

CONSPECTUS
The control of regiochemistry is a considerable challenge in the development of a wide array of catalytic processes. Simple π-components such as alkenes, alkynes, 1,3-dienes, and allenes are among the many classes of substrates that present complexities in regioselective catalysis. Considering an internal alkyne as a representative example, when steric and electronic differences between the two substituents are minimal, differentiating among the two termini of the alkyne presents a great challenge. In cases where the differences between the alkyne substituents are substantial, overcoming those biases to access the regioisomer opposite that favored by substrate biases often presents an even greater challenge.
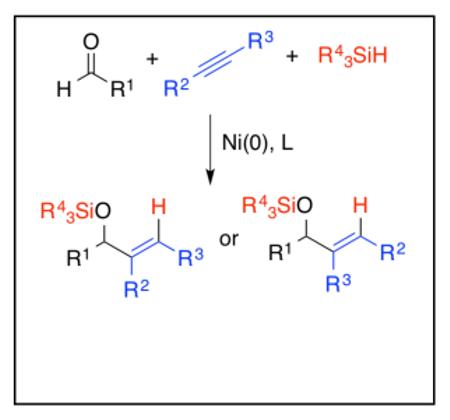
Nickel-catalyzed reductive couplings of unsymmetrical π-components make up a group of reactions where control of regiochemistry presents a challenging but important objective. In the course of our studies of aldehyde-alkyne reductive couplings, complementary solutions to challenges in regiocontrol have been developed. Through careful selection of the ligand and reductant, as well as the more subtle reaction variables such as temperature and concentration, effective protocols have been established that allow highly selective access to either regiosiomer of the the allylic alcohol products using a wide range of unsymmetrical alkynes. Computational studies and an evaluation of reaction kinetics have provided an understanding of the origin of the regioselectivity control. Throughout the various procedures described, the development of ligand-substrate interactions play a key role, and the overall kinetic descriptions were found to differ between protocols. Rational alteration of the rate-determining step plays a key role in the regiochemistry reversal strategy, and in one instance, the two possible regioisomeric outcomes in a single reaction were found to operate by different kinetic descriptions. With this mechanistic information in hand, the empirical factors that influence regiochemistry can be readily understood, and more importantly, the insights suggest simple and predictable experimental variables to achieving a desired reaction outcome.
These studies thus present a detailed picture of the influences that control regioselectivity in a specific catalytic reaction, but they also delineate strategies for regiocontrol that may extend to numerous classes of reactions. The work provides an illustration of how insights into the kinetics and mechanism of a catalytic process can rationalize subtle empirical findings and suggest simple and rational modifications in procedure to access a desirable reaction outcome. Furthermore, these studies present an illustration of how important challenges in organic synthesis can be met by novel reactivity afforded by base metal catalysis. The use of nickel catalysis in this instance not only provides an inexpensive and sustainable method for catalysis, but also enables unique reactivity patterns not accessible to other metals.
Keywords: Regiochemistry, stereochemistry, N-heterocyclic carbene, aldehyde, alkyne
Introduction
The control of selectivity in catalytic processes presents a considerable challenge in the development of new synthetic transformations. Different types of selectivity introduce unique challenges, including the control or enantioselectivity, regioselectivity, chemoselectivity, and site-selectivity.1 Among these, the development of regiodivergent strategies in catalyzed additions to π-components utilizing first row transition metals has been a particular emphasis of several projects in our laboratory in recent years. A regioselective process is defined as one where one regioisomer can be accessed in preference to another, and a regiodivergent processes is one where more than one regiochemical outcome can be selectively accessed for a single combination of substrates by tailoring the properties and behavior of the catalyst system. Developing regiodivergent processes presents considerably greater challenges than accessing a single regiochemical outcome. As depicted below (Scheme 1), catalytic additions to unsymmetrical alkenes and alkynes can afford two possible regioisomers, whereas additions to allenes can afford 4 possible regioisomeric outcomes. The various possible stereochemical relationships of the alkenes and stereogenic centers in the products add further to the number and types of isomers that may be obtained. Gaining access to all of the possible isomers in additions to unsymmetrical π-systems such as these presents a major challenge for nearly all classes of chemical reactions.2-4
Scheme 1.
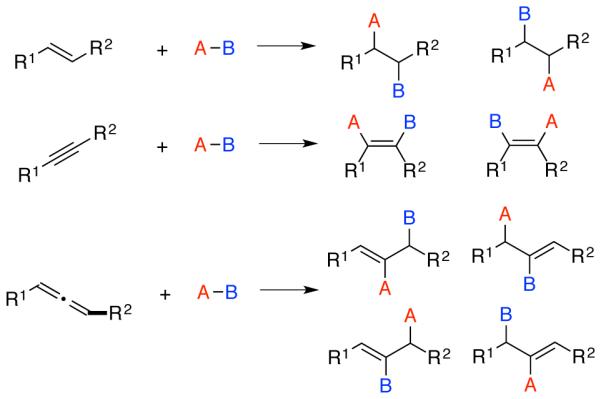
Regioselectivity outcomes in additions to alkenes, alkynes, and allenes.
One of the most widely employed strategies for control of catalytic, regioselective additions to π-components involves the use of directing groups on the alkyne.5 Strategies of this type can be very effective in the control of regiochemistry, where a remote directing group can bind to a catalyst and favor one orientation of the substrate in the regiochemistry-determining step. As installation and removal of the directing group can add additional steps to the overall synthetic operation, cases in which the directing group is either desired in the final product structure or is easily converted to a desired functional group provide the most useful contexts for directed regiocontrol strategies. On the other hand, scenarios where directing groups are not easily installed or removed present a different set of challenges and require the development of non-directed approaches.1,6-10 In these cases involving non-directed additions, π-components in regioselective and regiodivergent processes can typically be classified as biased or unbiased as illustrated for alkenes, alkynes, and allenes (Scheme 2). With biased substrates such as monosubstituted π-systems 1a-c, or alternatively disubstituted π-systems 2a-c where R1 and R2 markedly differ in steric or electronic properties, structural features of the substrates influence the properties of the π-system. This bias often promotes a single operative mechanistic pathway, leading to a strong preference for the formation of one regioisomer over another. In cases where two different catalysts can promote the same net addition process by fundamentally different mechanisms, the inherent structural biases in the substrate can often allow a regiodivergent outcome for the two catalyst systems. A classic illustration of this type of regiodivergency is the copper- and ruthenium-catalyzed click reactions of azides and alkynes to access either 1,4- or 1,5-triazole products.11-12 Alternatively, accessing regiodivergent outcomes with biased substrates for a single catalyst type that operates by a single mechanism presents a considerable challenge. In these cases, either significant variation in ligand structure or fundamental changes in the kinetics (and thus identity of the regiochemistry-determining step) are typically required.
Scheme 2.
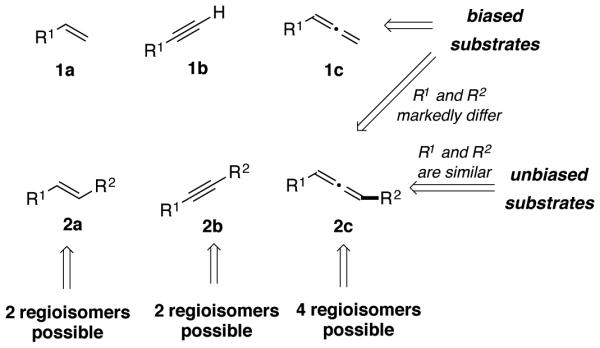
Biased and unbiased substrates in catalytic, regioselective operations.
The development of regioselective or regiodivergent processes with unbiased substrates such as disubstituted π-systems 2a-c, where the R1 and R2 groups possess similar steric and electronic properties, presents a different set of challenges (Scheme 2). In these cases, accessing any of the possible regioisomers selectively is typically quite challenging, since the contributing influence of the substrate is quite small, and any substrate-ligand interactions would likely be similar for the two regioisomeric pathways. The ability to develop regioselective or regiodivergent catalytic processes of π-components that possess only negligible steric or electronic biases are extremely challenging in nearly all contexts.
In recent years, our laboratory has developed nickel-catalyzed processes that allow the production of allylic alcohols through the reductive coupling of aldehydes and alkynes.13-15 With both biased and unbiased alkynes, highly selective, regiodivergent pathways have been developed. Numerous factors, including structures of the reductant and ligand, concentration, temperature, and rate of addition, all have an important influence on the regiochemical outcomes with both biased and unbiased classes of alkynes. Through a series of kinetic and computational investigations, considerable insight into the origin of regiocontrol in several of the optimized protocols has been developed. This review summarizes our insights to date on regiocontrol in this reaction class, with a focus on describing how mechanistic insights provided a rational and predictable path towards the development of highly effective synthetic procedures.
Mechanisms of Nickel-Catalyzed Aldehyde-Alkyne Reductive Couplings
A number of effective protocols have been developed for the reductive coupling of aldehydes and alkynes. These methods involve either temporary conversion of the alkyne to a nucleophilic alkenyl metal through processes such as hydroboration or hydrozirconation followed by aldehyde addition,16-17 or alternatively through metallacyclic intermediates derived from oxidative cyclization of a low-valent metal with a bound aldehyde and alkyne to produce a five-membered metallacyclic intermediate. The latter pathway has been developed for metals across the period table, including methods involving titanium, rhodium, iridium, and nickel. The control of regiochemistry has seen significant advances, particularly in directed processes from Jamison with nickel,18-20 Krische in iridium and rhodium-catalyzed processes,21-22 and Micalizio with titanium.23-34 Additionally, related methods from Krische developing transfer hydrogenation methods are especially attractive in that the alcohol oxidation state may be directly employed as the source of the electrophilic partner.25 The specific variant of aldehyde-alkyne reductive couplings that has been most extensively developed and studied in our laboratory involves silane reductants paired with phosphine or N-heterocyclic carbene (NHC) complexes of nickel(0), with NHC’s being the most effective in addressing challenging problems in regiocontrol.26-28 A brief perspective on the phosphine-promoted intramolecular addition studies from our laboratory will first be described, since this process was the focus of our initial evaluation of kinetics in this reaction class. More recent mechanistic insights in the NHC-promoted versions, which provide the highest degree of regiodivergency, will then be discussed.
The simplest framework for considering the metallacycle-based mechanism of aldehyde-alkyne reductive couplings involves coordination of the aldehyde and alkyne to Ni(0) to generate π-complex 3, followed by oxidative cyclization to metallacycle 4, σ-bond metathesis of 4 with the silane reductant to produce 5, and finally reductive elimination to form the C-H bond of product 6 (Scheme 3). The initial proposal of this mechanistic pathway29 was based on related metallacycle-forming processes involving nickel, and a dimeric nickel metallacycle directly obtained from an aldehyde-alkyne complex was subsequently reported by Ogoshi.30 Additionally, a number of theoretical studies have examined different combinations of metals, ligands, and reductants, with metallacycle formation uniformly being found as a key component of the operative pathway.31-35
Scheme 3.
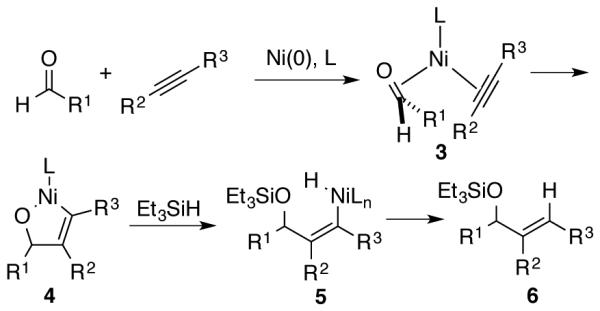
Metallacycle pathway for aldehyde-alkyne reductive couplings
The kinetics of this process was first studied on the reductive cyclization of ynal 7 using Et SiH as reductant and a catalyst derived from Ni(COD) and PCy (Scheme 4).36 In reactions conducted at −25 °C and monitored by in situ IR, the kinetics displayed a clear first-order dependence on both the ynal and the nickel catalyst, and a zero order dependence on silane. Most notably, plots of reaction progression across a four-fold increase in silane concentration nearly perfectly overlaid, illustrating the absence of a rate dependence on silane concentration. Further study illustrated that silane addition to the nickel catalyst was extremely slow in the presence of either aldehyde or alkyne, ruling out the possibility that an initial burst of Ni(0) oxidative addition to the silane followed by rate-determining addition of a nickel hydride intermediate to the aldehyde or alkyne could be operative. On this basis, the metallacycle-based mechanism, where oxidative cyclization is rate determining, is fully consistent with the observed kinetic profile.
Scheme 4.

Initial rates analysis of an intramolecular process.
The initial computational studies of Et3B-mediated intermolecular aldehyde-alkyne reductive couplings from Houk and Jamison found that metallacycle formation was rate-limiting,31-32 followed by a fast borane-mediated step involving cleavage of the Ni-O bond of the metallacycle. Houk and Montgomery also conducted a computational study of the intermolecular process involving NHC ligands and silane reductants, and similarly found that the rate-determining step was again metallacycle formation, followed by a fast σ-bond metathesis reaction of the silane.33 Most recently, post rate-limiting dimerization of a metallacycle derived from an intramolecular processes using silanes with a nickel(0)-phosphine catalyst was found to be competitive with the simple mononuclear pathway depicted above (Scheme 3), however, in this study as well, metallacycle formation was proposed to be the rate-determining step.35 In summary, across a broad range of combinations of substrate classes, including inter- and intramolecular processes, phosphine and NHC-derived catalyst systems, and silane and borane reductants, evidence from kinetics and computational studies of the most widely used protocols all pointed towards a metallacycle-based pathway involving a rate-determining oxidative cyclization to produce metallacycle 4 followed by a fast process involving metallacycle consumption by the reductant.
In our most recent efforts, we made a surprising empirical observation that the regiochemical outcome of intermolecular couplings varies according to silane structure if a bulky silane and bulky NHC ligand are simultaneously employed.37 The effect is not observed if only one of the two structural changes (ligand or silane) is made. The influence on regiochemistry is most pronounced at high temperature and when the reaction is conducted at high dilution or with low silane concentrations. Against the backdrop described above of extensive evidence that the silane involvement strictly occurs after the rate- and regiochemistry-determining step, this result was entirely unexpected. However, since this new protocol presented considerable preparative advantages in regiocontrol (vide infra), we conducted a mechanistic study to elucidate the observed effects.
In studying the reductive coupling of benzaldehyde with phenylpropyne, using (i-Pr)3SiH as reductant and the hindered NHC sIPr as ligand at rt, only a slight rate dependence on silane concentration was observed for the consumption of starting material. However, upon measuring the rate dependence for the two regioisomeric products separately, we observed that the rate of formation of the major regioisomer 8a was independent of silane concentration, whereas the rate of formation of the minor regioisomer 8b was strongly influenced by silane concentration (Scheme 5, panels a and b). By plotting the initials rates of the formation of the major and minor regioisomers as a ratio (Scheme 5, panel c), the markedly different dependence of the two regioisomeric pathways on silane concentration is easily visualized.
Scheme 5.

Initial rates analysis of a large NHC - large silane protocol.
The origin of this unusual affect appears to involve the reversible formation of the metallacycle 10b that leads to the minor regioisomer 8b and the irreversible formation of the metallacyle 10a that leads to the major regioisomer 8a (Scheme 6). The highly hindered nature of structure 10b, in particular the steric repulsions between the proximal phenyl group and the bulky ligand, slows the addition of the bulky silane, leading to the reversibility of its formation. The entropic penalty for the 10b to 11b conversion is maximized at elevated temperature, and the rate of the 10b to 11b conversion (and thus the rate of formation of 8b) is further slowed by maintaining a low concentration of silane. While changes between reversible and irreversible pathways are commonly invoked in discussions of regiodivergent processes, we are unaware of another case such as this where the kinetic profiles leading to two regioisomeric products in a single reaction are shown to differ to this clear extent.
Scheme 6.
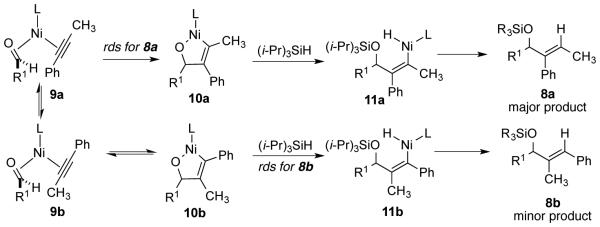
Mechanism of the large NHC - large silane protocol
Mechanistic Insights Inform Regiodivergent Strategies in Nickel-Catalyzed Aldehyde-Alkyne Couplings
The above mechanistic studies illustrate a number of opportunities for effecting regiocontrol and regiodivergency in aldehyde-alkyne reductive couplings. In considering the generality of regiocontrol strategies, it is instructive to consider not only the empirical observations that led to the development of effective strategies, but also the underlying mechanistic basis for why the strategies are effective. Such mechanistic insights can lead to rapid and rational optimization of reaction conditions when a new combination of substrates is encountered. While several scenarios of reaction kinetics were documented in the studies outlined above, the underlying goal was to be able to use these insights in the development of highly selective, regiodivergent procedures for the formation of either product 12a or 12b across a range of substrates (Scheme 7).
Scheme 7.
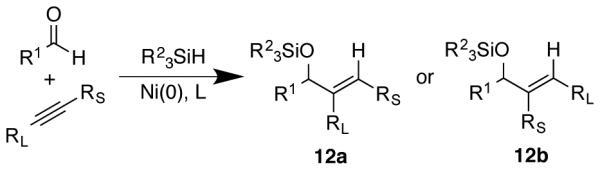
Desired regiodivergent outcome in aldehyde-alkyne couplings.
As the above kinetic studies demonstrated, most of the above-mentioned protocols involve the irreversible formation of metallacycles 14a and 14b.28,33 These strategies, summarized in the upper portion of Scheme 8, involve metallacycle formation as the rate- and regiochemistry-determining steps, where regioselectivity is determined by the relative rates of the 13a to 14a and the 13b to 14b conversions. The identity and concentration of the silane within this manifold is of no consequence, since the σ-bond metathesis steps involving silane (i.e. the formation of 15a and 15b) occur after the rate- and regiochemistry-determining steps. This manifold can be generally be favored by using unhindered silanes. With small NHC ligands, even bulky silanes such as (i-Pr)3SiH undergo fast additions, rendering metallacycle formation irreversible.
Scheme 8.

Implications of Observed Mechanisms in Regiocontrol Strategies
In contrast, a very specialized protocol can lead to the reversible formation of the more hindered metallacycle 14b, while the formation of metallacycle 14a under these conditions is formed irreversibly (at least in the non-syringe drive protocols where the kinetics studies were conducted).37 This strategy, summarized in the lower portion of Scheme 8, is governed by the relative rates of the formation of metallacycle 14a and the formation of intermediate 15b produced by σ-bond metathesis reaction of 14b with the silane. The change in silane order for the formation of the major and minor regioisomers noted above (Scheme 5) provides evidence for this conclusion. The more hindered metal-carbon bond of metallacycle 14b (nickel proximal to RL) compared with the less hindered environment of metallacycle 14a (nickel proximal to RS) is the origin of this change in kinetic description. The protocol that favors this outcome requires bulky silanes and bulky NHC ligands and is favored by low concentration of silane and elevated temperature. The temperature influence likely derives from the significant entropic penalty of the bimolecular addition required in the 14b to 15b conversion compared with unimolecular conversion of 13a to 14a. Notably, this protocol is only useful in accessing the more hindered regioisomer 12a.
Small Ligand Protocol
The identification of small ligands that are effective in accessing regioisomer 12b was conducted using 2-hexyne as a prototypical unbiased alkyne.28 With more biased alkynes such as terminal alkynes, aromatic alkynes, and conjugated enynes, the alkyne electronic and steric biases naturally favor regioisomer 12b, so simple ligands routinely employed such as IMes38-40 are entirely satisfactory for achieving high regioselectivity in these cases (Table 1, entries 1-3). However, with 2-hexyne, more specialized ligands such as ITol and IPr-Bac were found to be essential for the regioselective production of isomer 12b. For example, in comparing the additions of 2-hexyne, regioselectivities with IMes (16) or sIMes (17) were a modest 67:33 and 61:39 ratio, whereas ITol (18) and IPr-BAC (19) achieved a synthetically useful regioisomeric ratio of 87:13 to 88:12 (Table 1, entries 4-7) respectively. Of these ligands, IPr-BAC41 generally afforded the highest yield favoring the production of desired product 12b. While regioselectivities are unaffected by silane structure, with small ligand classes, bulky silanes effectively diminish the amount of aldehyde or alkyne hydrosilylation that can sometimes become competitive.
Table 1.
Regioselectivity of the small ligand protocol.
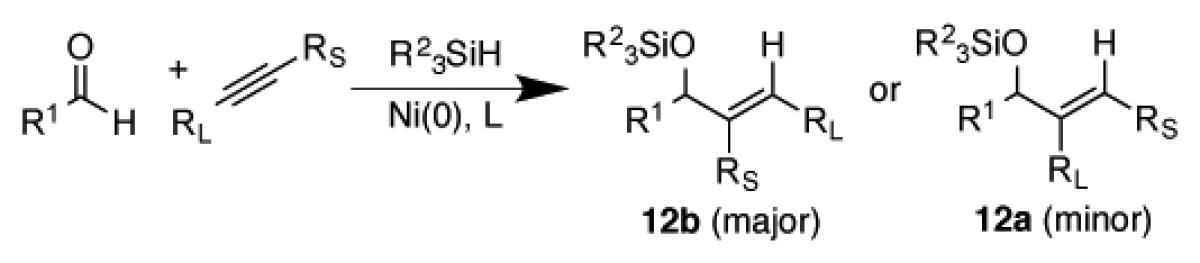
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | R1 | RS | RL | R23SiH | L | % yield | (12b:12a) |
| 1 | n-Hex | H | i-Pr | (i-Pr)3SiH | 16 | 74 | >98:2 |
| 2 | Ph | Me | Ph | (i-Pr)3SiH | 16 | 74 | >98:2 |
| 3 | n-Hex | Me | c-Hexenyl | (i-Pr)3SiH | 16 | 99 | 97:3 |
| 4 | n-Hex | Me | n-Pr | (i-Pr)3SiH | 16 | 83 | 67:33 |
| 5 | n-Hex | Me | n-Pr | (i-Pr)3SiH | 17 | 73 | 61:39 |
| 6 | n-Hex | Me | n-Pr | (i-Pr)3SiH | 18 | 18 | 87:13 |
| 7 | n-Hex | Me | n-Pr | (t-Bu)2SiH2 | 19 | 78 | 88:12 |
Computational studies provided considerable insight into the origin of the regioselectivities with small ligands such as ITol (18) and IPr-BAC (19).33 These unhindered ligand were examined in the coupling of 2-hexyne with propionaldehyde to evaluate the features that impact regioselectivity in the rate-determining metallacycle formation (Scheme 9). In this case, few steric interactions between the ligand and alkyne substituents are observed for either of the regioisomeric pathways, given the small ligand size and flexibility of the N-aryl orientation. However, strong steric repulsions experienced in the forming C-C bond in the proximal TS leading to the generation of the minor regioisomer 12a ultimately favor the formation of the major regioisomer 20. The difference in steric repulsions with the similar-sized Me and n-Pr groups is sufficiently large to afford the high level of regioselectivity, due to the relatively short forming C-C bond distances (c.a. 1.7 Å) in the oxidative cyclization transition states. In summary, as these studies illustrate, IMes serves an effective ligand in cases where the substrate biases favor the formation of regioisomer 12b, but with more unbiased classes of alkynes, the IPr-BAC ligand effectively differentiates similar groups (such as Me vs n-Pr) and allows highly regioselective production of regioisomer 12b through subtle substrate-ligand interactions.
Scheme 9. Oxidative Cyclization Transition State Energies for the Small Ligand Pathway with ITol as ligand.a.
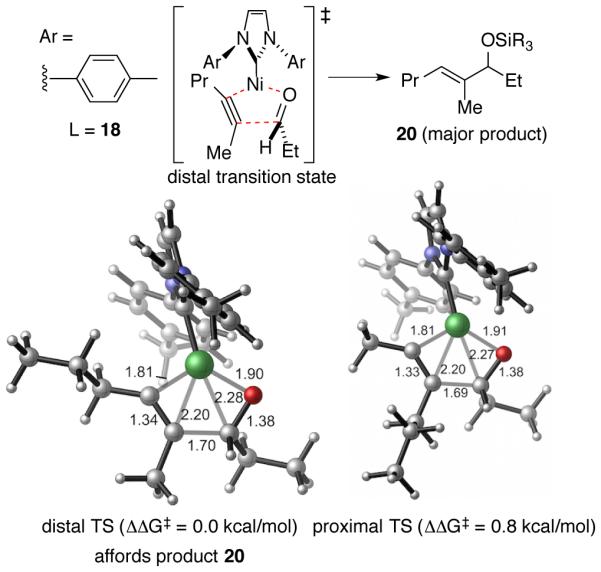
a Calculations were performed at the M06/SDD−6-311+G(d,p)//B3LYP/LANL2DZ−6-31G(d) level of theory. Here distal refers to the TS structure in which the bulkier alkyne substituent (n-Pr) is distal to the forming C-C bond.
Large Ligand / Small Silane Protocol
With the above advances in accessing the regioisomer that involves formation of the least-crowded C-C bond, the sensitivity of the regioselectivity to small changes in NHC structure suggested that the opposite regioisomeric outcome could potentially be accessed. Towards this objective, an exploration of bulky ligand structures was conducted in the benchmark case of additions of phenylpropyne to heptaldehyde.28 In progressing from IMes to IPr to sIPr to DP-IPr, the regiochemistry steadily improved to 94:6, now favoring the more hindered regioisomer 12a (Table 2, entries 1-4). For the relatively unbiased alkyne 2-hexyne, the combination of IPr-BAC (19) as a small ligand and either sIPr (22) or DP-IPr (23) as a large ligand thus provides access to a highly regiodivergent outcome, with regioselectivities from 12:88 with IPr-BAC favoring 12b to 94:6 with DP-IPr favoring 12a. Avoiding undesired aldehyde or alkyne hydrosilylation was the primary motivation for the use of bulkier silanes in these initial studies. However, additional implications of the silane structural change are described in the development of a large ligand – large silane discussed in the final section of this review.
Table 2.
Initial explorations with large ligand.
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | R1 | RL | RS | R23SiH | L | % yield | (12a:12b) |
| 1 | n-Hex | n-Pr | Me | (i-Pr)3SiH | 16 | 83 | 67:33 |
| 2 | n-Hex | n-Pr | Me | (i-Pr)3SiH | 21 | 84 | 80:20 |
| 3 | n-Hex | n-Pr | Me | (i-Pr)3SiH | 22 | 85 | 93:7 |
| 4 | n-Hex | n-Pr | Me | Et3SiH | 23 | 94 | 94:6 |
| 5 | n-Hex | i-Pr | H | (i-Pr)3SiH | 22 | 40 | 41:59a |
| 6 | n-Hex | i-Pr | H | Et3SiH | 23 | 76 | 95:5 |
| 7 | Ph | Ph | Me | Et3SiH | 22 | 65 | 58:42 |
| 8 | Ph | Ph | Me | Et3SiH | 23 | 96 | 69:31a |
Data from H. A. Malik thesis, University of Michigan
These successes then raised the possibility of accessing regiodivergent outcomes with highly biased alkynes such as terminal alkynes and aromatic alkynes. In these cases, sIPr and DP-IPr were the most effective in reversing the regiochemistry outcome that is naturally favored by substrate biases. In the case of a terminal alkyne (Table 2, entries 5-6), the use of sIPr was ineffective, leading to a 41:59 ratio favoring the same regioisomer 12b favored by IMes (16). However, the use of DP-IPr, which possesses the same N-aryl substituents as sIPr but also incorporates a C2-disubstituted ligand backbone, resulting in a dramatic improvement, providing the more hindered regioisomer 12a with 95:5 regioselectivity. Bulkier silanes generally diminished the observation of aldehyde or alkyne hydrosilylation minor byproducts; however, the steric demand of ligand 23 typically required the use of a small silane such as Et3SiH. With phenyl propyne, DP-IPr was slightly more effective than sIPr using Et3SiH, but even DP-IPr resulted in only a modest regioselectivity of 69:31 (Table 2, entries 7-8).
The unique characteristics of DP-IPr paired with Et3SiH in reversing the regiochemistry of terminal alkyne couplings were explored in a challenging macrocyclization process in the course of developing a total synthesis of the macrolide 10-deoxymethynolide.42 Utilizing the inherent substrate biases of the terminal alkyne of 24, the use of IMes ligand proved satisfactory in an endocyclization process to access product 25 in 58% yield as a 4:1 mixture of diastereomers with only endocyclization products being observed (Scheme 10). Alternatively, the unique properties of DP-IPr with terminal alkynes are illustrated by the highly selective exocyclization of the same substrate to afford 26 in 59% yield as a single regio- and stereoisomer. Recent efforts have further developed this promising macrocyclization method across a broader range of substrates and with more readily available ligand motifs.43
Scheme 10.

Regiodivergent reductive macrocyclizations.
Computational studies also provided considerable insight into the origin of the regioselectivities using large ligands such as IPr (21) (Scheme 11).33 Using this ligand, steric repulsions between the alkyne substituent and the ligand become significant during the formation of the metallacycle, and these effects override the aldehyde-alkyne steric interactions that govern the regiochemical outcome with smaller ligands. Additionally, 2D contour maps of the ligand van der Waals surface in these metal complexes revealed the precise positioning where ligand sterics can most effectively influence regiocontrol through interaction with the alkyne. The 2- and 6-positions of N-aryl NHC motifs were found to be especially effective in exerting steric control, explaining the special role of this NHC class compared with bulky but ineffective ligands such as N-adamantyl derivatives. In comparing ligands 21 and 22, computational studies found that the shorter Ni-C(carbene) bond distance (and the resulting greater effective steric bulk at the nickel center) with the saturated ligand 22 compared to the unsaturated ligand 21 was the origin of the improved large-ligand selectivity from ligand 22. In summary, as the above examples demonstrate, regiodivergent outcomes are thus possible for a number of unbiased (i.e. 2-hexyne) and strongly biased (i.e. terminal alkynes, aromatic alkynes, and conjugated enynes) using the combination of the complementary small ligand-small silane and large ligand-small silane pathways.
Scheme 11. Oxidative Cyclization Transition State Energies for Large Ligand / Small Silane Pathway with IPr as Ligand.a.
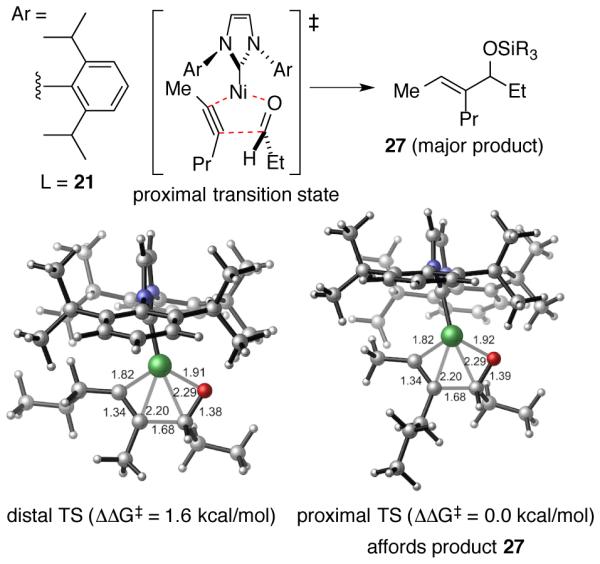
a Calculations were performed at the M06/SDD−6-311+G(d,p)//B3LYP/LANL2DZ−6-31G(d) level of theory. Here proximal refers to the TS structure in which the bulkier alkyne substituent (n-Pr) is proximal to the forming C-C bond.
Large Ligand / Large Silane Protocol
In our most recent work, an unusual influence of the silane structure on regiochemistry was noted when large ligands such as sIPr and large silanes such as (i-Pr)3SiH were simultaneously employed.37 An evaluation of kinetics suggested that the mechanism depicted earlier (Scheme 6), involving the reversibility of the metallacycle formation in the minor regioisomer pathway, was responsible for the effect. Several notable features were observed for this modified protocol, namely much improved regioselectivity for the production of regioisomer 12a with some substrate combinations compared with the large ligand-small silane protocol described above. For example, improved regiochemistry reversals could be accessed for a number of alkynes with a protocol optimized for internal alkynes, using (i-Pr)3SiH at 50 °C. In reversing the normal bias of phenyl propyne, the more hindered regioisomer 12a could be accessed in exceptional (>98:2) regioselectivities using either aromatic or aliphatic aldehydes (Table 3, entries 1-2). Very high regioselectivities favoring 12a could also be accessed with unbiased alkynes. For example, i-Bu and Et groups or i-Pr and Me substituents could be easily differentiated (Table 3, entries 3-4), whereas the very challenging case of 3-heptyne (Et vs Pr) proceeded to give a 68:32 mixture of regioisomers, thus defining the limits of the method (Table 3, entry 5). With a conjugated enyne, excellent regioselectivities were observed for the more hindered isomer 12a using sIPr even at rt to provide the desired isomer in 91:9 regioselectivity (Table 3, entry 6), thus reversing the regioselectivity normally accessed by this biased alkyne class (Table 1, entry 3). The elevated temperature protocol led to lower yields with terminal alkynes, and a procedure specifically used for terminal alkynes (rt, higher dilution, and (t-Bu)2MeSiH as reductant) provided good yields of 12a with excellent regiocontrol (Table 3, entries 7-8).
Table 3.
Regioselectivity of the large ligand / large silane protocol.
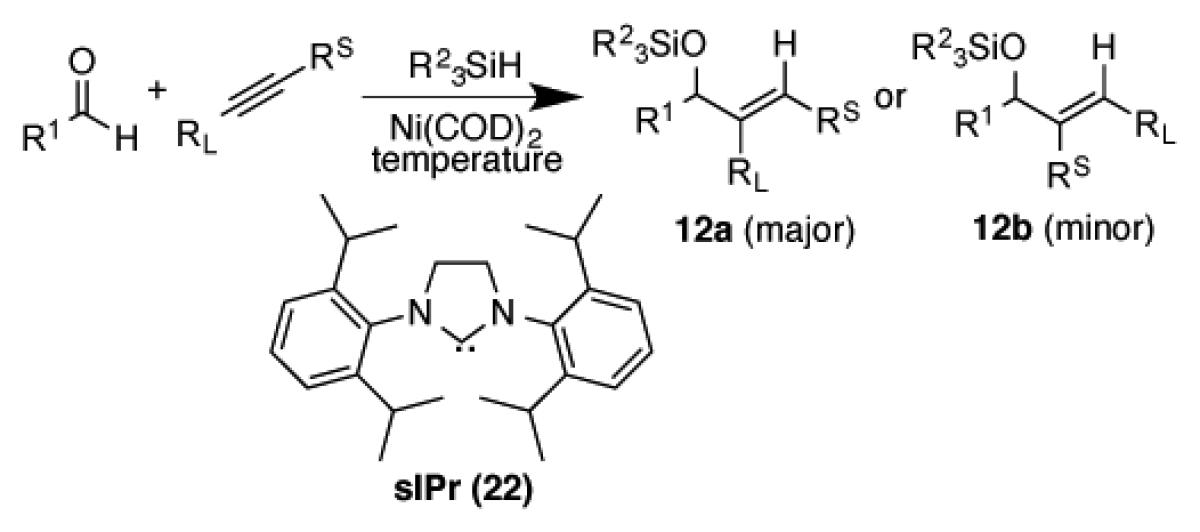
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | R1 | RL | RS | R23SiH | Temp (°C) | % yield | 12a:12b |
| 1 | Ph | Ph | Me | (i-Pr)3SiH | 25 | 82 | >98:2 |
| 2 | n-Hept | Ph | Me | (i-Pr)3SiH | 50 | 77 | >98:2 |
| 3 | n-Hept | i-Bu | Et | (i-Pr)3SiH | 50 | 66 | 93:7 |
| 4 | Ph | i-Pr | Me | (i-Pr)3SiH | 50 | 78 | >98:2 |
| 5 | Ph | n-Pr | Et | (i-Pr)3SiH | 50 | 56 | 68:32 |
| 6 | n-Hex | c-Hexenyl | Me | (i-Pr)3SiH | 25 | 77 | 91:9 |
| 7 | Ph | i-Pr | H | (t-Bu)2MeSiH | 25 | 61 | >98:2 |
| 8 | Ph | n-Hex | H | (t-Bu)2MeSiH | 25 | 69 | 95:5 |
Conclusions
In summary, a series of complementary protocols have been developed that allows highly selective access to either regioisomer in nickel-catalyzed reductive couplings. The strategy does not require directing groups, but rather relies upon precise control of the operative kinetic behavior of couplings and the development of ligand-substrate interactions to govern the regiochemical outcome. Regiodivergent outcomes are possible for unbiased internal alkynes as well as highly biased alkyne classes such as aromatic and terminal alkynes and conjugated enynes. By careful selection of ligand and reductant, modifications in the rate-determining step of the process can be engineered as a key component in the development of regiodivergent outcomes. The observation of different kinetic descriptions for the formation of two regioisomers in a single reaction is an underappreciated but highly effective strategy in accessing regiodivergent outcomes in catalytic processes.
Acknowledgements
Professor Ken Houk of UCLA is thanked for many helpful discussions during the course of this work. This work was funded by NIH grant (GM-57014, J.M., which supports the design of NHC ligands for regioselective transformations) and NSF grants (CHE-1265491, J.M., which supports the development of sustainable methods in catalysis, and CHE-1361104, K. N. Houk). G.J.S. acknowledges training grant support from NIH grant GM-007767. R.D.B. acknowledges a U.S. Department of Education GAANN Fellowship; A.R.S. acknowledges receipt of a Rackham Merit Fellowship and a Fred W. Lyons Fellowship.
Biographies
Evan P. Jackson, born in Portland, OR, obtained a B.A. degree from Willamette University in 2010 working with Sarah Kirk. He is currently completing his Ph.D. at the University of Michigan under the direction of John Montgomery studying nickel-catalyzed reductive coupling reactions.
Hasnain A. Malik obtained his B.S. degree from Lehigh University in 2005, where he conducted his undergraduate research with Professor Ned D. Heindel. In 2010 he earned his Ph.D. from the University of Michigan, Ann Arbor under the direction of Professor John Montgomery, working on strategies for regio- and enantiocontrol in Ni-catalyzed reductive couplings. Following postdoctoral studies with Professor Mark Lautens at the University of Toronto, he moved to the Department of Global Discovery Chemistry at the Novartis Institutes for BioMedical Research, Inc. (NIBRI) in Cambridge, Massachusetts where he is currently a Research Investigator.
Grant J. Sormunen, born in Duluth, MN, obtained a B.S. degree from the University of Wisconsin – Eau Claire in 2005 working with David Lewis. He received his Ph.D. degree at the University of Michigan in 2010 working with John Montgomery. In 2011 he became a Research Scientist at Promiliad Biopharma Inc.
Ryan D. Baxter obtained a B.Sc. degree from the University of Wisconsin Madison in 2005 working with Samuel H. Gellman, and received his M.Sc. (2007) and Ph.D. (2010) degrees at the University of Michigan working with John Montgomery. He then conducted postdoctoral research at the Scripps Research Institute as a National Institute of Health Postdoctoral Fellow with Donna Blackmond and Phil Baran before joining the faculty at UC Merced in 2014, where he is currently an Assistant Professor of Chemistry and Chemical Biology.
Peng Liu obtained his B.S. degree from the Peking University in 2003 and M.S. degree from the University of Guelph in 2006. He received his Ph.D. degree in 2010 and then performed postdoctoral studies with K. N. Houk at the University of California, Los Angeles. He joined the faculty at the University of Pittsburgh as an Assistant Professor of Chemistry in 2014.
Hengbin Wang, born in China, obtained a B.S. degree from Nankai University in 2001, and he received his Ph.D. degree at Emory University in 2012 working with Huw M.L. Daives. He then joined the Montgomery group at the University of Michigan as a postdoctoral researcher.
Abdur-Rafay Shareef, born in Richland, WA, obtained a B.S. and M.S. degree from Georgia State University working with David Boykin, and he received his Ph.D. degree at the University of Michigan working with David Sherman and John Montgomery. He is currently a chemist at the US Food and Drug Administration, where he develops Surface-enhanced Raman spectroscopy (SERS) and mass spectroscopy methods.
John Montgomery, born in Concord, NC, obtained an A.B. degree from the University of North Carolina in 1987 working with Maurice Brookhart and Joe Templeton, and he received his Ph.D. degree at Colorado State University in 1991 working with Lou Hegedus. He then conducted postdoctoral research at the University of California at Irvine as an American Cancer Society Postdoctoral Fellow with Larry Overman before joining the faculty at Wayne State University at 1993. He joined the faculty at the University of Michigan in 2005, where he is currently the Margaret and Herman Sokol Professor in Medicinal or Synthetic Chemistry.
References
- 1.Mahatthananchai J, Dumas AM, Bode JW. Catalytic Selective Synthesis. Angew. Chem. Int. Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- 2.Beller M, Seayad J, Tillack A, Jiao H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004;43:3368–3398. doi: 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]
- 3.Chopade PR, Louie J. 2+2+2 cycloaddition reactions catalyzed by transition metal complexes. Adv. Synth, Cat. 2006;348:2307–2327. [Google Scholar]
- 4.Saito S, Yamamoto Y. Recent advances in the transition-metal-catalyzed regioselective approaches to polysubstituted benzene derivatives. Chem. Rev. 2000;100:2901–2915. doi: 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]
- 5.Hoveyda AH, Evans DA, Fu GC. Substrate-Directable Chemical Reactions. Chem. Rev. 1993;93:1307–1370. [Google Scholar]
- 6.Ohmura T, Oshima K, Taniguchi H, Suginome M. Switch of Regioselectivity in Palladium-Catalyzed Silaboration of Terminal Alkynes by Ligand-Dependent Control of Reductive Elimination. J. Am. Chem. Soc. 2010;132:12194–12196. doi: 10.1021/ja105096r. [DOI] [PubMed] [Google Scholar]
- 7.Gao F, Hoveyda AH. alpha-Selective Ni-Catalyzed Hydroalumination of Aryl- and Alkyl-Substituted Terminal Alkynes: Practical Syntheses of Internal Vinyl Aluminums, Halides, or Boronates. J. Am. Chem. Soc. 2010;132:10961–10963. doi: 10.1021/ja104896b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu JY, Moreau B, Ritter T. Iron-Catalyzed 1,4-Hydroboration of 1,3-Dienes. J. Am. Chem. Soc. 2009;131:12915–12917. doi: 10.1021/ja9048493. [DOI] [PubMed] [Google Scholar]
- 9.Michel BW, McCombs JR, Winkler A, Sigman MS. Catalyst-Controlled Wacker-Type Oxidation of Protected Allylic Amines. Angew. Chem.-Int. Edit. 2010;49:7312–7315. doi: 10.1002/anie.201004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller ZD, Li W, Belderrain TR, Montgomery J. Regioselective Allene Hydrosilylation Catalyzed by N-Heterocyclic Carbene Complexes of Nickel and Palladium. J. Am. Chem. Soc. 2013;135:15282–15285. doi: 10.1021/ja407749w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hein JE, Fokin VV. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao HT, Lin ZY, Jia GC, Fokin VV. Ruthenium-catalyzed azide-alkyne cycloaddition: Scope and mechanism. J. Am. Chem. Soc. 2008;130:8923–8930. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 13.Montgomery J. Organonickel Chemistry. In: Lipshutz BH, editor. Organometallics in Synthesis. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2013. pp. 319–428. [Google Scholar]
- 14.Montgomery J. Nickel-catalyzed reductive cyclizations and couplings. Angew. Chem. Int. Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. [DOI] [PubMed] [Google Scholar]
- 15.Moslin RM, Miller-Moslin K, Jamison TF. Regioselectivity and enantioselectivity in nickel-catalysed reductive coupling reactions of alkynes. Chem. Commun. 2007:4441–4449. doi: 10.1039/b707737h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oppolzer W, Radinov RN. Catalytic Asymmetric Synthesis of Secondary (E)-Allyl Alcohols from Acetylenes and Aldehydes via (1-Alkenylzinc) Intermediates. Helv. Chim. Acta. 1992;75:170–173. [Google Scholar]
- 17.Wipf P, Xu WJ. Preparation of Allylic Alcohols by Alkene Transfer from Zirconium to Zinc. Tetrahedron Lett. 1994;35:5197–5200. [Google Scholar]
- 18.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. Alkene-directed, nickel-catalyzed alkyne coupling reactions. J. Am. Chem. Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735. [DOI] [PubMed] [Google Scholar]
- 19.Miller KM, Jamison TF. Ligand-switchable directing effects of tethered alkenes in nickel-catalyzed additions to alkynes. J. Am. Chem. Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 20.Moslin RA, Miller KM, Jamison TF. Directing effects of tethered alkenes in nickelcatalyzed coupling reactions of 1,6-enynes and aldehydes. Tetrahedron. 2006;62:7598–7610. [Google Scholar]
- 21.Jang HY, Krische MJ. Catalytic C-C bond formation via capture of hydrogenation intermediates. Accounts Chem. Res. 2004;37:653–661. doi: 10.1021/ar020108e. [DOI] [PubMed] [Google Scholar]
- 22.Ngai MY, Kong JR, Krische MJ. Hydrogen-mediated C-C bond formation: A broad new concept in catalytic C-C coupling. J. Org. Chem. 2007;72:1063–1072. doi: 10.1021/jo061895m. [DOI] [PubMed] [Google Scholar]
- 23.Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Regioselective Reductive Cross-Coupling Reactions of Unsymmetrical Alkynes. Eur. J. Org. Chem. 2010:391–409. doi: 10.1002/ejoc.200901094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahadoor AB, Flyer A, Micalizio GC. A pentenyl dianion-based strategy for convergent synthesis of ene-1,5-diols. J. Am. Chem. Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. [DOI] [PubMed] [Google Scholar]
- 25.Bower JF, Kim IS, Patman RL, Krische MJ. Catalytic Carbonyl Addition through Transfer Hydrogenation: A Departure from Preformed Organometallic Reagents. Angew. Chem. Int. Ed. 2009;48:34–46. doi: 10.1002/anie.200802938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang XQ, Montgomery J. Nickel-catalyzed preparation of bicyclic heterocycles: Total synthesis of (+)-allopumiliotoxin 267A, (+)-allopumiliotoxin 339A, and (+)-allopumiliotoxin 339B. J. Am. Chem. Soc. 2000;122:6950–6954. [Google Scholar]
- 27.Mahandru GM, Liu G, Montgomery J. Ligand-dependent scope and divergent mechanistic behavior in nickel-catalyzed reductive couplings of aldehydes and alkynes. J. Am. Chem. Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]
- 28.Malik HA, Sormunen GJ, Montgomery J. A General Strategy for Regiocontrol in Nickel-Catalyzed Reductive Couplings of Aldehydes and Alkynes. J. Am. Chem. Soc. 2010;132:6304–6305. doi: 10.1021/ja102262v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oblinger E, Montgomery J. A new stereoselective method for the preparation of allylic alcohols. J. Am. Chem. Soc. 1997;119:9065–9066. [Google Scholar]
- 30.Ogoshi S, Arai T, Ohashi M, Kurosawa H. Nickeladihydrofuran. Key intermediate for nickel-catalyzed reaction of alkyne and aldehyde. Chem. Commun. 2008:1347–1349. doi: 10.1039/b717261c. [DOI] [PubMed] [Google Scholar]
- 31.McCarren PR, Liu P, Cheong PHY, Jamison TF, Houk KN. Mechanism and Transition-State Structures for Nickel-Catalyzed Reductive Alkyne-Aldehyde Coupling Reactions. J. Am. Chem. Soc. 2009;131:6654–6655. doi: 10.1021/ja900701g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu P, McCarren P, Cheong PHY, Jamison TF, Houk KN. Origins of Regioselectivity and Alkene-Directing Effects in Nickel-Catalyzed Reductive Couplings of Alkynes and Aldehydes. J. Am. Chem. Soc. 2010;132:2050–2057. doi: 10.1021/ja909562y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu P, Montgomery J, Houk KN. Ligand Steric Contours To Understand the Effects of N-Heterocyclic Carbene Ligands on the Reversal of Regioselectivity in Ni-Catalyzed Reductive Couplings of Alkynes and Aldehydes. J. Am. Chem. Soc. 2011;133:6956–6959. doi: 10.1021/ja202007s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu P, Krische MJ, Houk KN. Mechanism and Origins of Regio- and Enantioselectivities in Rh-I-Catalyzed Hydrogenative Couplings of 1,3-Diynes and Activated Carbonyl Partners: Intervention of a Cumulene Intermediate. Chem. Eur. J. 2011;17:4021–4029. doi: 10.1002/chem.201002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haynes MT, II, Liu P, Baxter RD, Nett AJ, Houk KN, Montgomery J. Dimer Involvement and Origin of Crossover in Nickel-Catalyzed Aldehyde-Alkyne Reductive Couplings. J. Am. Chem. Soc. 2014;136:17495–17504. doi: 10.1021/ja508909u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baxter RD, Montgomery J. Mechanistic Study of Nickel-Catalyzed Ynal Reductive Cyclizations through Kinetic Analysis. J. Am. Chem. Soc. 2011;133:5728–5731. doi: 10.1021/ja200867d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson EP, Montgomery J. Regiocontrol in Catalytic Reductive Couplings through Alterations of Silane Rate Dependence. J. Am. Chem. Soc. 2015;137:958–963. doi: 10.1021/ja511778a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arduengo AJ, Gamper SF, Calabrese JC, Davidson F. Low-Valent Coordinate Carbene Complexes of Nickel(0) and Platinum(0) J. Am. Chem. Soc. 1994;116:4391–4394. [Google Scholar]
- 39.Herrmann WA. N-heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem.-Int. Edit. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 40.Haynes MT, Jackson EP, Montgomery J. Nickel Complexes of N-Heterocyclic Carbenes. In: Nolan SP, editor. N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis. Wiley-VCH; 2014. pp. 371–396. [Google Scholar]
- 41.Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Cyclopropenylidenes: From interstellar space to an isolated derivative in the laboratory. Science. 2006;312:722–724. doi: 10.1126/science.1126675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shareef AR, Sherman DH, Montgomery J. Nickel-catalyzed regiodivergent approach to macrolide motifs. Chem. Sci. 2012;3:892–895. doi: 10.1039/C2SC00866A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Negretti S, Knauff AR, Montgomery J. Exo-Selective Reductive Macrocyclization of Ynals. Org. Lett. 2015;17:1493–1496. doi: 10.1021/acs.orglett.5b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]