ito K, Zou Y, Fukuhara S, Kunimoto S, Fukuzaki K, Sato T, Ge J, Mochizuki N, Nakaya H, Saku K, Komuro I. Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep. 2008;9:179–186. doi: 10.1038/sj.embor.7401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010:3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. Allosteric modulation of β-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem. 2014;289:28271–28283. doi: 10.1074/jbc.M114.585067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- 10.Pironti G, Strachan RT, Abraham D, Yu SM-W, Chen M, Chen W, Hanada K, Mao L, Watson LJ, Rockman HA. Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II type 1 receptors. Circulation. 2015;131:XX–XXX. doi: 10.1161/CIRCULATIONAHA.115.015687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadoshima J, Qiu Z, Morgan JP, Izumo S. Tyrosine kinase activation is an immediate and essential step in hypotonic cell swelling-induced ERK activation and c-fos gene expression in cardiac myocytes. EMBO J. 1996;15:5535–5546. [PMC free article] [PubMed] [Google Scholar]
- 12.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 13.Amedeo Modesti P, Zecchi-Orlandini S, Vanni S, Polidori G, Bertolozzi I, Perna AM, Formigli L, Cecioni I, Coppo M, Boddi M, Serneri GG. Release of preformed Ang II from myocytes mediates angiotensinogen and ET-1 gene overexpression in vivo via AT1 receptor. J Mol Cell Cardiol. 2002;34:1491–1500. doi: 10.1006/jmcc.2002.2095. [DOI] [PubMed] [Google Scholar]
- 14.Baskin KK, Grueter CE, Kusminski CM, Holland WL, Bookout AL, Satapati S, Kong YM, Burgess SC, Malloy CR, Scherer PE, Newgard CB, Bassel-Duby R, Olson EN. MED13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol Med. 2014;6:1610–1621. doi: 10.15252/emmm.201404218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarreal FJ, Kim NN, Ungab GD, Printz MP, Dillmann WH. Identification of functional angiotensin II receptors on rat cardiac fibroblasts. Circulation. 1993;88:2849–2861. doi: 10.1161/01.cir.88.6.2849. [DOI] [PubMed] [Google Scholar]
- 16.Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, Takahashi T, Makino S, Kato T, Ogawa S. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem. 2000;275:29717–29723. doi: 10.1074/jbc.M003128200. [DOI] [PubMed] [Google Scholar]
- 17.Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung ME, Leonard JN. Stabilization of Exosome-targeting Peptides via Engineered Glycosylation. J Biol Chem. 2015;290:8166–8172. doi: 10.1074/jbc.M114.621383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian T, Zhu YL, Zhou YY, Liang GF, Wang YY, Hu FH, Xiao ZD. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J Biol Chem. 2014;289:22258–22267. doi: 10.1074/jbc.M114.588046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hazan-Halevy I, Rosenblum D, Weinstein S, Bairey O, Raanani P, Peer D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015 doi: 10.1016/j.canlet.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM, Tesmer JJG, Koch WJ. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med. 2015;7:277ra231. doi: 10.1126/scitranslmed.aaa0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Homan KT, Wu E, Cannavo A, Koch WJ, Tesmer JJG. Identification and characterization of amlexanox as a G protein-coupled receptor kinase 5 inhibitor. Molecules. 2014;19:16937–16949. doi: 10.3390/molecules191016937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi-Doku C, Arjun S, Zheng Y, Riquelme JA, Kearney J, Sharma V, Multhoff G, Hall AR, Davidson SM. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J Am Coll Cardiol. 2015;65:1525–1536. doi: 10.1016/j.jacc.2015.02.026. [DOI] [PubMed] [Google Scholar]