

Abstract
Camptothecin (CPT) is a potent antitumor agent and functions via inhibiting the activity of topoisomerase I during DNA replication. However, the clinical application of CPT has been greatly hindered by its extremely poor solubility, the instability of its active lactone ring in blood stream, as well as the non-specific toxicity to normal tissues. In addition, most of the formulations developed so far are not applicable for formulating CPT. In this study, two novel CPT prodrugs were developed by conjugating CPT to α-tocopherol via a carbonate ester bond (CPT-VE) or disulfide linkage (CPT-S-S-VE). Both CPT prodrugs were able to self-assemble into nanofibers with the facilitation of a PEG5K-Fmoc-VE2-based nanomicellar carrier. Both prodrug nanoassemblies exhibited excellent stability. Fluorescence quenching, UV absorbance, and FT-IR studies demonstrated strong interactions between carrier and prodrugs, including hydrophobic interaction, π-π stacking, as well as hydrogen bonding. NMR studies suggested that prodrugs were successfully incorporated into PEG5K-Fmoc-VE2 during self-assembly process. In vitro, PEG5K-Fmoc-VE2/CPT-S-S-VE presented significantly higher level of cytotoxicity on tumor cells compared to PEG5K-Fmoc-VE2/CPT-VE. Biodistribution study showed that CPT-S-S-VE formulated in PEG5K-Fmoc-VE2 micelles was effectively converted to parent CPT following delivery to tumor tissues. Finally, PEG5K-Fmoc-VE2/CPT-S-S-VE nanofibers showed superior tumor growth inhibition in an aggressive murine breast cancer model (4T1.2).
Keywords: Camptothecin, Nanofibers, Tocopherol-based prodrug, Disulfide-responsive release, Chemotherapy
Introduction
Formulations represent an important strategy to improve the performance of many anticancer agents via improving their water solubility and facilitating their selective delivery to tumors. Most of the current approaches are focused on the improvement of the carriers so as to improve their compatibility with the input drugs. Although these strategies work well for many anticancer agents with relatively simple structures, they cannot well accommodate some agents with complex structures, such as camptothecin (CPT). CPT, a potent anticancer agent, targets a wide spectrum of cancers. It functions via inhibiting the activity of topoisomerase I during DNA replication in the S-phase of the cell cycle, leading to cell death [1]. The active CPT requires a lactone form for the terminal ring of CPT, which is better conserved under acidic condition (pH<5) [2]. Under physiologic condition, the active lactone form can be slowly converted to carboxylate form, rendering CPT inactive [2, 3]. Various strategies have been attempted to protect the active lactone structure but only met with limited success [2–4].
In addition to its instability, CPT is poorly soluble in aqueous solutions [5]. It even shows low solubility in many organic solvents such as chloroform and ethanol [6, 7]. Therefore, CPT is hard to be formulated into some commonly used carriers via a simple method of solvent evaporation.
Recently, polymer-drug conjugates have received increasing attention as a strategy to improve the water solubility and circulation time of anticancer agents. Caiolfa et al reported that a conjugate of CPT with HPMA polymer via a proteinase sensitive linker showed dramatically improved antitumor activity in HT29 human colon carcinoma xenograft model [1]. McRae Page et al also reported promising results with a poly(MPC-DHLA)-CPT conjugate with a disulfide linkage in MCF-7 and COLO-205 tumor models [8]. Despite these encouraging results, there is a potential issue of limited efficacy of CPT release from the macromolecular conjugates due to the steric hindrance.
Prodrug approach represents another promising strategy to improve the carrier/drug compatibility. It has been reported that doxorubicin (DOX) conjugated with D-α-tocopherol succinate (N-DOX-TOS) was able to self-assemble in aqueous solution upon stabilized by D-α-tocopherol poly(ethylene glycol) 2000 succinate. A drug loading capacity of 34% was achieved and the resulting nanoassembly led to significantly enhanced antitumor activity over free DOX [9].
In the present study, we investigate whether Vitamin E-derivatized prodrug could be effectively loaded into PEG5K-Fmoc-VE2 micelles, a nanocarrier that was recently developed in our lab [10, 11]. Our previous study showed that the conjugate of PEG5K with two molecules of Vitamin E (PEG5K-VE2) was more effective than other three conjugates (PEG5K-VE, PEG2K-VE2, and PEG2K-VE) with respect to both drug loading capacity and formulation stability [12]. In addition, introduction of a drug-interactive Fmoc motif into PEG5K-VE2 led to significantly enhanced carrier/drug interaction, resulting in further improvement in the performance of the drug-loaded micelles [10, 11]. Two types of CPT-VE prodrugs were synthesized in the present study, one with an ester linkage and the other with a disulfide bond. The two prodrugs were compared with respect to DLC, the efficiency of drug release, in vitro cytotoxicity and in vivo antitumor activity. The carrier/drug interaction was also investigated.
Materials and Methods
Materials
D-α-Tocopherol succinate, Triphosgene, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), trypsin-EDTA solution, Triton X-100, and Dulbecco’s Modified Eagle’s Medium (DMEM) were all purchased from Sigma-Aldrich (MO, USA). D-alpha-tocopherol was purchased from Tokyo Chemical Industry (OR, USA). Camptothecin, Bis (2-hydroxyethyl) disulfide, and N,N′-dicyclohexylcarbodiimide (DCC) were purchased from Alfa Aesar (MA, USA). 4-Dimethylaminopyridine (DMAP) was purchased from Calbiochem-Novabiochem Corporation (CA, USA). Fetal bovine serum (FBS) and penicillin-streptomycin solution were from Invitrogen (NY, USA). LysoTracker was purchased from Life Technologies (Carlsbad, CA). All solvents used in this study were HPLC grade.
Synthesis of Camptothecin-Vitamin E conjugate with a carbonate ester bond (CPT-VE)
CPT (1 eq. molar) and DMAP (2 eq. molar) were mixed in 20 mL DCM. Triphosgene (0.35 eq. molar) was then added dropwise into the yellowish solution. The reaction was allowed for 20 min at room temperature. Afterwards, Vitamin E (2 eq. molar) was added into the solution and the reaction continued overnight. The resulting CPT-VE was purified via column with a mobile phase of MeOH/DCM (2:98) and a yield of 68%. 1H-NMR (Fig. S1). 13C-NMR (CDCl3-d, ppm) 167.12, 157.30, 152.90, 152.26, 149.69, 148.99, 146.57, 145.77, 140.96, 131.11, 130.63, 129.80, 128.43, 128.21, 128.02, 120.20, 95.65, 78.23, 75.09, 67.06, 50.00, 39.36, 37.42, 37.37, 37.27, 32.75, 32.67, 31.74, 31.02, 27.96, 24.80, 24.78, 24.41, 22.73, 22.64, 21.00, 20.48, 19.75, 19.69, 19.63, 19.61, 12.72, 11.87, 11.71, 7.72. ESI-MS: C50H64N2O7 (M+1+) 805.4714, found at 805.4721.
Synthesis of Camptothecin-Vitamin E conjugate with a disulfide bond (CPT-S-S-VE)
Vitamin E succinate (1 eq. molar) was reacted with Bis (2-hydroxyethyl) disulfide (2 eq. molar) overnight with the assistance of DCC (2 eq. molar) and DMAP (0.2 eq. molar). The resulting VE-S-S-OH was purified through column with a mobile phase of ethyl acetate/petroleum ether (2:3) and a yield of 75%. 1H-NMR (Fig. S2). 13C-NMR (CDCl3-d, ppm) 172.12, 171.07, 149.46, 140.46, 126.65, 124.92, 123.03, 117.42, 75.07, 62.73, 60.36, 41.51, 39.40, 37.47, 37.44, 37.31, 36.95, 32.80, 32.71, 31.16, 29.05, 28.76, 28.00, 24.83, 24.46, 22.78, 22.69, 21.04, 20.62, 19.81, 19.71, 12.98, 12.12, 11.85. ESI-MS: C37H62O6S2 (M+Na+) 689.3885, found at 689.3882.
Next, CPT (1 eq. molar) and DMAP (2 eq. molar) were dissolved in 20 mL DCM with stirring. Triphosgene (0.35 eq. molar) was added dropwise into the yellowish solution. The reaction was allowed for 20 min at room temperature. Then, VE-S-S-OH (2 eq. molar) was added into the solution and the reaction continued overnight. The final product (CPT-S-S-VE) was purified via column with a mobile phase of ethyl acetate/petroleum ether (7:4) and a yield of 58%. 1H-NMR (Fig. S3). 13C-NMR (CDCl3-d, ppm) 171.90, 170.86, 167.25, 157.30, 153.45, 152.31, 149.43, 148.91, 146.52, 145.61, 140.43, 131.17, 130.73, 129.70, 128.49, 128.19, 128.09, 126.66, 124.92, 123.01, 120.27, 117.38, 95.96, 78.05, 77.35, 77.03, 76.71, 75.05, 70.57, 69.27, 67.08, 66.53, 62.50, 50.01, 39.37, 37.45, 37.43, 37.29, 37.12, 36.56, 32.80, 32.71, 31.90, 29.00, 28.75, 27.98, 24.80, 24.45, 22.72, 22.63, 21.02, 20.58, 19.76, 19.66. ESI-MS: C58H76N2O11S2 (M+H+) 1041.4891, found at 1041.4887.
Biophysical characterization of CPT, CPT-VE, and CPT-S-S-VE nanoparticles
CPT-VE or CPT-S-S-VE dissolved in DCM was mixed with predetermined amount of PEG5K-Fmoc-VE2 or PEG5K-VE2. The solvent was removed by nitrogen flow followed by vacuum for four hours [13–15]. PBS solution was added to the dried film followed by gentle votexing and the resulting formulations were filtered through a 0.22 μm filter prior to characterization. PEG5K-VE2/CPT and PEG5K-Fmoc-VE2/CPT were prepared through dialysis method. First, both CPT and PEG5K-Fmoc-VE2 (PEG5K-VE2) were dissolved in DMSO and mixed well. The mixture was then transferred into a dialysis bag (MWCO 12,000) and dialyzed against 200 mL deionized water for 4 days during which the medium was replaced freshly everyday with stirring at room temperature. The diameter and size distribution of the nanoformulations were evaluated by dynamic light scattering (DLS). The morphology was determined by cryo-electron microscopy (cryoEM) and transmission electron microscopy (TEM). The concentrations of CPT, CPT-VE, and CPT-S-S-VE in nanoparticles were measured by UPLC-QTOFMS Analysis. The drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated as described previously [10].
Fluorescence quenching
PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE were similarly prepared as described above. The samples were put into a 96-well plate, and the fluorescence intensity of PEG5K-Fmoc-VE2 was recorded on a Synergy H1 Hybrid reader (BioTek), employing an excitation wavelength of 270 nm and emission wavelength from 300–500 nm. The fluorescence intensity of prodrugs was examined using an excitation wavelength of 370 nm and emission wavelength from 400–700 nm.
UV absorbance spectroscopy
The absorption spectra of CPT (DMSO), CPT-VE (DMSO), CPT-S-S-VE (DMSO), PEG5K-Fmoc-VE2/CPT-VE (water) and PEG5K-Fmoc-VE2/CPT-S-S-VE (water) were collected using Varian Cary 50 Bio UV-Visible Spectrophotometer over a wavelength ranging from 310 to 420 nm. All samples were loaded into a quartz cell and measured against distilled water or DMSO as the reference.
Fourier transform infrared spectroscopy (FT-IR)
FT-IR of CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE, PEG5K-Fmoc-VE2/CPT-S-S-VE and PEG5K-Fmoc-VE2 was determined using a VERTEX 70/70v FT-IR spectrometer (Bruker) to determine the potential hydrogen bonding of carrier/drug in the frequency of 4997–500 cm−1 (KBr pellet).
Cell culture
4T1.2, a mouse breast cancer cell line, was used in this work and cultured in DMEM containing 10% FBS and 1% penicillin-streptomycin in a humidified environment at 37 °C with 5% CO2.
Animals
Female BALB/c mice of 6–8 weeks of age were purchased from Charles River (Davis, CA). Animals were housed under pathogen-free conditions according to AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) guidelines. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh.
In vitro cytotoxicity
The anti-proliferation activity of CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE with or without 10 mM GSH was evaluated in 4T1.2 in comparison to free CPT. Briefly, 2000 cells/well were seeded into 96-well plates and cultured overnight to allow attachment. Cells were then challenged with various concentrations (equivalent CPT concentration) of CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE with or without 10 mM GSH. Free PEG5K-Fmoc-VE2, at concentrations equivalent to the amount of carriers in drug/carrier mixed micelles, was also added into cells. Cells were incubated for 72 h and cell viability was examined by MTT assay.
CPT release inside cells
1×106 4T1.2 cells were grown into 6-well plates and cultured overnight to allow attachment. Cells were then treated with CPT-VE and CPT-S-S-VE (100 ng/mL in terms of CPT) for 24 h. Cells were washed with ice-cold PBS three times and extracted in MeOH. The extraction was dried under gentle stream of clean dry air, then redissolved in a mixture of MeOH:H2O (1:1, v/v). The samples were vortexed and then centrifuged at 14,000 rpm for 10 min at 4°C. The supernatants were transferred to another set of 1.5 mL microtubes and stored at −80°C for MS analysis. For UPLC-QTOFMS analysis, chromatographic separation of CPT was performed on an Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm; Waters, Milford, MA). The flow rate of the mobile phase was 0.5 ml/min using a gradient ranging from 2% to 98% acetonitrile/water containing 0.1% formic acid in 6 minutes. The column temperature was maintained at 50°C. Waters SYNAPT G2S TOF-MS (Waters, Milford, MA) was operated in positive mode with electrospray ionization. The source and desolvation temperatures were set at 150°C and 500°C, respectively. Nitrogen was applied as the cone gas (50 L/h) and desolvation gas (800 L/h). Data were processed using QuanLynx (v 4.1, Waters). Extracted ion chromatograms (EICs) were extracted using a 20 mDa window centered on the expected m/z 349.11 for CPT.
Uptake study
2,000 4T1.2 cells/well were seeded into 96-well plates and incubated overnight. Cells were then treated with PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE for 2 h at 4 or 37 °C. Afterwards, the cells were washed thrice by cold PBS to remove the drug that was not taken up by cells. Cells were further cultured in drug-free medium for 24 h and cytotoxicity was examined via MTT assay.
Mechanistic investigation of the uptake pathway was further performed by using confocal laser scanning microscopy (CLSM, FluoView 1000, Olympus, Japan). Briefly, 3×105 4T1.2 cells/well were seeded into 6-well plates and incubated overnight. Cells were then challenged with PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE for 30 min at 37 °C at a CPT concentration of 6 μg/mL. After that, cells were washed three times with cold PBS and fixed with 4% paraformaldehyde for 30 min. Then, lysotracker was applied to cells for 5 min. Subsequently, cells were washed thrice with cold PBS prior to the observation under CLSM.
Biodistribution
PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE (carrier/drug: 0.75:1) were intravenously injected into 4T1.2 tumor-bearing mice at a dose of 5 mg CPT/kg, respectively (n=3). At 24 h post-injection, tumors, liver, spleen, lung, heart, kidneys and blood were collected from the mice. Tissues were homogenized using Power Gen 500 homogenizer (Fisher Scientific) with 100 mg tissues mixed with 900 μL methanol, and the CPT was extracted overnight at −20 °C. The samples were then centrifuged at 14,000 rpm for 10 min at 4°C and the supernatant was measured by UPLC-QTOFMS as described above.
In vivo antitumor therapeutic study
The antitumor efficacy of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE was investigated in 4T1.2 tumor-bearing mice. 2 x 105 4T1.2 cells in 200 μL saline were inoculated subcutaneously at the right flank of female BALB/c mice. Mice were randomly assigned to five groups (n = 5), when tumors in the mice reached a volume of 50–100 mm3, and this day was designated as day 1. From day 1, mice were i.v. administered with saline, PEG5K-Fmoc-VE2/CPT-VE (5 mg CPT/kg), PEG5K-Fmoc-VE2/CPT-S-S-VE (5 mg CPT/kg), and PEG5K-Fmoc-VE2/CPT-S-S-VE (10 mg CPT/kg) on days 1, 4, and 7, respectively. Free CPT was given to mice i.p. (5 mg/kg) because of its low solubility [16]. Tumor sizes were measured with a digital caliper on days 1, 4, 7, 9, 12, 15, 18, 21 and calculated according to the following formula: (L×W2)/2, where L and W are length and width of each tumor. Meanwhile, the tumor growth inhibition rate (IR) was assessed and defined as: IR % = (1 – tumor volume in the treated group/tumor volume in the saline group) × 100%. Mouse body weight was also monitored as an indication of toxicity. Tumors from all groups were weighted and imaged at the termination of the study. In addition, the tumors were subjected to the H&E staining and pathohistological examination.
Statistical analysis
In all statistical analyses, the significance level was set at a probability of P < 0.05. All results were reported as the mean ± standard deviation (SD) unless otherwise indicated. Statistical analysis was performed by using the Student’s t-test for two groups, and one-way ANOVA for multiple groups, followed by Newman-Keuls test if P < 0.05.
Results
Synthesis of VE-derivatized CPT prodrugs
Two CPT prodrugs were developed, one with a carbonate ester bond (CPT-VE) and the other one with a disulfide linkage (CPT-S-S-VE). The synthesis routes for the two prodrugs were illustrated in Schemes 1 & 2. For the synthesis of CPT-VE, CPT was first reacted with triphosgene for 20 min with the assistance of DMAP to generate CPT-COCl, which was further reacted with VE overnight to yield CPT-VE. For the synthesis of CPT-S-S-VE, tocopherol-succinate was initially coupled to Bis (2-hydroxyethyl) disulfide to obtain VE-S-S-OH. Meanwhile, CPT-COCl was obtained by reacting CPT with triphosgene. VE-S-S-OH was then coupled to CPT-COCl to yield CPT-S-S-VE. The chemical identities of the two prodrugs were confirmed by 1H-NMR (Figs. S1&3), 13C-NMR, and ESI-MS (Synthesis section in Methods).
Scheme 1.

Synthetic route of CPT-VE (carbonate ester bond)
Scheme 2.

Synthetic route of CPT-S-S-VE (disulfide bond)
Biophysical characterizations of CPT, CPT-VE, and CPT-S-S-VE nanoassemblies
Free CPT has a low solubility in DCM (< 2 mg/mL). Coupling with VE via either carbonate ester or disulfide linkage led to a significant improvement in the solubility of the resulting conjugates in DCM (>100 mg/mL in terms of CPT). Both CPT-VE and CPT-S-S-VE could be readily loaded into PEG5K-Fmoc-VE2 in aqueous solution and the resulting complexes showed a nanofiber-like structure in cryoEM (Fig. 1). In addition, the nanofiber structure appeared to be more extended for PEG5K-Fmoc-VE2/CPT-S-S-VE than that for PEG5K-Fmoc-VE2/CPT-VE. Similar nanofiber structures were observed for the TEMs of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE (Fig. S4). In contrast, loading of CPT-VE or CPT-S-S-VE into PEG5K-VE2 led to formation of spherical structures (Fig. 1). Spherical structures were also observed for CPT formulated in PEG5K-VE2 or PEG5K-Fmoc-VE2 micelles (Fig. 1).
Fig. 1.

Electron microscopy imaging of PEG5K-VE2/CPT (A&B), PEG5K-VE2/CPT-VE (C&D), PEG5K-VE2/CPT-S-S-VE (E&F), PEG5K-Fmoc-VE2/CPT (G&H), PEG5K-Fmoc-VE2/CPT-VE (I&J), and PEG5K-Fmoc-VE2/CPT-S-S-VE (K&L). EM images in panel A, C, E, and G are from negatively stained samples. Panels B, D, F, H-L are cryoEM images recorded from frozen-hydrated samples. Panel I and K are enlarged views of J and L, respectively. All panels, except I-K, are at the same scale as in A. Scale bars, 100 nm in A&J, 50 nm in I&K.
Table 1 shows that the DLCs of PEG5K-VE2 and PEG5K-Fmoc-VE2 nanomicelles for CPT were only 0.30% and 0.65%, respectively. In contrast, the DLCs for both prodrugs were improved significantly. The DLCs of PEG5K-VE2 for CPT-VE and CPT-S-S-VE were 4.7% and 6.2%, respectively. Incorporation of Fmoc into PEG5K-VE2 led to a further improvement in the DLC for CPT-VE (6.6%) and CPT-S-S-VE (9.2%). Derivatization of CPT with VE also led to a significant improvement in the colloidal stability of the drug-loaded micelles, particularly for the PEG5K-Fmoc-VE2 micellar system (Table 1). The sizes and size distributions of CPT, CPT-VE, and CPT-S-S-VE nanoassemblies as determined by DLS (Table 1 & Fig. S5–S10) were consistent with the EM results (Fig. 1).
Table 1.
Biophysical characterization of CPT, CPT-VE, and CPT-S-S-VE nanoformulations
| Molar ratio | DLC (%) | DLE (%) | Size (nm) | PDI | Stability | Zeta potential (mv) | |
|---|---|---|---|---|---|---|---|
| PEG5K-VE2/CPT | 15:1 | 0.30 | 79.6 | 52.3±2.8 | 0.24 | 3 days | 1.35±0.02 |
|
| |||||||
| PEG5K-Fmoc-VE2/CPT | 7.5:1 | 0.65 | 85.3 | 76.4±3.6 | 0.25 | 8 days | 0.87±0.06 |
|
| |||||||
| PEG5K-VE2/CPT-VE | 0.75:1 | 4.7 | 72.5 | 85.4±3.5 | 0.23 | 5 days | −1.22±0.32 |
| 1:1 | 4.3 | 83.8 | 52.3±2.2 | 0.21 | 7 days | −1.03±0.13 | |
|
| |||||||
| PEG5K-VE2/CPT-S-S-VE | 0.5:1 | 6.2 | 71.7 | 62.2±2.4 | 0.24 | 7 days | −0.66±0.08 |
| 0.75:1 | 5.3 | 85.4 | 48.6±1.8 | 0.22 | 12 days | 0.16±0.03 | |
|
| |||||||
| PEG5K-Fmoc-VE2/CPT-VE | 0.5:1 | 6.6 | 76.2 | 93.2±4.5 | 0.26 | 21 days | −1.29±0.18 |
| 0.75:1 | 5.4 | 89.1 | 87.4±4.6 | 0.22 | 30 days | −0.95±0.21 | |
|
| |||||||
| PEG5K-Fmoc-VE2/CPT-S-S-VE | 0.25:1 | 9.2 | 70.6 | 81.5±3.9 | 0.20 | 31 days | −1.03±0.25 |
| 0.5:1 | 6.8 | 83.5 | 57.6±2.1 | 0.17 | 62 days | −0.78±0.14 | |
| 0.75:1 | 5.7 | 96.3 | 49.7±1.8 | 0.15 | 75 days | 0.64±0.12 | |
DLC (%) = [weight of CPT loaded ÷ (weight of carrier + weight of CPT or CPT prodrugs loaded)]
DLE (%) = (weight of loaded drug ÷ weight of input drug) X 100%
PDI: polydispersity index
Stability: no significant size change was observed during the time period tested in PBS solution at room temperature.
NMR analysis of loading of CPT prodrugs into micelles
To confirm whether CPT-VE or CPT-S-S-VE was indeed incorporated into the PEG5K-Fmoc-VE2 micelles. A series of 1NMR studies were conducted, including CPT-VE in CDCl3, CPT-S-S-VE in CDCl3, PEG5K-Fmoc-VE2 in CDCl3 or D2O, PEG5K-Fmoc-VE2/CPT-VE in CDCl3 or D2O, and PEG5K-Fmoc-VE2/CPT-S-S-VE in CDCl3 or D2O. CPT-VE and PEG5K-Fmoc-VE2 showed their respective peaks in CDCl3 (Fig. S1), which were consistent with their structures. Mixed PEG5K-Fmoc-VE2/CPT-VE in CDCl3 also clearly showed the typical peaks for PEG5K-Fmoc-VE2 and CPT-VE, respectively (Fig. S1). However, when PEG5K-Fmoc-VE2/CPT-VE was examined in D2O, the peaks for PEG5K-Fmoc-VE2 and CPT-VE completely disappeared, except the PEG and solvent peaks. Similar data were obtained for CPT-S-S-VE system (Fig. S3). All of the NMR samples were subjected to overnight scanning in order to rule out the possibility that the disappearances of the peaks were due to the lack of sufficient scanning.
Fluorescence quenching assay was performed to gain a mechanistic insight into the carrier/drug interactions. Fig. 2A shows that CPT, CPT-VE, and CPT-S-S-VE exhibited a similar fluorescence pattern upon excitation at 370 nm. This fluorescence spectrum appears to be specific for CPT as PEG5K-VE2 and PEG5K-Fmoc-VE2 did not give any fluorescence signals under the same condition. Fig. 2A also shows that the fluorescence intensity of CPT-VE or CPT-S-S-VE was drastically decreased when loaded into carriers. The fluorescence quenching was more dramatic when either prodrug was incorporated into PEG5K-Fmoc-VE2 in comparison to PEG5K-VE2 (Fig. 2A & B). The blue curve in Fig. 2C shows a fluorescence spectrum that was consistent with that of Fmoc as reported in literature [17]. It is apparent that incorporation into PEG5K-Fmoc-VE2 a prodrug, particularly CPT-S-S-VE, led to significant quenching of Fmoc fluorescence. The significant fluorescence quenching in both experiments suggests a strong π-π interaction between the carrier and the prodrug.
Fig. 2.

A: fluorescence quenching study of CPT-VE or CPT-S-S-VE nanoformulations. B: quantitation of the fluorescence decrease in CPT-VE or CPT-S-S-VE nanoassemblies compared to the free CPT-VE or CPT-S-S-VE. C: Fmoc Fluorescence. D: reduction of Fmoc fluorescence intensity after incorporating CPT-VE or CPT-S-S-VE into PEG5K-Fmoc-VE2.
UV absorbance analysis
Fig. 3 shows the respective UV-absorbance of free CPT, CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE. A significant decrease of UV-absorbance was shown for the PEG5K-Fmoc-VE2/prodrug mixed micelles, especially the CPT-S-S-VE system. In addition, a clear red shift of UV-absorbance was observed in both mixed micelles, further suggesting that π-π stacking contributed significantly to the overall carrier/drug interactions.
Fig. 3.

UV-absorbance of CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE, and PEG5K-Fmoc-VE2/CPT-S-S-VE.
Fourier transform infrared spectroscopy (FT-IR) analysis
FT-IR was conducted to determine the hydrogen bonding between the carrier and prodrug. As depicted in Fig. 4A&B, after subtracting the IR of PEG5K-Fmoc-VE2 from that of PEG5K-Fmoc-VE2/CPT-VE, two new peaks at 1463.11 cm−1 and 1472.51 cm−1 were identified (Fig. 4B). The decrease of absorbance at 1472.51 cm−1 and a concurrent increase of absorbance at 1463.11 cm−1 suggested that incorporation of CPT-VE resulted in the disruption of the hydrogen bonding among carrier molecules and the formation of new hydrogen bonding between carrier and CPT-VE molecules. Similar results were obtained for PEG5K-Fmoc-VE2/CPT-S-S-VE system (Fig. 4C&D).
Fig. 4.

A: FT-IR of PEG5K-Fmoc-VE2 and PEG5K-Fmoc-VE2/CPT-VE. B: Difference of FT-IR between PEG5K-Fmoc-VE2 and PEG5K-Fmoc-VE2/CPT-VE. C: FT-IR of PEG5K-Fmoc-VE2 and PEG5K-Fmoc-VE2/CPT-S-S-VE. D: Difference of FT-IR between PEG5K-Fmoc-VE2 and PEG5K-Fmoc-VE2/CPT-S-S-VE.
In vitro cytotoxicity and CPT intracellular release
To examine whether VE-derivatized CPTs still retained the biological function of its parental CPT, cytotoxicity of free CPT, CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE was assessed in 4T1.2 cells. To further examine the reduction-mediated release of CPT for CPT-S-S-VE, GSH, a disulfide bond reducer, was added in some of the treatment groups. As shown in Fig. 5A, free CPT inhibited the cell growth in a concentration-dependent manner. CPT-S-S-VE was more active than CPT-VE but both were significantly less effective than free CPT in inhibiting the cell growth. It is also apparent that addition of 10 mM GSH into CPT-S-S-VE led to a significant improvement in the cytotoxicity. In contrast, GSH had no effect on the cytotoxicity of CPT-VE. Delivery of CPT-S-S-VE or CPT-VE via PEG5K-Fmoc-VE2 led to a level of cytotoxicity that was similar to that of free prodrug. Again, PEG5K-Fmoc-VE2/CPT-S-S-VE was more active than PEG5K-Fmoc-VE2/CPT-VE in inhibiting the tumor cell growth.
Fig. 5.
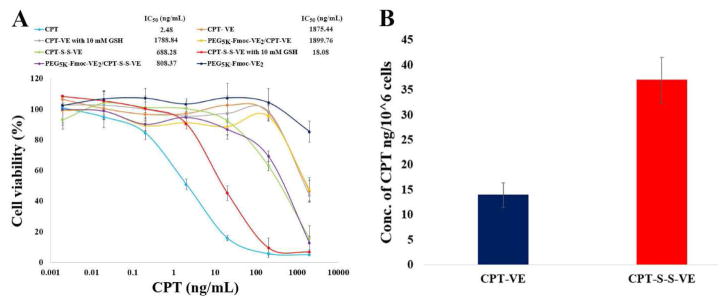
A: The cell-killing effect of CPT-VE, CPT-S-S-VE, PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE with or without GSH (10 mM) in 4T1.2 cancer cell. Cells were treated with indicated concentrations of different CPT formulations for 72 h and cytotoxicity was then assessed by MTT assay. Values reported are the means ± SD for triplicate samples. B: Intracellular release of CPT in 4T1.2 cells treated with CPT-VE and CPT-S-S-VE (100 ng/mL in terms of CPT) for 24 h.
To confirm that the enhanced cytotoxicity for CPT-S-S-VE was indeed ascribed to the more effective release of CPT, the amounts of parent CPT inside cells were measured after treating cells with CPT-S-S-VE or CPT-VE (100 ng/mL in terms of CPT) for 24 h. As shown in Fig. 5B, the amount of released CPT in CPT-S-S-VE-treated cells was about twice as much as that in CPT-VE-treated cells.
Mechanism of intracellular uptake of CPT-VE and CPT-S-S-VE nanoassemblies
To examine whether the intracellular uptake of PEG5K-Fmoc-VE2/CPT-VE or CPT-S-S-VE mixed micelles was mediated by endocytosis, the temperature dependence of uptake was evaluated by comparing the cytotoxicity of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE following treatment at 4 and 37 °C, respectively. In agreement with the data in Fig. 5B, PEG5K-Fmoc-VE2/CPT-S-S-VE was more effective than PEG5K-Fmoc-VE2/CPT-VE in inhibiting the tumor cell growth (Fig. S11A). It is also apparent that cells exposed to PEG5K-Fmoc-VE2/CPT-S-S-VE or PEG5K-Fmoc-VE2/CPT-VE at 4 °C showed better viability compared to cells exposed to the mixed micelles at 37 °C. These data are in consistent with the notion that pinocytic/endocytic uptake is inactivated at 4 °C [18]. To further elucidate the underlying mechanism for the intracellular uptake of the nanomicelles, their intracellular trafficking was investigated via confocal laser scanning microscope. The images were collected at an excitation wavelength of 360 nm and an emission wavelength of 440 nm as CPT is known to emit fluorescence at 440 nm [19]. As shown in Fig. S11B, a perinuclear punctate distribution was observed for both PEG5K-Fmoc-VE2/CPT-S-S-VE and PEG5K-Fmoc-VE2/CPT-VE. The fluorescence from CPT prodrug nanofibers was well colocalized with that from LysoTracker, indicating that endocytic pathway was involved in the uptake of the mixed micelles. Moreover, significantly enhanced fluorescence signal was detected in cells treated with PEG5K-Fmoc-VE2/CPT-S-S-VE compared to that in PEG5K-Fmoc-VE2/CPT-VE-treated cells.
In vivo biodistribution
Fig. 6 shows the in vivo tissue distribution of CPT in 4T1.2 tumor-bearing mice 24 h following i.v. administration of PEG5K-Fmoc-VE2/CPT-VE or PEG5K-Fmoc-VE2/CPT-S-S-VE at a dose of 5 mg CPT/kg. Due to the presence of various metabolites of CPT and CPT prodrugs, we focused on the detection and analysis of parent CPT. Significant amounts of free CPT were detected in tumor tissues for both PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE, particularly for the latter formulation. The amounts of CPT in tumor tissues for PEG5K-Fmoc-VE2/CPT-S-S-VE were about 3 times as much as that for PEG5K-Fmoc-VE2/CPT-VE. In addition, the amounts of CPT in tumor tissues for PEG5K-Fmoc-VE2/CPT-S-S-VE were significantly higher than that in most normal tissues examined including heart, lung, spleen, and kidneys. Significant amounts of CPT were also detected in liver for both PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE.
Fig. 6.

Tissue biodistribution of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE (5mg CPT/kg) in 4T1.2-tumor bearing mice. *p < 0.001, compared to PEG5K-Fmoc-VE2/CPT-VE. Values reported are the means ± SD for triplicate samples.
Antitumor activity
In vivo antitumor effect of CPT prodrug nanoassemblies was examined in 4T1.2 tumor bearing mice (Fig. 7). As expected, mice quickly developed tumors in an uncontrolled manner in saline group (Fig. 7A). Free CPT showed a moderate effect in suppressing the tumor growth at a dose of 5 mg CPT/kg. Significantly improved antitumor activity was achieved in mice treated with PEG5K-Fmoc-VE2/CPT-VE compared to free CPT group. The antitumor activity was further enhanced in the mice treated with PEG5K-Fmoc-VE2/CPT-S-S-VE with an IR of 81.90% (Table 2). Doubling the dose to 10 mg CPT/kg led to almost complete remission with an IR of 93.48%. All of the treatments were well tolerated by mice as evidenced by no significant weight changes of mice during the experimental period (Fig. 7B). Fig. 7C & D show the images and weights of the tumors that were collected following the completion of the study, which were consistent with the tumor growth curves (Fig. 7A). The tumor tissues were further subject to H&E staining and pathohistological examinations. As depicted in Fig. 7E, tumors from the mice treated with PEG5K-Fmoc-VE2/CPT-S-S-VE exhibited significantly more necrotic/apoptotic tumor cells, particularly in the higher dose group (10 mg CPT/kg). Tumor tissue damage was also noticed in the group treated with free CPT or PEG5K-Fmoc-VE2/CPT-VE but to a lesser degree (Fig. 7E).
Fig. 7.

Antitumor efficacy of various CPT prodrug nanoformulations in 4T1.2 breast tumor model. Solid arrows indicate the i.v. injection. A: tumor volume. *p < 0.01, compared to PEG5K-Fmoc-VE2/CPT-S-S-VE (5 mg/kg); αp < 0.001, compared to PEG5K-Fmoc-VE2/CPT-VE (5 mg/kg) and CPT (5 mg/kg); βp < 0.001, compared to saline. B: mouse body weight. C: tumor images. D: tumor weight (g). E: Pathological examination of tumor tissues resected from the mice. Nuclei were stained by hematoxylin (blue), and extracellular matrix and cytoplasm were doped by eosin (red). Significant amount of necrotic/apoptotic cells were discerned in tumor tissues treated with PEG5K-Fmoc-VE2/CPT-S-S-VE over the other CPT formulations
Table 2.
Tumor growth inhibition rate in 4T1.2 tumor bearing mice
| Treatments | IR (%) |
|---|---|
| CPT 5 mg/kg | 43.69 |
| PEG5K-Fmoc-VE2/CPT-VE 5 mg/kg | 56.06 |
| PEG5K-Fmoc-VE2/CPT-S-S-VE 5 mg/kg | 81.90 |
| PEG5K-Fmoc-VE2/CPT-S-S-VE 10 mg/kg | 93.48 |
IR % = (1 – tumor volume in the treated group/tumor volume in the saline group) × 100%
IR % was calculated based on the tumor volume on day 21
Discussion
In the present study, two VE-based CPT prodrugs were developed to facilitate the incorporation of CPT into PEG5K-Fmoc-VE2 nanomicellar formulation. One is conjugation of CPT with VE via a carbonate ester bond to generate CPT-VE prodrug, in which the CPT is expected to be released following the breakdown of the ester bond by the esterase in cells. In order to further facilitate the liberation of the CPT from prodrug, disulfide linkage was introduced to bridge CPT and VE, which resulted in the formation of CPT-S-S-VE. It has been well established that reducing agents, such as GSH are capable of effectively breaking the disulfide bond through thiol exchange process [20–25]. Therefore, active CPT is likely to be readily released following intracellular delivery, particularly in cancerous cells where the concentration of GSH is significantly increased (~ 10 mM) [23–26].
For the synthesis of CPT-VE, we initially started with an attempt to directly conjugate CPT with VE-succinate. It was anticipated that the carboxylate group in VE-succinate can be readily coupled to the hydroxyl group in CPT with the catalysis of DCC and DMAP. However, several attempts only met with limited success, which may be due to the inadequate accessibility of the hydroxyl group in CPT, resulting in inefficient conjugation between –OH and –COOH. Triphosgene was then applied to convert the –OH of CPT to –COCl that is more active in reacting with other nucleophiles including –OH [27]. Subsequent reaction of CPT-COCl with VE (-OH) successfully yielded CPT-VE with a carbonate ester bond. For the synthesis of CPT-S-S-VE, an input ratio (m/m) of 1:2 between VE-succinate and HOCH2CH2-S-S-CH2CH2OH was used in the 1st step. Under this condition, the major product was VE-S-S-OH and only a very small amount of VE-S-S-VE was present. VE-S-S-OH was then coupled to CPT-COCl to yield CPT-S-S-VE with a disulfide bond. Reduction-sensitive linkage has been extensively employed as a strategy to facilitate the drug release or activation of prodrugs at tumor site [20–25]. Following the breakage of the disulfide linkage in CPT-S-S-VE upon exposure to GSH, the parent CPT is expected to be effectively generated by a next step of intramolecular cyclization (Scheme 2). Indeed, CPT-S-S-VE was much more effective than CPT-VE in releasing the CPT as demonstrated in both cultured tumor cells and tumor tissues in tumor-bearing mice (Figs. 5&6).
Both CPT-VE and CPT-S-S-VE exhibited significantly improved solubility in DCM over free CPT, which was essential in streamlining the micelle preparation procedure and facilitating our solvent evaporation method. In contrast, dialysis method was needed in formulating free CPT, which is tedious and time-consuming. Derivatizing CPT with VE led to dramatically augmented DLC in PEG5K-Fmoc-VE2/CPT-VE (6.6%) and PEG5K-Fmoc-VE2/CPT-S-S-VE (9.2%), even in PEG5K-VE2/CPT-VE (4.7%) and PEG5K-VE2/CPT-S-S-VE (6.2%), which compared favorably to the very low CPT loading in PEG5K-VE2/CPT (0.3%) and PEG5K-Fmoc-VE2/CPT (0.65%) (Table 1). Meanwhile, the colloidal stability of the prodrug-formulated nanofibers was also significantly enhanced (PEG5K-Fmoc-VE2/CPT-VE > 21 days and PEG5K-Fmoc-VE2/CPT-S-S-VE > 31 days), in comparison to the CPT-loaded nanoparticles. The drastically improved DLC and formulation stability in both prodrug formulations could be ascribed to the enhanced interactions between the prodrugs and the carriers as evidenced by fluorescence quenching (Fig. 2), UV absorbance (Fig. 3), as well as FT-IR studies (Fig. 4). After coupling with VE, the compatibility of CPT prodrugs with VE-containing carriers was greatly ameliorated. The VE molecules in CPT prodrugs are likely to function as an anchor to facilitate the insertion in between VE chains in PEG5K-Fmoc-VE2 (Fig. 8). In addition, the presence of Fmoc is expected to contribute to the enhanced carrier/drug interactions [10, 28–31]. The strong carrier/carrier and carrier/drug interactions are likely to contribute significantly to the formation of nanofiber structures of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE (Fig. 8).
Fig. 8.

Proposed mechanism for the interaction of PEG5K-Fmoc-VE2 with CPT-VE or CPT-S-S-VE and the formation of nanofiber structures
The significantly enhanced cell viability in vitro at 4 °C implied that VE-derivatized CPT nanofibers internalize cells via endocytosis (Fig. S11A). This was further corroborated by the results from the confocal imaging, in which the nanofibers were well colocalized with the lysotracker (Fig. S11B). This mechanism of cellular uptake may allow our nanoformulations to bypass the P-gp-mediated efflux pump and overcome the multidrug resistance [32, 33].
Both PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE nanofibers showed significantly enhanced antitumor efficacy over PEG5K-Fmoc-VE2/CPT in a breast tumor model (Fig. 7). Multiple mechanisms might contribute to the overall enhanced tumor growth inhibition. First of all, conjugation of VE molecule to the hydroxyl group on CPT can enhance CPT stability in vivo [2–4]. Second, when the CPT-VE or CPT-S-S- VE is loaded into PEG5K-Fmoc-VE2, the degradation of CPT in the blood could be further reduced, resulting in the improved bioavailability of CPT. It has also been reported that the elongated nanofibers are able to circulate for a much longer period of time than the particles of spherical morphology [34]. Thus, CPT prodrug nanofibers will have a better chance to be preferentially deposited in tumors. Once the nanoassemblies arrive at the tumors, the small size of the particles will allow effective penetration into the tumor tissues [35]. Moreover, PEG5K-Fmoc-VE2 has been shown to be capable of inhibiting the function of p-gp ATPase, which may slow down or prevent the drug resistance that may develop over the course of treatment [10]. The further improved antitumor activity of CPT-S-S-VE nanofibers over CPT-VE nanofibers is likely due to the fact CPT is more readily released from the former formulation (Fig. 6). More studies are needed to better understand the underlying mechanism for the improved antitumor activity of PEG5K-Fmoc-VE2/CPT-S-S-VE.
Conclusion
Two VE-derivatized CPT prodrugs were developed by conjugating CPT to VE via a carbonate ester bond (CPT-VE) or disulfide linkage (CPT-S-S-VE). Both CPT prodrugs were able to self-assemble into nanofibers upon stabilization by PEG5K-Fmoc-VE2, resulting in a significantly improved DLC over free CPT. Both nanofibers exhibited excellent stability profiles. More importantly, PEG5K-Fmoc-VE2/CPT-S-S-VE nanoformulation showed superior antitumor efficacy, resulting in almost complete tumor regression in an aggressive murine breast cancer model. To reiterate, VE-conjugated prodrugs may represent an effective approach for managing the therapeutic agents that are complex in structures and hard to formulate with current delivery systems.
Supplementary Material
Fig. S1. NMR spectra of CPT-VE in CDCl3, PEG5K-Fmoc-VE2 in CDCl3, PEG5K-Fmoc-VE2/CPT-VE in CDCl3, and PEG5K-Fmoc-VE2/CPT-S-S-VE in D2O.
Fig. S2. 1H-NMR spectrum (400 MHz) of VE-S-S-OH in CDCl3.
Fig. S3. NMR spectra of CPT-S-S-VE in CDCl3, PEG5K-Fmoc-VE2 in CDCl3, PEG5K-Fmoc-VE2/CPT-S-S-VE in CDCl3, and PEG5K-Fmoc-VE2/CPT-S-S-VE in D2O.
Fig. S4. TEM images of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE.
Figure S5. Size distribution of PEG5K-VE2/CPT.
Figure S6. Size distribution of PEG5K-Fmoc-VE2/CPT.
Figure S7. Size distribution of PEG5K-VE2/CPT-VE.
Figure S8. Size distribution of PEG5K-VE2/CPT-S-S-VE.
Figure S9. Size distribution of PEG5K-Fmoc-VE2/CPT-VE.
Figure S10. Size distribution of PEG5K-Fmoc-VE2/CPT-S-S-VE.
Fig. S11. A: The cell viability in 4T1.2 cells after being treated with PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE for 2 h at 4 or 37 °C, followed by 24 h incubation at a CPT concentration of 6 μg/mL. B: Confocal laser scanning microscopy (CLSM) images of 4T1.2 cells incubated with PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE for 30 min at 37 °C at a CPT concentration of 6 μg/mL.
Acknowledgments
This work was supported in part by NIH Grants RO1CA173887, RO1GM102989, and R21CA173887.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d’Argy R, et al. Polymer-bound camptothecin: initial biodistribution and antitumour activity studies. Journal of controlled release. 2000;65:105–19. doi: 10.1016/s0168-3659(99)00243-6. [DOI] [PubMed] [Google Scholar]
- 2.Barreiro-Iglesias R, Bromberg L, Temchenko M, Hatton TA, Concheiro A, Alvarez-Lorenzo C. Solubilization and stabilization of camptothecin in micellar solutions of pluronic-g-poly(acrylic acid) copolymers. Journal of controlled release. 2004;97:537–49. doi: 10.1016/j.jconrel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Shenderova A, Burke TG, Schwendeman SP. Stabilization of 10-hydroxycamptothecin in poly(lactide-co-glycolide) microsphere delivery vehicles. Pharmarceutical Research. 1997;14:1406–14. doi: 10.1023/a:1012172722246. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Jiang Z, Zhang S, Saltzman WM. Poly(omega-pentadecalactone-co-butylene-co-succinate) nanoparticles as biodegradable carriers for camptothecin delivery. Biomaterials. 2009;30:5707–19. doi: 10.1016/j.biomaterials.2009.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modi S, Xiang TX, Anderson BD. Enhanced active liposomal loading of a poorly soluble ionizable drug using supersaturated drug solutions. Journal of controlled release. 2012;162:330–9. doi: 10.1016/j.jconrel.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Harada Y, Yamamoto T, Sakai M, Saiki T, Kawano K, Maitani Y, et al. Effects of organic solvents on drug incorporation into polymeric carriers and morphological analyses of drug-incorporated polymeric micelles. International journal of pharmaceutics. 2011;404:271–80. doi: 10.1016/j.ijpharm.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Yang L, Cui F, Cun D, Tao A, Shi K, Lin W. Preparation, characterization and biodistribution of the lactone form of 10-hydroxycamptothecin (HCPT)-loaded bovine serum albumin (BSA) nanoparticles. International journal of pharmaceutics. 2007;340:163–72. doi: 10.1016/j.ijpharm.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 8.McRae Page S, Martorella M, Parelkar S, Kosif I, Emrick T. Disulfide cross-linked phosphorylcholine micelles for triggered release of camptothecin. Molecular pharmaceutics. 2013;10:2684–92. doi: 10.1021/mp400114n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duhem N, Danhier F, Pourcelle V, Schumers JM, Bertrand O, Leduff CS, et al. Self-assembling doxorubicin-tocopherol succinate prodrug as a new drug delivery system: synthesis, characterization, and in vitro and in vivo anticancer activity. Bioconjugate chemistry. 2014;25:72–81. doi: 10.1021/bc400326y. [DOI] [PubMed] [Google Scholar]
- 10.Lu J, Zhao W, Liu H, Marquez R, Huang Y, Zhang Y, et al. An improved d-alpha-tocopherol-based nanocarrier for targeted delivery of doxorubicin with reversal of multidrug resistance. Journal of controlled release. 2014;196:272–86. doi: 10.1016/j.jconrel.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Huang Y, Zhao W, Lu J, Zhang P, Zhang X, et al. Fmoc-conjugated PEG-vitamin E2 micelles for tumor-targeted delivery of paclitaxel: enhanced drug-carrier interaction and loading capacity. The AAPS journal. 2014;16:1282–91. doi: 10.1208/s12248-014-9651-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Huang Y, Zhao W, Chen Y, Li J, Gao X, et al. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Molecular pharmaceutics. 2013;10:2880–90. doi: 10.1021/mp300729y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Lu J, Gao X, Li J, Zhao W, Sun M, et al. PEG-derivatized embelin as a dual functional carrier for the delivery of paclitaxel. Bioconjugate chemistry. 2012;23:1443–51. doi: 10.1021/bc3000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, Huang Y, Zhao W, Marquez RT, Meng X, Li J, et al. PEG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials. 2013;34:1591–600. doi: 10.1016/j.biomaterials.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu J, Zhao W, Huang Y, Liu H, Marquez R, Gibbs RB, et al. Targeted delivery of Doxorubicin by folic acid-decorated dual functional nanocarrier. Molecular pharmaceutics. 2014;11:4164–78. doi: 10.1021/mp500389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai W, Mo R, Lu Y, Jiang T, Gu Z. Folding graft copolymer with pendant drug segments for co-delivery of anticancer drugs. Biomaterials. 2014;35:7194–203. doi: 10.1016/j.biomaterials.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yun MH, Kwon KI. High-performance liquid chromatography method for determining alendronate sodium in human plasma by detecting fluorescence: application to a pharmacokinetic study in humans. Journal of pharmaceutical and biomedical analysis. 2006;40:168–72. doi: 10.1016/j.jpba.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 18.Tomoda H, Kishimoto Y, Lee YC. Temperature effect on endocytosis and exocytosis by rabbit alveolar macrophages. The Journal of biological chemistry. 1989;264:15445–50. [PubMed] [Google Scholar]
- 19.Tsai TH, Chen YF, Chou CJ, Chen CF. Measurement and pharmacokinetics of unbound 20(S)-camptothecin in rat blood and brain by microdialysis coupled to microbore liquid chromatography with fluorescence detection. Journal of chromatography A. 2000;870:221–6. doi: 10.1016/s0021-9673(99)00854-7. [DOI] [PubMed] [Google Scholar]
- 20.Brulisauer L, Kathriner N, Prenrecaj M, Gauthier MA, Leroux JC. Tracking the bioreduction of disulfide-containing cationic dendrimers. Angewandte Chemie. 2012;51:12454–8. doi: 10.1002/anie.201207070. [DOI] [PubMed] [Google Scholar]
- 21.Lee SY, Kim S, Tyler JY, Park K, Cheng JX. Blood-stable, tumor-adaptable disulfide bonded mPEG-(Cys)4-PDLLA micelles for chemotherapy. Biomaterials. 2013;34:552–61. doi: 10.1016/j.biomaterials.2012.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lv S, Tang Z, Zhang D, Song W, Li M, Lin J, et al. Well-defined polymer-drug conjugate engineered with redox and pH-sensitive release mechanism for efficient delivery of paclitaxel. Journal of controlled release. 2014;194:220–7. doi: 10.1016/j.jconrel.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Suma T, Miyata K, Anraku Y, Watanabe S, Christie RJ, Takemoto H, et al. Smart multilayered assembly for biocompatible siRNA delivery featuring dissolvable silica, endosome-disrupting polycation, and detachable PEG. ACS nano. 2012;6:6693–705. doi: 10.1021/nn301164a. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Yang G, Guo X, Tang Z, Zhong Z, Zhou S. Redox-responsive polyanhydride micelles for cancer therapy. Biomaterials. 2014;35:3080–90. doi: 10.1016/j.biomaterials.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Liu K, Huang Y, Xu J, Li J, Ma X, et al. Reduction-sensitive dual functional nanomicelles for improved delivery of paclitaxel. Bioconjugate chemistry. 2014;25:1689–96. doi: 10.1021/bc500292j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. The Journal of nutrition. 2004;134:489–92. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 27.Lee MH, Kim JY, Han JH, Bhuniya S, Sessler JL, Kang C, et al. Direct fluorescence monitoring of the delivery and cellular uptake of a cancer-targeted RGD peptide-appended naphthalimide theragnostic prodrug. Journal of the American Chemical Society. 2012;134:12668–74. doi: 10.1021/ja303998y. [DOI] [PubMed] [Google Scholar]
- 28.Gao X, Huang Y, Makhov AM, Epperly M, Lu J, Grab S, et al. Nanoassembly of surfactants with interfacial drug-interactive motifs as tailor-designed drug carriers. Molecular pharmaceutics. 2013;10:187–98. doi: 10.1021/mp300319m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang P, Huang Y, Liu H, Marquez RT, Lu J, Zhao W, et al. A PEG-Fmoc conjugate as a nanocarrier for paclitaxel. Biomaterials. 2014;35:7146–56. doi: 10.1016/j.biomaterials.2014.04.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P, Lu J, Huang Y, Zhao W, Zhang Y, Zhang X, et al. Design and evaluation of a PEGylated lipopeptide equipped with drug-interactive motifs as an improved drug carrier. The AAPS journal. 2014;16:114–24. doi: 10.1208/s12248-013-9536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Huang Y, Zhao W, Liu H, Marquez R, Lu J, et al. Targeted delivery of anticancer agents via a dual function nanocarrier with an interfacial drug-interactive motif. Biomacromolecules. 2014;15:4326–35. doi: 10.1021/bm501339j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Emanuele A, Jevprasesphant R, Penny J, Attwood D. The use of a dendrimer-propranolol prodrug to bypass efflux transporters and enhance oral bioavailability. Journal of controlled release. 2004;95:447–53. doi: 10.1016/j.jconrel.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. The Journal of pharmacology and experimental therapeutics. 2006;317:1372–81. doi: 10.1124/jpet.106.101154. [DOI] [PubMed] [Google Scholar]
- 34.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nature nanotechnology. 2007;2:249–55. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan YH, et al. Well-defined, size-tunable, multifunctional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjugate chemistry. 2010;21:1216–24. doi: 10.1021/bc1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. NMR spectra of CPT-VE in CDCl3, PEG5K-Fmoc-VE2 in CDCl3, PEG5K-Fmoc-VE2/CPT-VE in CDCl3, and PEG5K-Fmoc-VE2/CPT-S-S-VE in D2O.
Fig. S2. 1H-NMR spectrum (400 MHz) of VE-S-S-OH in CDCl3.
Fig. S3. NMR spectra of CPT-S-S-VE in CDCl3, PEG5K-Fmoc-VE2 in CDCl3, PEG5K-Fmoc-VE2/CPT-S-S-VE in CDCl3, and PEG5K-Fmoc-VE2/CPT-S-S-VE in D2O.
Fig. S4. TEM images of PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE.
Figure S5. Size distribution of PEG5K-VE2/CPT.
Figure S6. Size distribution of PEG5K-Fmoc-VE2/CPT.
Figure S7. Size distribution of PEG5K-VE2/CPT-VE.
Figure S8. Size distribution of PEG5K-VE2/CPT-S-S-VE.
Figure S9. Size distribution of PEG5K-Fmoc-VE2/CPT-VE.
Figure S10. Size distribution of PEG5K-Fmoc-VE2/CPT-S-S-VE.
Fig. S11. A: The cell viability in 4T1.2 cells after being treated with PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE for 2 h at 4 or 37 °C, followed by 24 h incubation at a CPT concentration of 6 μg/mL. B: Confocal laser scanning microscopy (CLSM) images of 4T1.2 cells incubated with PEG5K-Fmoc-VE2/CPT-VE and PEG5K-Fmoc-VE2/CPT-S-S-VE for 30 min at 37 °C at a CPT concentration of 6 μg/mL.