

Abstract
Neurodegenerative disorders are a heterogeneous group of chronic progressive diseases and have pathological mechanisms in common. A certain causative gene identified for a particular disease may be found to play roles in more than one neurodegenerative disorder. We analyzed the GGGGCC repeat expansions of C9orf72 gene in patients with SCA3/MJD from mainland China to determine whether the C9orf72 gene plays a role in the pathogenesis of SCA3/MJD. In our study, there were no pathogenic repeats (>30 repeats) detected in either the patients or controls. SCA3/MJD patients with intermediate/intermediate or short/intermediate genotype (short: <7 repeats; intermediate: 7-30 repeats) of the GGGGCC repeats had an earlier onset compared with those with short/short genotype. The presence of the intermediate allele of the GGGGCC repeats in the patients decreased the age at onset by nearly 3 years. Our study firstly demonstrate that the development of SCA3/MJD may involve some physiological functions of the C9orf72 gene and provide new evidence to the hypothesis that a specific mutation identified in one of the neurodegenerative disorders may be a modulator in this class of diseases.
Introductions
Many neurodegenerative disorders, including the following, arise from abnormal protein interactions in the central nervous system: Parkinson’s disease (PD); Alzheimer’s disease (AD); frontotemporal lobar degeneration (FTLD); amyotrophic lateral sclerosis (ALS); and the polyglutamine (polyQ) diseases, including spinocerebellar ataxias (SCA) types 1, 2, 3, 6, 7, and 17, dentatorubral-pallidoluysian atrophy (DRPLA), Huntington’s disease (HD), spinal bulbar muscular atrophy (SBMA), and the recently identified Huntington disease-like 2 (HDL2). These disorders are characterized by adult onset, a chronic progressive course, distinct clinical phenotypes, specific cellular abnormalities, and eventually fatal outcomes. Mutations in the same gene or even the same mutation may perform variable phenotypes among individuals [1]. However, the clinical syndromes from the varied mutations may be strikingly similar to each other [2, 3]. Previous study [4–6] suggested that the CAG repeats mutation in SCA2 or SCA3/Machado-Joseph disease (MJD) may be associated with Parkinsonism. A recent study [7] showed a common molecular mechanism between SCA3/MJD and ALS due to the similar ubiquitination and degradation of ataxia-3 and SOD. A subsequent investigation [8] reported that a patient with a family history of ALS presented symptoms of cerebellar ataxia, which indicates some unclear but subsistent association between SCA and ALS/FTLD. This suggests that there are biological relationships among these genes.
SCA3/MJD, which is the most common dominantly inherited ataxia in China and other countries [9–11], is caused by an unstable CAG trinucleotide repeat expansions in the ATXN3 gene. SCA3/MJD patients have a clinically heterogeneous presentation with an extreme range of age at onset of 4 years to 70 years [12]. The clinical variability of SCA3/MJD is only partially explained by the CAG repeats in the expanded ATXN3 alleles, which indicates that the residual variance is likely explained by other unknown factors. GGGGCC repeat expansions, which locates in the first intron of the C9orf72 gene, was recently identified as a major cause of ALS and FTLD [13]. The pathogenic mechanism of hexanucleotide repeat expansions include interfering with the normal expression of the encoded protein and the loss of protein function through the generation of abnormal toxic RNA foci and the subsequent disruption of normal cellular pathways [14].
Motivated by the observation that different neurodegenerative disorders share some of the same clinical and pathological features, we hypothesized that the genes involved in ALS/FTLD might also play roles in other neurodegenerative disorders. In this study, we investigated a large cohort of SCA3/MJD cases under the hypothesis that the GGGGCC repeat expansions of the C9orf72 gene may also contribute to the pathogenesis of SCA3/MJD.
Materials and Methods
Subjects
A total of 127 SCA3/MJD patients were screened for GGGGCC repeats mutations of the C9orf72 gene. A total of 314 unrelated, healthy individuals were selected as controls. Both of the groups were enrolled from Xiangya Hospital of Central South University from January 1995 to August 2012. The clinical diagnoses for all SCA3/MJD patients were determined according to the Harding criterion and were confirmed by molecular diagnosis [15]. The age at onset was defined as the age at which the patient, or a close person, noticed the first symptoms (usually gait unbalance). Patients with long-term duration were asked for the age at onset of the mentioned symptoms. To obtain a more accurate age at onset, additional information from previous records and the scores of the International Cooperative Ataxia Rating Scale (ICARS) from at least two experienced neurologists were also taken into account. None of the subjects had cardiac disease, tumors, or other neurological disease. Written informed consent was obtained from all of the individuals. The study was approved by the Expert Committee of Xiangya Hospital of Central South University in China (equivalent to an Institutional Review Board).
Genetics analysis
Peripheral venous blood samples were drawn from the SCA3/MJD patients and controls and were processed within 4–6 hours to extract the genomic DNA, which was stored at –20°C until further analysis. The genomic DNA was purified from whole blood leukocytes using a DNA extraction kit (QIAGEN, Germany). Recombinant DNA technology, including T-vector cloning, followed by direct DNA sequencing was used to evaluate the size of the CAG repeats in the ATXN3 gene. We screened the presence of the GGGGCC hexanucleotide expansion of C9orf72 using a 2-step polymerase chain reaction protocol as previously reported [16]. First, we used a previously reported repeat-primed polymerase chain reaction assay to detect the size of the larger expanded alleles. Then, we performed a classical FAM-fluorescent labeled PCR assay to detect the accurate genotype of the non-pathogenic mutation carriers. The analysis of the PCR fragment length was performed using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA) and visualized with the GeneMapper software (version 3.2, Applied Biosystems, Foster City, CA, USA). The pathogenic threshold of the GGGGCC repeats was defined as more than 30 [13, 17]. The positive control, which was used to verify the trustworthiness and reliability of our experiment, included a DNA sample from an ALS patient (>30 repeat expansions) recruited from Xiangya Hospital [18].
Statistics analysis
Univariate linear regression analyses were performed to determine the effect on age at onset of the expanded ATXN3 allele (linear and quadratic effect) in SCA3/MJD patients. The logarithmically (decimal) transformed age at onset was treated as dependent variables. The differences in the distribution of the GGGGCC repeats size between the SCA3/MJD patients and the controls was tested using the Mann-Whitney U test. The GGGGCC repeats mutations with >30 repeats have also been reported outside of the FTLD and ALS spectrum, such as healthy individuals [19, 20]. Previous study reported that ≥7 units in non-pathogenic carriers are strongly correlated with C9orf72 expression [21]. Later, a study [16] identified a statistically significant association between the intermediate repeats and PD risk. Thus, in our study, individuals were classified into three genotypes, including S/S, S/I, and I/I (S: short allele<7 units; I: intermediate allele≥7 units) according to an individual’s two repeat alleles. Owing to the small number of individuals with I/I genotypes, we classified subjected into two groups according to the longer allele of the GGGGCC repeats: short allele (the longer allele <7 units, including S/S genotypes) and intermediate allele (the longer allele ≥7 units, including S/I, and I/I genotypes). We employed two analytical approaches for association testing, the larger repeat allele as a continuous variable and two genotyping categorical variables based on the individual’s short or intermediate alleles. Firstly, a partial correlation was used to analyze the role of the GGGGCC repeats size on the age at onset of SCA3/MJD and to control the influence of the expanded CAG in the ATXN3 allele. Then, a covariance analysis model was used to adjust the effect of expended CAG repeats after interaction analysis. Also, we used multivariate linear regression analyses to test the effect of several potential variables on age at onset, including the CAG repeat size, the C9orf72 genotypes, and gender. The statistical analyses were performed using the SPSS (version19.0).
Results
Table 1 presents the demographic information for this study. The accurate numbers of CAG repeats in the SCA3/MJD patients ranged from 64 to 84, as determined by the direct sequencing of recombinant DNA. No pathological repeat expansions of the C9orf72 gene was detected in any of the individuals. The range of the repeat expansions in the SCA3/MJD patients was 2–18 units, and the most frequent number of repeats in all of the subjects was 2, followed by 6, 7, and 8 (S1 File). There was a difference when compared with that of Europe [21], where the most frequent repeats in Europe was 2 units, followed by 5 and 8. No significant difference was found in the distribution of repeats numbers in an individual’s larger allele between SCA3/MJD patients and normal controls (Fig 1 and Fig 2; more details in S1 File). We found that 142 of the 314 healthy controls (45.22%) harbored the intermediate sized C9orf72 allele (range, 7–29), whereas 67 of the 127 patients with SCA3/MJD (52.76%) also harbored the intermediate-sized allele; this difference is not statistically significant.
Table 1. Demographic information and average number of repeats between SCA3/MJD patients and controls.
| Variables | SCA3/MJD patients | Controls | p value |
|---|---|---|---|
| Case, n | 127 | 314 | - |
| Male, n (%) | 72 (56.7) | 173 (55.1) | 0.83 a |
| Age, Mean ± SD (years) | 37 ± 9 | 36 ± 9 | 0.08 b |
| Number of G4C2 repeats, Mean ± SD (size) | 6 ± 3 | 5 ± 3 | 0.47 c |
| Intermediate allele, n (%) | 67 (52.76) | 142 (45.22) | 0.17 a |
a x 2 test.
b t-tests.
c Mann-Whitney U test.
Fig 1. Distribution of the GGGGCC repeats size in SCA3/MJD patients and controls.

There was no significant difference in the distribution of the GGGGCC repeats length between SCA3/MJD patients and controls.
Fig 2. Frequencies of C9orf72 genotypes.

A presents the frequencies in SCA3/MJD patients. B presents the frequencies in controls.
The log age at onset was determined by the size of the expanded allele in SCA3/MJD patients, the determinant coefficient was 0.435. Significant quadratic effects of the ATXN3 expanded alleles were found (negative effect P<0.0001, Fig 3). When larger repeat allele was used as a continuous variable and the expanded CAG repeats size in the ATXN3 allele as a control variable, the partial correlation analysis showed that the size of the GGGGCC repeats was not correlated with the adjusted age at onset. No significant interaction between GGGGCC repeats and expanded CAG repeats was observed before performing covariance analysis. Interestingly, we observed patients with the intermediate sized allele of the GGGGCC repeats presented a 3-year earlier onset compared with that obtained with the short-sized allele group (Table 2). In multivariate linear regression analyses, when C9orf72 genotype was taken into consideration the percentage of explanation of the onset variance increased from 45.7% to 46.4%. However, when gender was included as a variable, the model was not significantly improved.
Fig 3. Modification of the age at onset due to expanded CAG repeats in ATXN3 of SCA3/MJD patients.
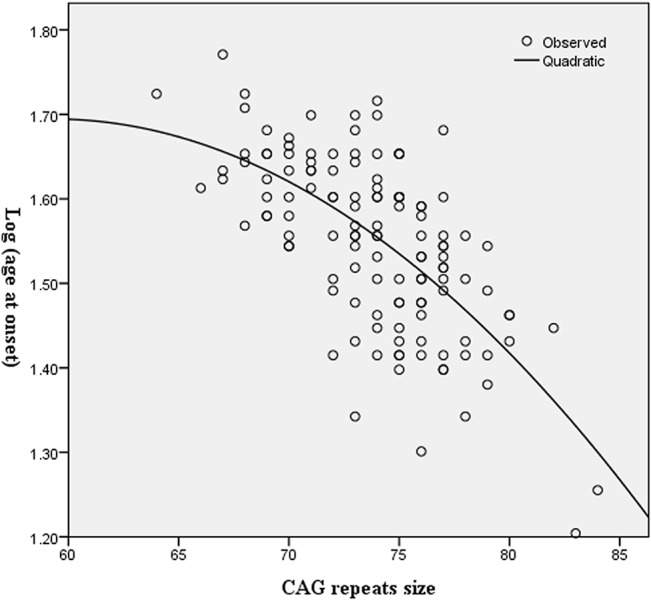
The X-axis denotes the expanded CAG repeat lengths and the Y-axis indicates logarithmically transformed ages at onset. Model parameters: Log (age at onset) = -0.571 + 0.076 Exp—0.001 Exp2. With Exp = Expanded CAG repeats in ATXN3.
Table 2. Descriptive statistics of the age at onset analysis of SCA3/MJD patients.
| Variables | C9orf72 genotypes | p value | |
|---|---|---|---|
| intermediate allele * | short allele * | ||
| Patients, n (%) | 67 (52.76) | 60 (47.24) | - |
| Male, n (%) | 36 (28.35) | 36 (28.35) | 0.59 a |
| Age at onset, Mean± SD (years) | 36 ± 8 | 35 ± 8 | 0.46 b |
| Adjusted age at onset**, Mean ± SD (years) | 34 ±1 | 37 ± 1 | 0.00 b |
| Expanded CAG repeat length, Mean ± SD, (size) | 74 ± 3 | 75 ± 3 | 0.06 b |
a x 2 test.
b t test.
* The SCA3/MJD patients were classified into two genotypes according to the longer allele of the GGGGCC repeat: intermediate alleles (the repeat size ≥ 7 units) and short alleles (the repeat size < 7 units).
** Adjusted for the size of the expanded CAG repeats in SCA3/MJD patients.
Discussion
The GGGGCC repeats in intron 1 of the C9orf72 gene were recently identified not only as a major cause of ALS and FTLD but also as a modifier in the pathogenesis of PD and AD [19, 20, 22, 23]. Previous studies indicated that neurodegenerative disorders exhibit clinical phenotype overlap and share common molecular mechanisms. In this study, we investigated the GGGGCC repeats mutation of the C9orf72 gene and its association with SCA3/MJD patients. No large expansion was identified in our cohort, which suggests that large GGGGCC repeats of the C9orf72 gene do not play a causative role in the pathogenesis of SCA3/MJD. Preliminary evidence suggested that the C9orf72 mutation rates in patients with clinically diagnosed ALS/FTD in China, Japan, Korea, and Taiwan were much lower than that observed in Caucasian populations [16]. Here, we found a difference of the intermediate allele distribution between China and Europe. It is implied that the number of repeats varied greatly due to different nationalities and ethnicities.
The so-called polyglutamine diseases share the feature with each other: a negative relation between age at onset and the number of repeats in the expansion. However, the repeats length only explains 50–80% of the variability of age at onset, suggesting that other genetic factors contribute to the variability [24]. As the variability in age at onset is not completely explained by the effects of the causative and modifier sister genes, other genetic or environmental factors must also play a role. Great efforts have been devoted to the study of genetic links between neurodegenerative disorders. Previous study [2] reported biological interactions between some of the proteins involved in ataxias. The CAG tract of SCA2 gene interferes with MJD phenotype [25]. Recently, after a study of a large cohort of different SCAs, the authors reported that the polyglutamine genes interact with each other in SCA diseases to modify age at onset even when they contain a number of repeats considered to be normal [26]. In our study, we employed two analytical approaches for association testing, the larger repeat allele as a continuous variable and two genotyping categorical variables based on the individual’s short or intermediate alleles. However, only when the C9orf72 gene as categorical variables can a significant result be found. There might be two reasons for the difference. Firstly, the sample of SCA3/MJD is not large enough to find the positive results. Secondly, the intermediate alleles act as a contributor to decrease the age at onset of SCA3/MJD patients. Then, how the intermediate alleles act as predisposing alleles and what is the pathogenic pathway of them in SCA3/MJD patients should be addressed in further studies. We found that the presence of the intermediate allele of the GGGGCC repeats in the SCA3/MJD patients decreased the age at onset by nearly 3 years. These results show that the length of the GGGGCC repeats in the C9orf72 gene influence the SCA3/MJD phenotypes, i.e., larger repeats may cause earlier onset, which indicates that the expansion of the GGGGCC repeats exerts a modifying effect on SCA3/MJD disease diversity. That is to say, our results likely suggest a potential adjustment function between different genome structures to influence the disease. The data from this study support the hypothesis that the pathogenesis of diverse neurodegenerative disorders arises via interactive mechanisms, which may yield common therapeutic targets. This interaction thus provides new information that may aid both the understanding of the common pathogenic mechanisms associated with this class of neurodegenerative disorders and the identification of candidate modulators for these diseases.
In conclusion, our study provides the first demonstration that the larger GGGGCC repeats are associated with an earlier age at onset of SCA3/MJD. These data indicate that the development of SCA3/MJD may involve some physiological functions of the C9orf72 gene and support the hypothesis that a specific mutation identified in one of the neurodegenerative disorders may be a modulator in this disease class [3, 20, 27]. Further studies, particularly on the interaction between C9orf72 and ataxin-3, are needed to verify our results.
Supporting Information
(XLS)
Acknowledgments
We are grateful to all of the subjects for their participation in our study.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by the National Basic Research Program (973 Program) (Nos. 2012CB944601, 2012CB517902 and 2011CB510002 to Hong Jiang), the New Century Excellent Talents in University (No. NCET-10-0836 to Hong Jiang), the National Natural Science Foundation of China (Nos. 81410308019, 81471156, 81271260, 30971585 to Hong Jiang), Hunan Funds for Distinguished Young Scientists (No. 14JJ1008 to Hong Jiang), Xinjiang Natural Science Foundation (No. 201318101-4 to Hong Jiang), the Undergraduate Innovation Project of Central South University (No. YB13028, 201410533324 to Hong Jiang), Fundamental Research Funds for the Central Universities of Central South University (No. 2014zzts078 to Zhao Chen) and High-level medical personnel of Hunan province 225 Project.
References
- 1. Hardy J, Gwinn-Hardy K (1998) Genetic classification of primary neurodegenerative disease. Science 282(5391): 1075–1079. [DOI] [PubMed] [Google Scholar]
- 2. Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, et al. (2006) A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125(4): 801–814. [DOI] [PubMed] [Google Scholar]
- 3. Humbert S, Saudou F (2006) The ataxia-ome: connecting disease proteins of the cerebellum. Cell 125(4): 645–647. [DOI] [PubMed] [Google Scholar]
- 4. Wang JL, Xiao B, Cui XX, Guo JF, Lei LF, Song XW, et al. (2009) Analysis of SCA2 and SCA3/MJD repeats in Parkinson's disease in mainland China: genetic, clinical, and positron emission tomography findings. Movement Disorders 24(13): 2007–2011. 10.1002/mds.22727 [DOI] [PubMed] [Google Scholar]
- 5. Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, et al. (2011) The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Human Molecular Genetics 20(1): 141–154. 10.1093/hmg/ddq452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Socal MP, Emmel VE, Rieder CR, Hilbig A, Saraiva-Pereira ML, Jardim LB (2009) Intrafamilial variability of Parkinson phenotype in SCAs: novel cases due to SCA2 and SCA3 expansions. Parkinsonism & Related Disorders 15(5): 374–378. [DOI] [PubMed] [Google Scholar]
- 7. Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, et al. (2009) Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Human Molecular Genetics 18(22): 4268–4281. 10.1093/hmg/ddp380 [DOI] [PubMed] [Google Scholar]
- 8. Lindquist SG, Duno M, Batbayli M, Puschmann A, Braendgaard H, Mardosiene S, et al. (2013) Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clinical Genetics 83(3): 279–283. 10.1111/j.1399-0004.2012.01903.x [DOI] [PubMed] [Google Scholar]
- 9. Jiang H, Tang B, Xia K, Zhou Y, Xu B, Zhao G, et al. (2005) Spinocerebellar ataxia type 6 in Mainland China: molecular and clinical features in four families. Journal of the Neurological Sciences 236(1–2): 25–29. [DOI] [PubMed] [Google Scholar]
- 10. Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu Y, et al. (2011) Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. Zhong Nan Da Xue Xue Bao. Yi Xue Ban. Journal of Central South University. Medical Sciences 36(6): 482–489. 10.3969/j.issn.1672-7347.2011.06.003 [DOI] [PubMed] [Google Scholar]
- 11. Yi J, Zhang L, Tang B, Han W, Zhou Y, Chen Z, et al. (2013) Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PloS One 8(1): e54792 10.1371/journal.pone.0054792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bettencourt C, Raposo M, Kazachkova N, Cymbron T, Santos C, Kay T, et al. (2011) The APOE epsilon2 allele increases the risk of earlier age at onset in Machado-Joseph disease. Archives of Neurology 68(12): 1580–1583. 10.1001/archneurol.2011.636 [DOI] [PubMed] [Google Scholar]
- 13. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2): 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125): 1335–1338. 10.1126/science.1232927 [DOI] [PubMed] [Google Scholar]
- 15. Salanti G, Amountza G, Ntzani EE, Ioannidis JP (2005) Hardy-Weinberg equilibrium in genetic association studies: an empirical evaluation of reporting, deviations, and power. European Journal of Human Genetics 13(7): 840–848. [DOI] [PubMed] [Google Scholar]
- 16. Jiao B, Guo JF, Wang YQ, Yan XX, Zhou L, Liu XY, et al. (2013) C9orf72 mutation is rare in Alzheimer's disease, Parkinson's disease, and essential tremor in China. Frontiers in Cellular Neuroscience 7: 164 10.3389/fncel.2013.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2): 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiao B, Tang B, Liu X, Yan X, Zhou L, Yang Y, et al. (2014) Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiology of Aging 35(4): 936 e919-922. 10.1016/j.neurobiolaging.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 19. Xi Z, Zinman L, Grinberg Y, Moreno D, Sato C, Bilbao JM, et al. (2012) Investigation of c9orf72 in 4 neurodegenerative disorders. Archives of Neurology 69(12): 1583–1590. 10.1001/archneurol.2012.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, et al. (2013) Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. American Journal of Human Genetics 92(3): 345–353. 10.1016/j.ajhg.2013.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S, et al. (2013) A Pan-European Study of the C9orf72 Repeat Associated with FTLD: Geographic Prevalence, Genomic Instability, and Intermediate Repeats. Human Mutation 34(2): 363–373. 10.1002/humu.22244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, et al. (2012) Repeat expansion in C9ORF72 in Alzheimer's disease. New England Journal of Medicine 366(3): 283–284. 10.1056/NEJMc1113592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kohli MA, John-Williams K, Rajbhandary R, Naj A, Whitehead P, Hamilton K, et al. (2013) Repeat expansions in the C9ORF72 gene contribute to Alzheimer's disease in Caucasians. Neurobiology of Aging 34(5): 1519 e1515-1512. 10.1016/j.neurobiolaging.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Dellen A, Hannan AJ (2004) Genetic and environmental factors in the pathogenesis of Huntington's disease. Neurogenetics 5(1): 9–17. [DOI] [PubMed] [Google Scholar]
- 25. Jardim L, Silveira I, Pereira ML, do Ceu Moreira M, Mendonca P, Sequeiros J, et al. (2003) Searching for modulating effects of SCA2, SCA6 and DRPLA CAG tracts on the Machado-Joseph disease (SCA3) phenotype. Acta Neurologica Scandinavica 107(3): 211–214. [DOI] [PubMed] [Google Scholar]
- 26. Tezenas du Montcel S, Durr A, Bauer P, Figueroa KP, Ichikawa Y, Brussino A, et al. (2014) Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain 137(Pt 9): 2444–2455. 10.1093/brain/awu174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Es MA, Diekstra FP, Veldink JH, Baas F, Bourque PR, Schelhaas HJ, et al. (2009) A case of ALS-FTD in a large FALS pedigree with a K17I ANG mutation. Neurology 72(3): 287–288. 10.1212/01.wnl.0000339487.84908.00 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.