

Abstract
Background
DNA repair mechanisms are crucial for maintenance of the genome in all organisms, including parasites where successful infection is dependent both on genomic stability and sequence variation. MSH2 is an early acting, central component of the Mismatch Repair (MMR) pathway, which is responsible for the recognition and correction of base mismatches that occur during DNA replication and recombination. In addition, recent evidence suggests that MSH2 might also play an important, but poorly understood, role in responding to oxidative damage in both African and American trypanosomes.
Methodology/Principal Findings
To investigate the involvement of MMR in the oxidative stress response, null mutants of MSH2 were generated in Trypanosoma brucei procyclic forms and in Trypanosoma cruzi epimastigote forms. Unexpectedly, the MSH2 null mutants showed increased resistance to H2O2 exposure when compared with wild type cells, a phenotype distinct from the previously observed increased sensitivity of T. brucei bloodstream forms MSH2 mutants. Complementation studies indicated that the increased oxidative resistance of procyclic T. brucei was due to adaptation to MSH2 loss. In both parasites, loss of MSH2 was shown to result in increased tolerance to alkylation by MNNG and increased accumulation of 8-oxo-guanine in the nuclear and mitochondrial genomes, indicating impaired MMR. In T. cruzi, loss of MSH2 also increases the parasite capacity to survive within host macrophages.
Conclusions/Significance
Taken together, these results indicate MSH2 displays conserved, dual roles in MMR and in the response to oxidative stress. Loss of the latter function results in life cycle dependent differences in phenotypic outcomes in T. brucei MSH2 mutants, most likely because of the greater burden of oxidative stress in the insect stage of the parasite.
Author Summary
Trypanosoma brucei and Trypanosoma cruzi are protozoa parasites that cause sleeping sickness and Chagas disease, respectively, two neglected tropical diseases endemic in sub-Saharan Africa and Latin America. The high genetic diversity found in the T. cruzi population and the highly diverse repertoire of surface glycoprotein genes found in T. brucei are crucial factors that ensure a successful infection in their hosts. Besides responding to host immune responses, these parasites must deal with various sources of oxidative stress that can cause DNA damage. Thus, by determining the right balance between genomic stability and genetic variation, DNA repair pathways have a big impact in the ability of these parasites to maintain infection. This study is focused on the role of a DNA mismatch repair (MMR) protein named MSH2 in protecting these parasites’ DNA against oxidative assault. Using knock-out mutants, we showed that, besides acting in the MMR pathway as a key protein that recognizes and repairs base mismatches, insertions or deletions that can occur after DNA replication, MSH2 has an additional role in the oxidative stress response. Importantly, this extra role of MSH2 seems to be independent of other MMR components and dependent on the parasite developmental stage.
Introduction
Two members of the trypanosomatidae family, Trypanosoma cruzi and Trypanosoma brucei, are important human pathogens, since they cause, respectively, Chagas disease, or American trypanosomiasis, and African Sleeping Sickness, or Human African trypanosomiasis. Together, T. cruzi and T. brucei infections affect almost 20 million people [1, 2]. The life cycles of both these parasites involve two hosts: an invertebrate vector and a mammalian host. In the digestive tract of the insect vector T. cruzi multiplies as epimastigotes and differentiates into metacyclic trypomastigotes, which are expelled with the vector’s faeces. After a blood meal, trypomastigotes injected in the host bloodstream can invade different cell types, where they replicate as intracellular amastigotes that, after a number of replication cycles in the host cell cytoplasm, differentiate into trypomastigotes and lyse the host cell membrane. Despite being similar in general strategy, the life cycle of T. brucei is different to that of T. cruzi in several key details. Notably, T. brucei does not display any intracellular replicative stages. In the mammal, T. brucei is exclusively extracellular, replicating in the bloodstream and tissue fluids as bloodstream form (BSF) cells, which can be taken up by the tsetse fly vector during a bloodmeal. In the insect vector BSF cells differentiate into replicative procyclic forms (PCF), which then undergo several further differentiation events associated with migration to the fly salivary glands, where non-replicative metacyclic trypomastigotes are formed and can be passed into a new mammalian host through the proboscis when the infected fly is feeding [3]. Irrespective of the detailed differences in the life cycles, differentiation between the mammal-infective and vector-infective forms of both T. cruzi and T. brucei is accompanied by dramatic metabolic changes and morphological alterations [4].
The ability to multiply and survive inside a host or vector is crucial for the maintenance of a parasite infection and transmission, allowing continuation of the life cycle. As for any cell, unicellular parasites are exposed to potentially deleterious events during cell division. The by-products of cellular metabolism, allied to routine errors during DNA replication or recombination processes, represent endogenous sources of potential DNA damage and genome change. In addition, all organisms are subjected to exogenous genotoxic agents from the environment or, in the case of parasites, derived from host. In the mammalian host, T. cruzi invades non-phagocytic cells or can be internalized by macrophages by a phagocytosis-like process [5]. Inside macrophages T. cruzi triggers the activation of NADPH oxidase, which generates large amounts of reactive oxygen species (ROS) such as O2⠁-. Moreover, pro-inflammatory cytokines triggered by T. cruzi infection also stimulate infected macrophages to produce high amounts of nitric oxide (⠁NO) through the induction of inducible nitric oxide synthase (iNOS), which can react with O2⠁- producing peroxynitrite (ONOO-), a powerful oxidant and cytotoxic molecule [6, 7]. Similarly, the insect life forms of both parasites must also deal with the invertebrate oxidative stress response generated against the parasite. Upon challenge with T. brucei the tsetse fly activates iNOS, generating ⠁NO, and increases the levels of ROS such as hydrogen peroxide (H2O2) [8, 9]. To deal with all of these potentially genome damaging agents, trypanosomes, like any other organism, possess multiple DNA repair pathways [10, 11], though little work has detailed to what extent these pathways are needed in different parasite life cycle stages. One such DNA repair pathway is Mismatch Repair (MMR), which acts to detect and correct mispaired bases that escape the proofreading activity of DNA polymerases during replication or occur during recombination between non-identical DNA molecules. In eukaryotic cells, even small distortions in DNA caused by single-base mismatches or insertion/ deletion loops (IDLs) of 1–3 bases are recognised by the “sliding clamp” heterodimer formed by MSH2 and MSH6 proteins, which moves along DNA searching for errors [12]. Larger IDLs are recognised by a distinct heterodimer, composed of MSH2 and MSH3. Eukaryotes encode several further MSH proteins, all of which are homologous to bacterial MutS proteins, though many have adopted non-MMR roles [13]. Following mismatch recognition, an ATP-dependent conformation change in MSH2-6 or MSH2-3 promotes interaction with heterodimeric homologues of bacterial MutL proteins, bringing these factors to the site of the lesion. The excision step is catalyzed by an endonuclease activity within the MutL-related heterodimer formed between MLH1-PMS1 [14], and that activity is activated by PCNA (a component of replication machinery) [15] [16]. A defect in or directed inactivation of the MMR pathway leads to a 100–1000 fold increase in spontaneous mutation rate in all organisms examined, including Escherichia coli [17], Saccharomyces cerevisiae [18], Caenorhabditis elegans [19, 20], and T. brucei [21]. In human cells and murine models MMR deficiency is correlated with predisposition to cancer [22, 23]. MMR suppresses mutations by increasing DNA replication fidelity through preventing base substitutions or repeat sequence instability, events that can also occur during homologous recombination [24].
In addition to repair of non-identical DNA sequences, MMR has also been demonstrated to be involved in the response to DNA damage induced by genotoxic agents, among them the alkylator N-methyl-N’-nitro-N-nitrosoguanidine (MNNG), the anti-cancer drug cisplatin and H2O2 [25]. Methylation of DNA by MNNG gives rise to O 6-methylguanine (O 6-MeG), opposite which thymine can be misincorporated during replication, forming a base mismatch recognised by MMR. Defective MMR is associated with increased tolerance to MMNG [26], due to loss of what has been termed a futile cycle of repair, where MMR fails to remove O 6-MeG and instead re-inserts either cytosine or thymine opposite the lesion. In some organisms, active MMR at MNNG damage leads to persistent DNA breaks and increased cell death [27]. H2O2 generates reactive oxygen species (ROS) with high capacity to damage DNA [28]. The most common DNA lesion resulting from ROS is 7,8-dihydro-8-oxoguanine (8-oxoG) [29]. MMR has the ability to prevent oxidative mutagenesis in E. coli by removing 8-oxoG or adenine misincorporated paired with 8-oxoG [30]. In S. cerevisiae it was demonstrated that the MSH2-MSH6 heterodimer has a direct role in removing adenine in 8-oxoG:A mispaired bases [31]. More recently, Zlatanou et al. proposed a non-MMR role for MSH2-MSH6 in human fibroblasts in response to oxidative damage. The model suggests that in some circumstances DNA oxidative damage by MSH2-MSH6 does not recruit other MMR proteins, but instead repair is mediated through the action of monoubiquinated-PCNA (mUb-PCNA) and Polη [32].
With the availability of the complete genome sequences of T. brucei and T. cruzi the full repertoire of the parasite’s putative DNA MMR machinery was revealed to comprise homologues of MSH2, MSH3, MSH6 (originally named MSH8) [21], MLH1 and PMS1 [10, 33, 34]. However, functional characterization studies to date have been limited to a single life cycle stage in each parasite and have only tested the roles of MSH2 in T. brucei and T. cruzi, and MLH1 in T. brucei. Null mutants of T. brucei msh2 (Tbmsh2-/-) or mlh1 (Tbmlh1-/-) in BSF cells show several phenotypes characteristic of MMR-deficient cell lines: increased tolerance to MNNG, increased rates of sequence change in a number of microsatellite repeat loci, and elevated rates of homologous recombination-based integration of transformed DNA molecules carrying base mismatches relative to genomic target loci [21, 35, 36]. In addition, evidence that TbMSH2 and TcMSH2 play a role in in the oxidative stress response has been found in studies showing that Tbmsh2-/- BSF cells and Tcmsh2+/- epimastigotes are more susceptible to H2O2 treatment than wild type parasites [21, 37]. The role played by MSH2 in tackling oxidative damage appears not to involve a complete MMR reaction, since BSF Tbmlh1-/- mutants do not display equivalent increased H2O2 sensitivity to Tbmsh2-/- mutants [38]. Moreover, heterologous expression of T. cruzi MSH2 in T. brucei msh2-/- mutants shows that the American trypanosome MSH2 protein can functionally replace the endogenous MSH2 protein in the oxidative stress response, but cannot work with the T. brucei MMR machinery to successfully execute MMR [38]. Although only single allele knock-outs of msh2 have been described in T. cruzi epimastigotes, the T. cruzi msh2+/- mutants display increased sensitivity to H2O2 treatment [37], consistent with the null mutants in T. brucei BSF cells [38]. However, despite evidence in both T. brucei and T. cruzi that H2O2-induced damage predominantly affects the kinetoplast genome when MSH2 is lost or impaired [37], the exact role of MSH2 remains elusive. Here, we have sought to clarify the role of MSH2 in the oxidative stress response of T. brucei and T. cruzi, and to ask to what extent this putative MMR-related function is conserved or diverged in the two parasites. We describe the generation of msh2 null mutants in T. brucei PCF cells, which differ from BSF cells in the increased use of mitochondrial metabolism [39], with the consequence that there is greater endogenous reactive oxygen species. In addition, we describe, for the first time, the generation of msh2 null mutants in T. cruzi epimastigote cells, which also rely on aerobic metabolism [40]. In both cell types, msh2 null mutants are viable and are impaired in MMR. Unexpectedly, both mutants do not display increased sensitivity to H2O2 treatment, but instead, increased resistance, which we suggest to be due to parasite adaptation to the loss of a crucial molecule. Thus, we propose that MSH2 provides one of several interconnected mechanisms that are common to T. brucei and T. cruzi and allow both parasites to cope with oxidative stress.
Methods
Parasite cultures
T. brucei cultures of the Lister 427 strain were maintained as both bloodstream (BSF) and procyclic (PCF) forms. BSF parasites were maintained at 37°C, 5% CO2 in HMI-9 (GIBCO) medium supplemented with 20% fetal bovine serum (GIBCO). Cell passages were performed every 48 hours, with population density maintained between 1 x 105 and 2 x 106 cells.mL-1. PCF cells were maintained at 27°C in SDM-79 (GIBCO) medium supplemented with 10% fetal bovine serum (GIBCO). Weekly passages were performed, but cell density was never lower than than 5 x 105 cells.mL-1.
Epimastigote forms of the CL Brener clone of T. cruzi were maintained in logarithmic growth phase at 28°C in liver infusion tryptose (LIT) medium supplemented with 10% fetal bovine serum (GIBCO) and penicillin (10,000 U.mL-1)/Streptomycin (10,000 μg.mL-1) (GIBCO) as described by [41]. Metacyclic trypomastigotes, obtained after metacyclogenesis of epimastigotes cultures maintained in LIT medium for 15 to 20 days, were used to infect Vero cells cultured in DMEM medium (GIBCO) supplemented with 5% fetal bovine serum (GIBCO).
Parasite transfections
Transfection of PCF parasites were carried out at a density of 5 x106 cells.mL-1 resuspended in 0.5 mL of ice-cold Zimmerman medium (132 mM NaCl, 8 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, 0.5 mM MgAc2, and 0.06 mM CaAc2, pH 7.5) and 10 μg of DNA. The mixture was subjected to two rounds of electroporation with a Bio-Rad Gene Pulser II (1.5 kV current and 25 μF capacitance). The cells were then transferred into SDM79 medium and incubated at 27°C overnight. To select for antibiotic-resistant transfectants, parasites were diluted 1:100 and 1:10 in 96 well plates containing conditioned medium and the appropriate antibiotic and incubated for 10–14 days.
Transfection of BSF cells were carried out using AMAXA Nucleofactor (Amaxa Biosystems) with 4 x 107 cells resuspended in 100 μL of nucleofector solution (optimised for human T-cells) and mixed with 5–10 μg of DNA. After following the nucleofection protocol according to manufacturer instructions, the cells were serially diluted 1:10, 1:100 and 1:1000 in HMI-9 medium without antibiotic and incubated 6–12 hrs for recovery, after which 1 mL of HMI-9 medium containing antibiotic was added to each well. To select for MSH2 or MLH1 single allele mutants, PCF cells were selected with either 10 μg.mL-1 blasticidin or 1 μg.mL-1 puromycin; both drugs, at the same concentrations, were used to select for deletion of the second allele in the single allele mutants. To select for PCF msh2-/- cells in which MSH2 was re-expressed, transformants were grown in the presence of 2.5 μg.mL-1 phleomycin.
Transfection of T. cruzi epimastigotes was carried out with 107 cells.mL-1 collected during exponential growth phase and 100 μg of plasmid constructions as previously described [42]. Twenty-four hours after transfecting hygromycin resistant cell lines, 100 μg.mL-1 of selective antibiotic (G-418 Sulfate) was added to transfected cultures. Weekly passages were performed with increasing concentration of the selective drug (up to 200 μg.mL-1). After 30–40 days, a double-resistant population was selected and cloned cell lines were obtained by plating epimastigotes on semisolid blood agarose plates with 200 μg.mL-1 of G-418 Sulfate and Hygromycin B, after an additional 30 days of incubation at 28°C.
Plasmid constructs to generate knockout parasites, MSH2 re-expressers and to inhibit msh2 expression by RNAi
Knockout constructs to delete msh2 (TritrypDB Tb927.10.11020) or mlh1 (TritrypDB Tb927.8.6840) in T. brucei PCF cells have been described before [21] and were generated by PCR-amplifying the 5’ and 3’ UTRs corresponding to each gene from TREU 927 genomic DNA and cloning into pBluescript II KS and separated by antibiotic resistance gene (BSD and/or PUR), flanked by 230 bp of β-α tubulin and 330 bp of α-β tubulin processing signals. The constructs were linearized by digestion with XhoI and NotI restriction enzymes before transfecting into T. brucei PCF.
Epimastigote cultures of T. cruzi with one of the msh2 alleles disrupted by a Hygromycin resistance gene was generated as described by [37]. The deletion of the second allele was performed using a DNA construct containing the Neomycin phosphotransferase gene (Neo) flanked by intergenic sequences of the HX1 and GAPDH regions that were PCR-amplified from the pROCK-GFP vector [42] and cloned into pGEM T-easy vector (Promega). DNA sequence corresponding to ~500 bp of the 5’ and 3’ end of TcMSH2 (TritrypDB TcCLB.507711.320) coding region was inserted upstream and downstream of HX1 and GAPDH, respectively, in the pTopo_HX1_Neo_GAPDH and the plasmid linearized using the XbaI and SmaI restriction enzyme digestions before transfection.
T. brucei msh2-/- BSF and PCF were transfected with an MSH2 re-expression construct, which has been described before [21] and where the msh2 ORF integrates and replaces either the BSD or PUR resistance cassette in the mutated msh2-/- locus. This construct was generated using a 4.5 kb region containing the MSH2 gene flanked by MSH2 5’UTR and 3’UTR. The 4.5 kb fragment was cloned into pBluescript SK II, followed by a cloned phleomycin resistant cassette downstream of the 3’ UTR of the MSH2 gene. The construct was digested with HindIII prior to transfection.
To generate RNAi constructs for MSH2, a 513bp region of the gene was PCR-amplified from wild type Lister 427 genomic DNA and cloned into the vector pZJM [43] where they are flanked by opposing T7 promoters and Tet operator sequences. The MSH2 RNAi plasmid was linearized with NotI and used to transform T. brucei PCF strain 427 pLew29-pLew13 developed by Wirtz et al [44], constitutively co-expressing T7 RNA polymerase and Tet repressor. Clones were selected with 2.5 μg.ml-1 phleomycin.
RNA extraction, northern blot, cDNA generation, reverse transcriptase (RT) reaction and quantitative RT-PCR
Total RNA was isolated from T. brucei PCF form and T. cruzi epimastigote forms using the RNeasy kit (Qiagen). For cDNA generation RNA samples were treated, on column, with RNAse-free DNAse (Qiagen) as per the manufacturer’s instructions. cDNA was synthesized using High Capacity RNA-cDNA master mix (Applied Biosystems). cDNA from wild type (WT) cells, Tbmsh2+/- blasticidin (BSD) or puromycin (PUR) resistant clones, Tbmsh2-/-, Tbmlh1+/- BSD or PUR resistant clones and Tbmlh1-/- were used in reverse transcriptase (RT) PCR with specific primers for the TbMSH2 or TbMLH1 genes. As a positive control, primers to specifically PCR-amplify TbRAD51 were used. For northern blot analyses, 20 μg of total RNA of T. cruzi WT, Tcmsh2+/- and three different clones of Tcmsh2-/- were separated in a 1.2% agarose/MOPS/formaldehyde gel. The RNA was then transferred to hybond-N+membrane (GE Healthcare) and hybridized with a TcMSH2 specific PCR fragment previously labelled with [α-32P]-dCTP using the Amersham Ready-to-Go DNA Labelling Beads (GE Healthcare). The hybridization was carried out as previously described by [45]; briefly, hybridisation was in 50% formamide buffer overnight at 42°C, after which the blot was washed twice with 2× SSC/ 0.1% SDS at 65°C for 20 min. The membrane was then exposed to a phosphor screen of the STORM 820 phosphor image (GE Healthcare) and analysed by ImageQuant TL software (GE Healthcare). To quantify levels of mRNA by RT-PCR after RNAi induction, primers specific for msh2 and mlh1 genes were used. As a endogenous control primers for GPI8 gene (TritrypDB Tb427.10.13860) were used. SYBR Green PCR Master Mix (Applied Biosystems) was used for PCR in 96 well plates. Reactions were run on an ABI Prism 7000 thermocycler and mRNA levels quantified from amplification according to the manufacturer’s instructions;
Microsatellite instability
T. brucei PCF MMR mutants were tested for genetic instability by PCR amplification of the region containing the JS-2 microsatellite from parasites grown in the absence or presence of 20 μM H2O2 for 48 hours. The JS-2 microsatellite has been mapped to Chromosome IV and is composed of GT-dinucleotide repeats [46]. Genomic DNA was extracted from 10 independent clonal populations derived from PCF wild type and knockout mutants and PCR-amplified with JS2 microsatellite complementary primers. PCR products were resolved for 50 minutes at 100 V by electrophoresis in a 3% low melting agarose gel.
Treatment with genotoxic agents
Mid-log phase PCF T. brucei cultures were inoculated in SDM-79 (GIBCO) at density of 5 x 105 cells.mL-1 in the absence or presence of 2.5 μM or 5 μM N-methyl-N’-nitro-N-nitrosoguanidine (MNNG, Tokyo chemical industry Ltd). After 72 hours survival was determined using a haematocytometer. T. cruzi epimastigote form cells in the exponential growth phase were counted and diluted to 1x 107 cells.mL-1 in LIT medium in the absence or presence of 5 μM MNNG (Tokyo chemical industry Ltd). After 72 hours cell densities were determined by counting live cells with a haematocytometer using Erythrosin B exclusion.
Mid-log phase T. brucei PCF cells were diluted to 5 x 105 cells.mL-1 in SDM-79 (GIBCO) and then incubated with 10 μM or 20 μM H2O2 (VWR) at 27°C for 48–72 hours. BSF cells were diluted to 1 x 106 cells.mL-1 in HMI-9 medium (GIBCO) and incubated with 100 μM or 200 μM H2O2 (VWR) at 37°C, 5% CO2 for 48 hours. After growth, cell density was measured using a haematocytometer. T. cruzi epimastigote form in exponential growth phase were diluted to 1x 107 parasites.mL-1 in PBS 1x and incubated with 75 μM H2O2 (Sigma-Aldrich) for 20 minutes at 28°C. After this, the cells were centrifuged, washed once with PBS and allowed to recover in LIT medium for 48 hours before cell densities were determined by counting live cells with a haematocytometer using Erythrosin B exclusion.
Cell growth analyses after induction of RNAi
Two clones of PCF RNAi cell line targeting MSH2 were grown to log phase and diluted to a starting density of 5 x 105 cells.mL-1. Each culture was then split and grown in the absence of tetracycline or after the addition of tetracycline to 2 μg.mL-1. Cell density was determined every 24 hours up to 72 hours and then the cultures were diluted to their starting density. After adding again tetracycline to induce siRNA expression, parasite numbers were further determined every 24 hours for a further 72 hours, before diluting again the cultures back to starting densities.
T. cruzi in vitro infection of Vero cells and mouse macrophages
Trypomastigotes released in the supernatants of infected Vero cells were counted with a haemocytometer and an equivalent cell number for each culture was used to infect Vero cells or intraperitoneal macrophages (harvested from BALB/c mice) that had been allowed to adhere to glass cover slips in a 24 well plate. Trypomastigotes were incubated with the cell monolayers for 4 hours, after which non-internalized trypomastigotes were washed away with PBS 1x. The infected cells were incubated for an additional 48 hours at 37°C. The number of intracellular amastigotes was determined by staining cell nuclei with DAPI (1000 μg.mL-1) and visualised through fluorescence microscopy (Olympus). The average number of amastigotes was determined from analysing 1000 host cells, in different fields of the microscopy slide.
In vitro infection of BALB/c mice macrophages were carried out in strict accordance with the Brazilian laws regarding animal use (Law # 11.794, December 8, 2008), following a protocol approved by the Committee on the Ethics of Animal Experiments of UFMG (CETEA-UFMG) under the number 132/2014.
8-oxoguanine measurement
8-oxoG measurement was performed based on the principle of binding affinity of FITC-conjugated avidin to 8-oxoG [47] and an adapted protocol [48], which has been previously used to quantify oxidative DNA lesions in different T. cruzi strains and mutants for different DNA repair proteins [37, 49, 50]. Briefly, 20 μL aliquots of cell suspensions were distributed on a glass slide coated with poly-lysine (Sigma-Aldrich) and left to adhere for 5–10 min, after which cells were fixed with ice-cold methanol for 20 min at -20°C. The methanol-fixed cells were then washed with room temperature PBS 1x, cells permeabilized with 0.1% Triton X-100 for 3 min and incubated with RNase A (100 μg.mL-1) for 1 hour at 37°C. After another wash with PBS 1x, cells were treated with proteinase K (10 μg.mL-1) for 7 min at room temperature, followed by treatment with HCl (4N) for 7 min at room temperature and overnight blocking with 10% fetal bovine serum. FITC-avidin was diluted 1:200 in blocking buffer and incubated with the cells for 1 hour at room temperature. After extensive washing with PBS, cell nucleus and kDNA were stained with DAPI. Slides were mounted with prolong gold anti-fade solution (Molecular Probes/Life Technologies), visualized under a fluorescence microscope and images of at least 10 different fields were captured. After manually delimiting the nucleus and the kDNA of each parasite, as visualized by DAPI staining, FITC- fluorescence intensity restricted to these sites were determined with ImageJ software and plotted as the average fluorescence (arbitrary units) of 100 random cells.
Epitope tagging MSH2
T. brucei BSF, PCF and T. cruzi epimastigote cells were transfected with constructs to tag MSH2 C-terminally in the endogenous locus. Primers were generated against a C-terminal region of msh2 ORF, but excluding the stop codon. The region was selected to have a unique restriction enzyme recognition site within the ORF fragment that could be used to linearize the DNA. This fragment was cloned into the plasmid pNAT12myc [51] allowing in frame fusion of the ORF with 12 repeats of the c-MYC epitope. Similarly, the C-terminal region of Tcmsh2 ORF was cloned in pCR 2.1 Topo vector (Life Technologies) in frame with the 12 repeats of the c-MYC epitope derived from the T. brucei pNAT12myc plasmid. As a selective mark, neomycin resistance gene derived from the pROCK_Neo plasmid [42] was also cloned in the same pCR 2.1 Topo vector. The constructs were linearized with BmgBI (for T. brucei) or SalI (for T. cruzi) restriction enzymes and used to transfect heterozygous msh2 knockout mutants of T. brucei, and wild type T. cruzi epimastigotes; transfected T. brucei or T. cruzi were selected with 10 μg.mL-1 blasticidin or 200 μg.mL-1 G418 Sulfate, respectively.
Cellular localization
T. brucei BSF and PCF expressing MSH2 fused to 12x c-MYC epitopes were fixed in chilled methanol for 1 hour or overnight. Fixed slides were then blocked with 2% fetal calf serum (FCS), incubated with mouse anti-c-MYC antiserum (1:5000) (Millipore) for 1 hour, followed by incubation with Alexafluor 594 conjugated anti-mouse IgG secondary antiserum (Molecular Probes/ Life Technologies) in the dark. Slides were mounted with vectashield containing 4’,6-diamidino-2-phenylindole (DAPI; Vector labs) and visualized on a Zeiss Axioplan microscope. Images were captured using Hamamatsu ORCA-ER digital camera and Openlab software. T. cruzi epimastigote and amastigote cells expressing MSH2 fused to 12x c-MYC epitopes were fixed with 4% paraformaldehyde for 5 minutes, permeabilized with 0.1% Triton X-100 for 10 min, blocked with 1% BSA, 0.2% Tween 20 for 1 hour at room temperature and incubated with 1:500 anti-c-MYC antiserum conjugated to Alexa 488 (Milipore) for 1 hour. After washing with PBS, nuclei were stained with 1 μg.mL-1 of DAPI (Molecular Probes/ Life Technologies) for 5 min and cover slides mounted with prolong gold anti-fade solution (Molecular Probes/ Life Technologies). Images were captured on an Olympus BX60 microscope using Q-color 5 digital camera and Qcapture Pro 6.0 software.
Statistical analysis
Statistical analyses in this work were performed using GraphPad Prism version 5.00 (GraphPad Software, San Diego California USA). Data are presented as mean plus standard deviation, and all experiments were repeated at least three times. Results were analysed for significant differences using ANOVA followed by Bonferroni post-test. Statistical tests used are described at each figure legend. The level of significance was set at P < 0.05.
Results
T. brucei procyclic form and T. cruzi epimastigote msh2 null mutants have increased resistance to oxidative stress
The genomes of T. brucei and T. cruzi each contain a single-copy gene encoding MSH2 [33, 34]. To generate T. brucei PCF msh2 null (-/-) mutants, we relied upon the same constructs used previously to make BSF cells in which both msh2 alleles were deleted [21] (S1 Fig). In parallel, we also generated T. brucei PCF mlh1-/- mutants, again using pre-existing constructs. For both genes, the constructs replace the entire ORF (2856 bp and 2664 bp for MSH2 and MLH1, respectively) after homologous integration. Cells with a single allele deleted (heterozygous mutants; +/-) were first selected using either a construct encoding resistance to blasticidin (BSD) or puromycin (PUR) and confirmed by PCR (S2 Fig). The +/- cells were then transformed with the reciprocal construct to attempt to make-/- mutants lacking both alleles; in both cases, and for reasons that are unclear, this was only successful when transforming the PUR deletion constructs into the cells that had integrated BSD and, furthermore, required a number of transformation attempts using varying antibiotic selection concentrations. Correct integration of the constructs and loss of both TbMSH2 or TbMLH1 alleles in the-/- mutants were confirmed by PCR (S2 Fig). Deletion of both alleles in the PCF-/- mutants was further verified by RT-PCR amplification of RNA extracted from drug resistant parasites: as shown in Fig 1A, PCR products from the ORFs were detected in +/- cells as well as in wild type (WT) parasites, but not in the-/- mutants.
Fig 1. Generation of mismatch repair null mutants in T. cruzi and T. brucei.

(A) Agarose gel electrophoresis of reverse transcriptase (RT) PCR products to verify the absence of MSH2 or MLH1 mRNA in T. brucei procyclic form mutants. cDNA derived from wild type (WT) cells, blasticidin (BSD) or puromycin (PUR) resistant clones of Tbmsh2 single allele knockouts (+/-) and Tbmsh2 double allele knockouts (-/-) were PCR-amplified with MSH2 or MLH1- specific primers; the size of the PCR product is indicated and a control reaction without cDNA is indicated by C. The bottom panel shows, as positive controls, cDNAs from the same cells PCR-amplified (RT+) with primers specific for the RAD51 gene; a control for genomic DNA contamination, in which reverse transcriptase was excluded from the cDNA synthesis reaction (indicated by RT-) is also shown. (B) Total RNA extracted from T. cruzi epimastigote form wild type (WT) cells, a Tcmsh2 single allele knockout (+/-) mutant and three independent clones of Tcmsh2 double allele knockouts (-/-) were transferred to a nylon membrane and hybridized with [α-32P]-labeled probe specific for the T. cruzi MSH2 gene. The bottom panel shows hybridization with a probe for 24Sα rRNA, used as loading control.
To generate T. cruzi epimastigote msh2-/- mutants, we started with clones in which one MSH2 allele was deleted (+/- cells) [37] and replaced by the ORF for the hygromycin phosphotransferase (HYG) gene. It should be noted that in the msh2 heterozygous mutants (+/-) the HYG mRNA is generated by the predicted 5’ and 3’ processing signals flanking the MSH2 gene. In order to mutate the remaining allele, we found it was necessary to use a gene disruption construct (S1 Fig) in which a neomycin phosphotransferase gene ORF (NEO) was flanked upstream by processing signals derived from the T. cruzi intergenic region of the TcP2β gene [52], and downstream by the intergenic region derived from the gapdh gene [42]. To allow integration into the remaining MSH2 allele, homologous targeting was based on TcMSH2 ORF sequence, and not on the 5’ and 3’ UTRs, as used before to generate the msh2+/- mutants. Finally, in order to successfully select for-/- cells, in which the first allele was deleted and the second disrupted, transformed parasites had to be selected initially with a low drug concentration (100 μg.mL-1) that was gradually increased to 150 μg.mL-1 and 200 μg.mL-1 during a period of 4 weeks. PCR amplification of DNA extracted from the Tcmsh2+/- cells, and from three cloned putative Tcmsh2-/- cell lines showed the expected integration of the different MSH2 targeting constructs in the T. cruzi genome (S1 Fig). Northern blots using a probe specific for the TcMSH2 ORF and for the 24Sα rRNA as a loading control showed 52% less TcMSH2 mRNA in the +/- cells relative to WT, and no detectable MSH2 mRNA in the Tcmsh2-/- clones (Fig 1B), confirming the gene disruption. The apparent reduction in TcMSH2 mRNA levels in the +/- cells is consistent with the observation of altered phenotypes in these cells relative to WT (see below; and [37]). Similar to T. brucei msh2 mutants, no changes in population doubling times were detected when comparing growth of WT cells to the T. cruzi mutants with one or both msh2 alleles deleted (S3 Fig).
To ask if the mutations described above result in detectable loss of MMR, we measured the sensitivity of the parasites to MNNG [26]. T. brucei and T. cruzi mutants and WT cells were grown for 72 hours with increasing concentrations of MMNG. Survival of the cells was determined by measuring the cell density of the WT or mutant cells after MNNG treatment relative to untreated cells. In T. brucei, all cells showed increasing growth impairment as MNNG was increased from 2.5 to 5 μM. However, similar to what has been described in BSF cells [21], and consistent with the proposed futile cycle of alkylation repair in MMR-proficient cells [26, 27], deletion of one allele of Tbmsh2 or Tbmlh1 in the PCF+/- mutants caused increased tolerance to MNNG, and this tolerance increased yet further when both alleles were deleted in the-/- mutants (Fig 2A). Deletion of TbMSH2 or TbMLH1 in PCF cells also resulted in the-/- parasites displaying increased microsatellite instability, as was observed in T. brucei BSF mutants, indicating decreased replication fidelity (S4 Fig). In T. cruzi increased tolerance to 5 μM MNNG was seen in the Tcmsh2+/- cells and was not detectably increased in the-/- mutants (Fig 2B). Though this somewhat contrasts with the response of T. brucei MMR mutants to MNNG, these data nonetheless show that mutation of MMR genes in either parasite resulted in the expected enhanced survival in the presence of this alkylating drug.
Fig 2. Susceptibility of T. brucei and T. cruzi MMR knockout mutants to N-methyl-N’-nitro-N-nitrosoguanidine (MNNG).

(A) T. brucei wild type (WT) and procyclic form mutants (Tbmsh2+/-, Tbmsh2-/-, Tbmlh1+/- and Tbmlh1-/-) were grown in culture medium with 0 μM, 2.5 μM or 5 μM MNNG. Cell density was measured after 72 hours growth and is plotted as the percentage survival of the MNNG treated cells relative to untreated cultures. (B) WT T. cruzi epimastigotes and MSH2 mutants (Tcmsh2+/- and msh2-/-) were grown in culture medium with 0 μM or 5 μM MNNG. Cell viability was measured after 72 hours and is plotted as the percentage survival of the MNNG treated cells relative to untreated cultures. Vertical lines indicate standard deviation. ***p<0.001, **p<0.01, *p<0.05: determined by one-way ANOVA with Bonferroni post-test of knockout mutants relative to wild type cells.
To ask how the loss of the MMR genes affects the response of T. brucei and T. cruzi cells to oxidative stress, we compared growth of the WT cells and mutants in the presence of H2O2, again determining survival in the presence of the damaging agent relative to undamaged cells. In striking contrast to our previous observations in T. brucei, which showed that Tbmsh2-/- BSF cells are more susceptible to H2O2 [38], PCF T. brucei msh2 mutants were more resistant to H2O2 than WT cells after 48 hrs growth at either 10 μM or 20 μM (Fig 3A): improved survival was seen in both the msh2+/- and msh2-/- cells at this time point, and was also observed 24 and 72 hrs post-treatment (S5 Fig). In contrast, though T. brucei mlh1+/- mutants displayed a modest increase in H2O2 resistance (in particular upon treatment with 10 μM H2O2; Fig 3A), mlh1-/- mutants displayed no detectable difference from WT, indicating that the MMR response to H2O2 damage is predominantly mediated through MSH2 and not MLH1. This separation in function between the two MMR factors is consistent with observations in T. brucei BSF cells, though with distinct patterns of H2O2 sensitivity [38]. Analysis of the T. cruzi epimastigote mutants revealed that loss of both msh2 alleles also resulted in increased resistance to H2O2 in these parasite, though loss of one msh2 allele resulted in increased sensitivity to 75 μM H2O2, which is distinct from T. brucei msh2+/- mutants [38] and consistent with previous findings [37] (Fig 3B). Taken together, the enhanced survival of PCF T. brucei msh2 null mutants to H2O2 exposure suggests a life cycle difference in the consequence of this mutation relative to BSF cells. Moreover, this unexpected phenotype is conserved in epimastigote T. cruzi msh2 null mutants.
Fig 3. MSH2 knockout mutants are more resistant to oxidative stress generated by H2O2.

(A) T. brucei wild type (WT), msh2+/-, msh2-/-, mlh1+/- and mlh1-/- procyclic form cells were grown in culture medium with 0 μM, 10 μM or 20 μM H2O2. Cell density was measured after 48 hours and plotted as the percentage survival of the H2O2 treated cells relative to untreated. (B) T. cruzi epimastigote WT, msh2+/- and msh2-/- cells were incubated with or without 75 μM H2O2 for 20 minutes in PBS 1x and then allowed to grow in LIT medium for 48 hours, after which cell viability was determined and plotted as percentage survival of the treated cells relative to untreated. Vertical lines show standard deviation. ***p<0.001, **p<0.01, *p<0.05: determined by one-way ANOVA with Bonferroni post-test of mutants relative to wild type cells; ns indicates no signifcant difference.
T. cruzi msh2 null mutants have increased survival in macrophages
To investigate the role of MSH2 in protecting T. cruzi against oxidative stress generated by host cells, we compared the capabilities of T. cruzi WT and msh2-/- mutants to infect two different cell types: Vero cells and mouse macrophages. Equal numbers of trypomastigotes derived from Vero cells infected with WT and msh2-/- mutants were added to cultures of the two cell types and the numbers of intracellular amastigotes were determined 2 days later by staining parasite nuclei with DAPI. As shown in Fig 4A, no difference in the number of intracellular amastigotes was observed in Vero cells infected with WT or Tcmsh2-/- parasites. However, infections with each of the three Tcmsh2-/- clonal cell lines resulted in a 3-fold increase in the number of intracellular amastigotes relative to infection with WT cells in cultivated mouse macrophages (Fig 4B). As macrophages generate ROS, through an infection-induced respiratory burst [6], while Vero cells do not, these infection data appear consistent with the increased resistance of T. cruzi msh2-/- null mutants to H2O2 treatment (see Fig 3B). The infection data also indicate that the increased capacity to survive exposure to oxidative stress is maintained after T. cruzi differentiates into other life cycle stages.
Fig 4. Assessment of T. cruzi msh2 knockout mutant infectivity in vitro.
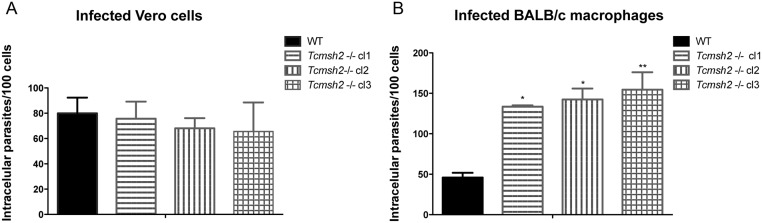
(A) T. cruzi trypomastigote cells released by Vero cells infected with either WT or with three cloned cell lines of Tcmsh2-/- mutants were counted and equal numbers were used to infect Vero cells attached to glass coverslips or (B) cultured intraperitoneal macrophages extracted from BALB/c mice. Graphs show the average number of intracellular parasites counted per 100 cells; vertical lines denote standard deviation. **p<0.01, *p<0.05: one-way ANOVA with Bonferroni post-test of knockout mutants relative to wild type.
T. brucei and T. cruzi msh2 mutants accumulate 8 oxoguanine in their DNA
The increased resistance to oxidative stress observed in T. brucei PCF cells and T. cruzi epimastigotes after MSH2 mutation could be a direct consequence of the loss of this MMR component, or could be due to indirect effects, such as a metabolic adaptation. To begin to address this, we determined the capacity of mutant cells to limit the accumulation of oxidized bases in their genome, which can arise from exposure to ROS generated by endogenous parasite metabolism or from the host. The primary DNA lesion generated by oxidative damage is 8-oxoG, which, if not removed by base-excision repair, can be recognized by MMR [26]. We therefore measured the levels of this oxidized base in the WT and msh2 mutant parasite genomes using avidin-conjugated FITC and measuring fluorescence [47]. As shown in Fig 5A and 5B, PCF T. brucei msh2+/- and msh2-/- mutants each displayed ~2 fold greater fluorescence in their nuclear DNA (nDNA) and kinetoplast DNA (kDNA) compared with WT cells. These fluorescence data indicate that the levels of 8oxoG were no greater in the T. brucei msh2-/- mutants than in the Tbmsh2+/- mutants, consistent with the observation that the two mutants display equivalent levels of resistance to H2O2 (Fig 3). In contrast, no difference was observed in the levels of fluorescence in the nDNA or kDNA of mlh1 mutants related to WT (Fig 5B), a distinction from T. brucei msh2 mutants again consistent with the separation of MMR functions observed when examining levels of resistance to H2O2. Increased levels of avidin-FITC fluorescence were also seen in the nDNA and kDNA of T. cruzi epimastigote msh2 mutants: 1.7 to 1.9 fold increase in fluorescence was seen in the nDNA and kDNA of both Tcmsh2+/- clones and both Tcmsh2-/- clones examined relative to WT (Fig 5C). These data suggest that loss or reduction of MSH2 expression (but not MLH1) in PCF T. brucei and in epimastigote T. cruzi results in the impairment of a pathway involved in the repair of DNA-directed oxidative damage, such as 8-oxoG. Thus, the increased resistance to H2O2 in the mutants is best explained by an indirect adaptation to cope with oxidative stress following MSH2 mutation.
Fig 5. 8-oxoguanine (8-oxoG) accumulation in MSH2 knockout cells.

FITC-avidin was used to estimate 8-oxoG levels based on the fluorescence intensity in the nuclear DNA (nDNA) and kinetoplast DNA (kDNA) of T. brucei procyclic forms and T. cruzi epimastigotes. (A) Representative images of FITC-avidin or DAPI stained T. brucei WT cells and Tbmsh2+/- or Tbmsh2-/- mutants are shown. (Bar = 1.9 μM) (B) Fluorescence intensity of FITC-avidin signals were quantified in the nDNA and kDNA using ImageJ software and plotted as arbitrary units; values shown are the average signal from 100 WT, Tbmsh2+/-, Tbmsh2-/-, Tbmlh1+/- or Tbmlh1-/- PCF cells; vertical lines show standard error (SEM). (C) FITC-avidin signal evaluated by the same process in T. cruzi epimastigote WT cells and in msh2+/- and msh2-/- knockout mutants. ***p<0.001: determined by one-way ANOVA with Bonferroni post-test of knockout mutants relative to wild type; ns indicates no signifcant difference.
Functional complementation confirms adaptation following loss of MSH2 in procyclic form T. brucei mutants
In order to test further if H2O2 resistance in msh2 mutants results from an adaptation process that occurred in insect life stages of T. brucei, we re-expressed MSH2 in both the BSF and PCF msh2-/- cells, using a previously described construct and conditions [21]. Integration of the MSH2 re-expression construct was confirmed by PCR (S6 Fig), and southern blot in BSF [21]. Both BSF and PCF msh2 mutants display increased tolerance to MNNG (Fig 2A), and re-expression of MSH2 in BSF msh2-/- mutants reverts this tolerance to the levels of msh2+/- mutants [21, 38]. As shown in Fig 6A, re-expressing MSH2 (Tbmsh2-/-/+) in the PCF msh2-/- mutants also resulted in levels of MNNG survival similar to msh2+/- cells. In addition, and as seen in T. brucei BSF msh2-/-/+ cells [21], PCF msh2-/-/+ cells no longer showed detectable microsatellite variation in clonal growth assays (S7 Fig). Taken together, these assays indicate that MMR function can be restored in T. brucei msh2 null mutants in both life cycle stages after re-introduction of MSH2 into the genome. In contrast, MSH2 re-expression had a different outcome for H2O2 sensitivity in the two life cycle stages. When T. brucei PCF msh2-/-/+ cells were grown in the presence of 10 μM or 20 μM H2O2 for 48 or 72 hrs, there was no significant difference in survival relative to the msh2-/- mutants, and survival was significantly different from the msh2+/- mutants (Fig 6B). However, in BSF T. brucei, the survival of the msh2-/-/+ cells (in this case after 48 hrs growth in either 100 or 200 μM H2O2) was indistinguishable from the msh2+/- cells and significantly greater than the msh2-/- mutants (Fig 6C). Thus, while re-expression of MSH2 in T. brucei msh2-/- null mutants was able to restore MMR function in both life cycle stages, the same re-expression was able to revert the increased sensitivity to H2O2 only in BSF msh2-/- mutants. In contrast, the increased tolerance to H2O2 observed in PCF after loss of MSH2 could not be reverted by re-expressing this gene in this life cycle form of T. brucei. This lack of MSH2 complementation is most simply explained by changes in expression or function of another factor(s) that allowed PCF MSH2 mutants to cope specifically with oxidative stress, though we cannot rule out the possibility of this specific phenotypic difference between BSF and PCF cells arising due to differing levels of MSH2 in the two life cycle stages and in the msh2-/-/+ re-expressers. As suggested by RNAi data (S8 Fig), the adaptation process that may have occurred during the cloning period needed to generate the msh2-/- mutants requires several generations. When MSH2 expression is abruptly inhibited by tetracycline induction of siRNA in T. brucei PCF cells, a discernible slowing of growth is observed as a consequence of the reduced msh2 mRNA expression.
Fig 6. Re-expression of MSH2 in T. brucei msh2 null mutants.

(A) Procyclic form (PCF) T. brucei (Tb) wild type (WT), Tbmsh2+/-, Tbmsh2-/- and Tbmsh2-/- cells in which MSH2 is re-expressed (Tbmsh2-/-/+) were grown in culture medium with 0 μM, 2.5 μM or 5 μM MNNG. Cell density was measured after 72 hours growth and is plotted as the percentage survival of the MNNG treated cells relative to untreated. (B) PCF WT, Tbmsh2+/-, Tbmsh2-/- and Tbmsh2-/-/+ cells were grown in culture medium with 0 μM, 10 μM or 20 μM H2O2 and cell density was determined 48 and 72 hours later; growth is shown percentage survival of the treated cells relative to untreated (C) Growth of wild type bloodstream form (BSF) T. brucei cells was compared to Tbmsh2+/-, Tbmsh2-/- and Tbmsh2-/-/+ BSF mutants in the presence of 100 μM or 200 μM H2O2 as described above; graph shows survival of the mutants after 48 hours growth plotting the density of the treated cells as a percentage of the untreated; vertical lines show standard deviation. ***p<0.001, **p<0.001: determined by one-way ANOVA with Bonferroni post-test of mutants relative to wild type; ns indicates no significant difference.
T. brucei and T. cruzi MSH2 display nuclear localization
In this study, and in previous work [37], we have shown that deletion of msh2 results in increased levels of 8-oxoG in both the nDNA and kDNA. Indeed, in BSF T. brucei the most pronounced cell cycle effect of MSH2 loss, which is exacerbated by H2O2 treatment, is the accumulation of cells with reduced amounts of kDNA (evaluated by DAPI staining) [37]. Because of these findings, and coupled with lack of a detectable kinetoplastid homologue of MSH1 (a MutS-like factor involved in mitochondrial genome repair, including oxidative damage repair) [53, 54], we hypothesized that MSH2 could provide a repair function for the trypanosomes’ mitochondrial (kDNA) genome. To test this hypothesis, we expressed T. brucei and T. cruzi MSH2 as C-terminal fusions with a 12X c-MYC epitope. For T. brucei, MSH2-myc was expressed from the endogenous locus in both BSF and PCF cells in which the other MSH2 allele was deleted; by evaluating MNNG sensitivity, we showed that the epitope tag did not affect the function of MSH2 in BSF cells (S9 Fig). As shown Fig 7A and 7B, immunolocalization of MSH2:myc with anti-c-MYC antiserum in T. brucei BSF and PCF cells revealed only a nuclear signal. The same nuclear localization was found in T. cruzi epimastigotes expressing MSH2:myc, as well as in amastigotes obtained after infection of Vero cells (Fig 7C and 7D). No changes in subcellular localization of the tagged MSH2 protein were observed in T. cruzi epimastigotes expressing MSH2:myc after exposure to H2O2 (Fig 7E) or other genotoxic agents, such as MNNG and cisplatin.
Fig 7. MSH2 displays nuclear localisation in T. brucei and T. cruzi.

Parasites expressing MSH2 C-terminally fused to 12 copies of the MYC epitope were used for immunolocalization of MSH2 using anti-MYC antiserum. (A) Transfected T. brucei bloodstream forms, (B) T. brucei procyclic forms, (C) T. cruzi epimastigotes, (D) T. cruzi intracellular amastigote cells inside infected Vero cels and (E) T. cruzi epimastigotes after 20 min treatment with 70μM H2O2 are shown. Representive images of DAPI staining of DNA, immunoflourescence with anti-MYC antiserum and phase contrast images of parasite cells are shown together with merged images. (Bars = 2.3 μm).
Discussion
In previous studies we have described the phenotypic effects of deleting both alleles of MSH2 or MLH1 in T. brucei BSF cells, and the effects of loss of a single allele of MSH2 in T. cruzi epimastigotes [21, 37]. Here, we provide several new insights into the function of MMR in African and American trypanosomes. First, we show that genetic ablation of MSH2 or MLH1 is possible in T. brucei PCF cells, as is MSH2 ablation in T. cruzi epimastigotes. In all cases, loss of MSH2 results in detectable impairment in MMR, as demonstrated by the increased tolerance to the alkylator MNNG, a phenotype specifically seen in MMR mutants in other eukaryotes [55] and in T. brucei BSF cells [21]. Second, we reveal a striking life cycle dependent difference in the effect of MSH2 ablation in T. brucei: in PCF cells loss of MSH2 results in increased tolerance to H2O2, whereas MSH2 loss results in increased sensitivity in BSF cells [38]. The same increased tolerance is also seen in T. cruzi epimastigote MSH2 mutants, where it impacts on the capacity of the parasite to grow in ROS-producing host macrophages. Importantly, altered H2O2 resistance was not observed when T. brucei BSF or PCF cells lacking MLH1 were examined. Finally, we provide evidence that the increased resistance to H2O2 in T. brucei after loss of MSH2 is due to a life cycle dependent adaptation in a facet of the cell that is distinct from MSH2, since re-expression of the protein restores MMR activity in PCF cells but does not alter the response to H2O2. In contrast, re-expression of MSH2 in T. brucei BSF msh2-/- mutants reverts both MMR impairment and H2O2 sensitivity.
Despite the complexity of phenotypes observed after MMR mutation, the findings indicate a common function of MSH2 in both trypanosomes: in addition to its role as a key component of MMR, MSH2 is also directly involved in the response to oxidative stress. Since loss of MLH1 did not result in increased resistance to H2O2 in PCF T. brucei, our new data confirms previous indications from BSF T. brucei [38] that although MSH2 acts in the parasite’s response to oxidative stress, such a role is unlikely to involve execution of MMR on oxidative lesions through MLH1-directed functions. Indeed, this is supported by the observation that PCF T. brucei MSH2 mutants, but not MLH1 mutants, display increased levels of 8-oxoG, a known form of oxidized base damage [56], in their nuclear and mitochondrial genomes. The same accumulation of 8-oxoG is also seen in T. cruzi msh2-/- and msh2+/- mutants [37], indicating that such a role for MSH2 is conserved in the two trypanosomes species. Most likely, a separation of function between MSH2 and MLH1 in this role will also be found in T. cruzi, though this has not to date been tested. The nature of MSH2’s role in responding the oxidative damage remains unclear, including whether or not it is limited to acting upon or repairing 8-oxoG. One possibility is that MSH2 acts in conjunction with MSH6 to repair 8-oxoG in DNA, a known activity of this heterodimer in other eukaryotes that can act independently of downstream MMR components [32]. However, we also observe accumulation of 8-oxoG in the kDNA of T. brucei and T. cruzi MSH2 mutants, consistent with previous descriptions of kDNA loss in T. brucei BSF msh2-/- mutants [37], and it is not clear that MSH2-6 could direct such a repair role in the mitochondrion. A broader role for MSH2 in the oxidative stress response might therefore need to be considered, perhaps related to findings in other eukaryotes that MSH2-6 can act to signal the presence of various forms of DNA damage, for instance through ATR [57–59]. It remains puzzling, however, that much of the signaling and wider activities attributed to MSH2-6 in other eukaryotes appear to reside in the extended N-terminal domain of MSH6 [59], which is notably truncated in trypanosomatid MSH6 orthologues [21]. Nonetheless, such a role for MSH2 perhaps most readily explains the adaptation we propose in T. brucei PCF, but not in BSF cells after loss of MSH2. PCF T. brucei have active mitochondrial metabolism, unlike BSF, and therefore greater levels of endogenous ROS, meaning that loss of MSH2 may be more detrimental in this life cycle stage [39]. T. cruzi epimastigotes also have an aerobic metabolism relying on mitochondrial respiration [60], and it is thus tempting to speculate that the same adaptation most likely occurred during the generation of msh2 null mutants in T. cruzi epimastigotes; though we have no direct evidence for such adaptation, the selection of cells lacking both alleles was notably problematic and necessitated an altered targeting transfection approach for removing the second allele.
To date, our analysis of the consequences of T. brucei MMR mutation have only been conducted in vitro, meaning it remains possible that the life cycle differences, and in particular the proposed PCF adaptation to MSH2 loss, are due to culture conditions rather than environmental and metabolic changes as the parasite cycles between host and vector. However, we provide direct evidence that the increased capacity to survive oxidative stress observed in T. cruzi msh2 -/- mutants may reflect challenges faced by this parasite during its life cycle. Although no difference in infectivity was observed between T. cruzi WT cells and msh2-/- mutants in Vero cells, macrophages infected with T. cruzi msh2-/- mutants contained almost 3-fold more intracellular amastigotes than macrophages infected with WT parasites. Due to their phagocytic nature, macrophages naturally produce ROS through the activation of NADPH oxidase during phagocytosis or by stimulation with a wide range of infective agents [61]. Although ROS are expected to be involved in pathogen elimination, increasing evidence suggests that ROS production actually could enhance T. cruzi infection in macrophages. For instance, it was recently proposed that oxidative stress mobilizes cellular iron and fuels T. cruzi infection [62, 63]. A role for ROS as a signalling molecule has also been highlighted in studies with Leishmania, where low ROS concentrations regulate proliferation and differentiation by modulating the activity of cellular targets by oxidation [64]. It seems reasonable to assume that MSH2 is part of the arsenal of functions that trypanosomatid parasites are equipped with to withstand and detect oxidative stress during their natural life cycles, and the absence of MSH2 can have wider effects than simply loss of MMR, as reflected in the adaptation we suggest in PCF T. brucei.
What is the nature of the adaptation following MSH2 mutation? Answering this question is complicated by the multiple potential pathways that might be affected. One possibility is that loss of MSH2 necessitates the up-regulation of an alternative repair pathway, such as base excision repair (BER). BER may be an attractive option because it is known to respond to oxidative stress and, indeed, a DNA glycosylase termed OGG1 is found in many organisms [65–67], including T. cruzi [68], that acts on 8-oxoG. However, increased BER activity seems incompatible with the increased levels of 8 oxoG we detect in T. brucei and T. cruzi MSH2 mutants. These data indicate that increased oxidative damage to nucleic acids occurs in the mutants and therefore suggests that the cells might adapt by limiting their exposure to oxidative stress in other ways. Trypanosomes have a complex set of enzymes involved in the oxidative stress response. One such pathway distinct from the mammalian host is based on trypanothione metabolism [69]. A paradox in our study is that we can only find evidence for a nuclear localization of MSH2, even after exposure to H2O2. Most oxidative damage is likely to occur in the trypanosome mitochondrion, as this is the major source of ROS. However, if MSH2 is limited to the nucleus, then how does loss of MSH2 result in (as noted above) increased levels of 8 oxoG in the kDNA? Moreover, if even some of the adaptation we see after loss of MSH2 affects trypanothione metabolism, then how is the loss of a nuclear protein communicated to these enzymes, which are thought to be mitochondrial or cytoplasmic [70–74]? One possibility is that C-terminal tagging impairs mitochondrial localization or MSH2 function. In BSF cells, we can rule this out because MSH2-myc was shown to be functional in the response to MNNG treatment (S9 Fig). However, the same analysis has not been conducted in PCF T. brucei, where perhaps mitochondrial localization is more important or pronounced. An alternative explanation is that trypanosome MSH2, perhaps in conjunction with MSH6, does indeed provide a role in signaling damage, similar to the activities detailed in other eukaryotes. Whether such signaling is limited to oxidative damage is unknown, but the signal provided by MSH2 may be communicated, in ways still to be determined, beyond the nucleus. Hence, if MSH2 is ablated, this element of the signaling cascade is lost. In the insect stages of at least T. brucei this loss is more deleterious than in the mammal, necessitating the selection for metabolic adaptation to deal with an altered oxidative stress response, with the consequence that the mutant becomes more resistant to ROS. The same hypothesis applies to T. cruzi since similar phenotypic changes were also seen in T. cruzi epimastigote MSH2 null mutants.
Clearly, further studies will be needed to understand how the parasites adapt to MSH2 mutation and it may be that metabolomic or proteomic approaches are the best strategies to take, in particular if no compensatory genetic changes follow MSH2 mutation. Nonetheless, this work reveals surprising complexity in the outcome of mutation to a core DNA repair pathway, suggesting interconnections with other cellular pathways. In addition, this study potentially reveals life cycle-dependent differences in MMR function. Loss of BRCA2 has also been shown to have different effects on genome stability in T. brucei BSF and PCF cells [75], but the extent to which the wide range of DNA repair pathways in trypanosomatids can vary in their activity or use through the life cycle remains little explored.
Supporting Information
(A) T.brucei (Tb) MSH2 or MLH1 genes were deleted by replacement of the coding sequence (CDS) of the gene with a DNA construct that consisted of Puromycin (PURR) or Blasticidin (BSDR) resistance genes flanked by processing sequences (providing signals for splicing and poliadenylation) derived from the αβ tubulin intergenic regions. Homologous integration was guided by 5’ and 3’ UTR sequences of either gene that surrounded the antibiotic resistance cassettes. The two alleles of T.cruzi MSH2 were each deleted by homologous recombination using different DNA constructs. The first allele was deleted by using 5’ and 3’ UTR sequences to guide integration of the CDS for the Hygromycin resistance gene (HygroR); in this case, splicing and polyadenylation of the resistance gene is assumed to be derived from endogenous MSH2 processing signals. For the second allele, the CDS was not deleted in its entirety, but instead it is was partly deleted by a construction containing a Neomycin resistance gene (NeoR) flanked by splicing and polyadenylation signal sequences derived from HX1 and GAPDH intergenic regions containing signals for splicing and poliadenylation. Integration of the resistance cassette here is guided by 5’ and 3’ MSH2 CDS sequence. (B) A PCR screen to identify double knockout clones (-/-) in which both alleles of TcMSH2 have been targeted was conducted with specific primers annealing in the HygroR or NeoR resistance genes together with common primers that recognised sequences that flanked the MSH2 fragment included in the the constructs. An agarose gel is shown of the PCR products generated using genomic DNA from T. cruzi wild type cells (WT), first allele mutant clones (cl1 and cl2 +/-) in which HygroR has been integrated, or double allele mutants (cl1 and cl2 -/-) in which both HygroR and NeoR have integrated. c- indicates a control reaction without any input genomic DNA.
(TIF)
PCR amplifications were carried out to verify the knockout of both TbMSH2 (A) or TbMLH1 alleles (B) using specific primers annealing in the target gene or the PURR or BSDR resistance genes together with common primers that recognised sequences that flanked the MSH2 or MLH1 fragment included in the the constructs. Arrows shown in the above panels denote regions of the wild type or mutated loci complementary to the different primers. PCR products generated using genomic DNA from T. brucei wild type cells (WT), PUR mutant clones (Tbmsh2/Tbmlh1 +/-) in which PURR has been integrated, BSD mutant clones (Tbmsh2/Tbmlh1 +/-) in which BSDR has been integrated or double allele mutants (Tbmsh2/Tbmlh1-/-) in which both PURR and BSDR have integrated are shown after separation on agarose gels.
(TIF)
WT T. cruzi epimastigote and two clonal cell lines with one (Tcmsh2+/-) or both msh2 alleles disrupted (Tcmsh2-/-) were diluted in LIT medium to 1x107 parasites.mL-1 and grown for additional 3 days. Cell densities were measured at 24 hours intervals; bars indicate standard deviation.
(TIF)
PCR amplification of the microsatellite locus JS2 from PCF WT, Tbmsh2+/-, Tbmsh2-/-, Tbmlh1+/-, and Tbmlh1-/- grown in the absence (-) and presence (+) of 20μM H2O2 for 48 hours and then cloned by limiting dilution in 96 well culture dishes. The JS2 locus was amplified from 10 clones from each cell line using primers JS2A and JS2B [21]. PCR products were separated on 3% agarose gesl; note, the two alleles of this JS2 locus have distinct sizes. PCR product from one of the wild type sub clones was also added in the first and last lane parallel to the mutants, for size comparison. Clones that show a difference in size relative to WT are indicated by an arrow; size markers are shown.
(TIF)
T. brucei wild type (WT), msh2+/-, msh2-/-, mlh1+/- and mlh1-/- procyclic form cells were grown in culture medium with 0 μM, 10 μM or 20 μM H2O2. Cell density was measured after 24 and 72 hours and plotted as the percentage survival of the H2O2 treated cells relative to untreated; vertical lines show standard deviation. ***p<0.001, **p<0.001, difference between mutants relative to wild type.
(TIF)
The upper diagram shows that the gene knockout strategy deletes the entire TbMSH2 ORF and indicates the position of primers (‘ORF’) used to test for the presence or absence of MSH2 ORF in genomic DNA of wild type (WT) cells, Tbmsh2+/- and Tbmsh2-/- mutants, and in three PCF Tbmsh2-/-/+ clones (1–3) in which the MSH2 ORF was reintegrated into the endogenous locus of the-/- mutant using a construct described previously [21]. The lower diagram shows two agarose gels, the uppermost displaying PCR products generated using the ORF primers and the different cells, relative to a control PCR (lower gel) in which the unaltered MSH2 5’ UTR was PCR-amplified (‘UTR’); size markers are shown (kb).
(TIF)
WT, Tbmsh2+/-, Tbmsh2-/- and Tbmsh2-/-/+ cells were grown in the absence (-) or presence (+) of 20 μM H2O2 for 48 hours and then cloned by limiting dilution in 96 well culture dishes. The JS2 locus was amplified from 10 clones from each cell line using primers JS2A and JS2B [21] and PCR products were separated on 3% agarose gel; note, the two alleles of the JS2 locus are distinct sizes and are marked by a dashed red line (‘c’ indicates a control PCR reaction run without genomic DNA). PCR product from one of the untreated wild type sub clones (wt-) was added in the first and last lane of the gels showing the mutants, for size comparison. Clones that show a difference in size relative to WT are indicated by an arrow; size markers are shown (bp).
(TIF)
(A) Growth curve of T. brucei PCF cells in which RNAi against MSH2 was induced by the presence of tetracycline (Tet+) (2 μg.ml-1) Cell density was measured every 24 hours over a period of 216 hours and compared between the Tet+ cells and PCF cells grown in the absence of tetracycline (Tet-).; the cultures were diluted to their starting density every 72 hours. The graph shows the average cell density of two independent clones, repeated twice; vertical lines indicate standard error. P<0.001 (B) mRNA levels of MSH2 and MLH1 were determined in one cloned T. brucei PCF cell line before and after RNAi induction by quantitative reverse-transcriptase PCR. Total RNA extracted from RNAi-induced (Tet+) and non-induced (Tet-) T. brucei PCF cells was reverse transcribed and quantified using GPI8 mRNA as an endogenous control. Error bars are standard deviations for four replicates. ***P<0.001
(TIF)
T. brucei BSF Tbmsh2+/- were transfected to express MSH2 C-terminally fused to a c-myc tag (Tbmsh2:myc) and the MMR efficiency was compared in these cells to T. brucei wild type (WT), Tbmsh2+/- and Tbmsh2-/- cells after treatment with MNNG. Cells were grown at a starting density of 1 x 105 cells.ml-1 in the presence of increasing concentrations of MNNG (from 0.39 μM—400 μM) in fluorescence-readable 96 well plates. After 48 hours, Alamar Blue was added to each well and fluorescence was measured after a further 24 hours of growth. IC50 values were determined and are shown as the mean of three experiments, with standard deviations indicated by vertical bars.
(TIF)
Acknowledgments
The authors would like to thank Wanderson daRocha for providing plasmid constructs and Craig Lapsley for technical support. This paper is dedicated to the memory of our colleague Juliano Leal de Paula, who performed sequence analyses of plasmid constructs.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
VGS was supported by a fellowship from CNPq [141073/2011-0] and a fellowship from Capes [8320/12-8] to work in the lab of RM, where the research was supported by the Wellcome Trust [083485] and the BBSRC [BB/K006495/1]. The Wellcome Trust Centre for Molecular Parasitology is supported by core funding from the Wellcome Trust [085349]. Work in SMRT’s lab is supported by grants from CNPq [305314/2013-0] and FAPEMIG [APQ-04132-10]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Chagas C. Nova tripanozomiaze humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. 1909.
- 2. Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375(9709):148–59. 10.1016/S0140-6736(09)60829-1 [DOI] [PubMed] [Google Scholar]
- 3. Ooi CP, Bastin P. More than meets the eye: understanding Trypanosoma brucei morphology in the tsetse. Front Cell Infect Microbiol. 2013;3:71 10.3389/fcimb.2013.00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Portman N, Gull K. Identification of paralogous life-cycle stage specific cytoskeletal proteins in the parasite Trypanosoma brucei. PLoS One. 2014;9(9):e106777 10.1371/journal.pone.0106777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walker DM, Oghumu S, Gupta G, McGwire BS, Drew ME, Satoskar AR. Mechanisms of cellular invasion by intracellular parasites. Cell Mol Life Sci. 2014;71(7):1245–63. 10.1007/s00018-013-1491-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piacenza L, Alvarez MN, Peluffo G, Radi R. Fighting the oxidative assault: the Trypanosoma cruzi journey to infection. Curr Opin Microbiol. 2009;12(4):415–21. 10.1016/j.mib.2009.06.011 [DOI] [PubMed] [Google Scholar]
- 7. Radi R. Peroxynitrite, a stealthy biological oxidant. J Biol Chem. 2013;288(37):26464–72. 10.1074/jbc.R113.472936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hao Z, Kasumba I, Aksoy S. Proventriculus (cardia) plays a crucial role in immunity in tsetse fly (Diptera: Glossinidiae). Insect Biochem Mol Biol. 2003;33(11):1155–64. [DOI] [PubMed] [Google Scholar]
- 9. Munks RJ, Sant'Anna MR, Grail W, Gibson W, Igglesden T, Yoshiyama M, et al. Antioxidant gene expression in the blood-feeding fly Glossina morsitans morsitans. Insect Mol Biol. 2005;14(5):483–91. [DOI] [PubMed] [Google Scholar]
- 10. Passos-Silva DG, Rajão MA, Nascimento de Aguiar PH, Vieira-da-Rocha JP, Machado CR, Furtado C. Overview of DNA Repair in Trypanosoma cruzi, Trypanosoma brucei, and Leishmania major. J Nucleic Acids. 2010;2010:840768 10.4061/2010/840768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Genois MM, Paquet ER, Laffitte MC, Maity R, Rodrigue A, Ouellette M, et al. DNA repair pathways in trypanosomatids: from DNA repair to drug resistance. Microbiol Mol Biol Rev. 2014;78(1):40–73. 10.1128/MMBR.00045-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peña-Diaz J, Jiricny J. Mammalian mismatch repair: error-free or error-prone? Trends Biochem Sci. 2012;37(5):206–14. 10.1016/j.tibs.2012.03.001 [DOI] [PubMed] [Google Scholar]
- 13. Clark N, Wu X, Her C. MutS Homologues hMSH4 and hMSH5: Genetic Variations, Functions, and Implications in Human Diseases. Curr Genomics. 2013;14(2):81–90. 10.2174/1389202911314020002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guarné A. The functions of MutL in mismatch repair: the power of multitasking. Prog Mol Biol Transl Sci. 2012;110:41–70. 10.1016/B978-0-12-387665-2.00003-1 [DOI] [PubMed] [Google Scholar]
- 15. Erie DA, Weninger KR. Single molecule studies of DNA mismatch repair. DNA Repair (Amst). 2014;20:71–81. 10.1016/j.dnarep.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pillon MC, Miller JH, Guarné A. The endonuclease domain of MutL interacts with the β sliding clamp. DNA Repair (Amst). 2011;10(1):87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glickman BW, Radman M. Escherichia coli mutator mutants deficient in methylation-instructed DNA mismatch correction. Proc Natl Acad Sci U S A. 1980;77(2):1063–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ji HP, Morales S, Welch K, Yuen C, Farnam K, Ford JM. Identification of a novel deletion mutant strain in Saccharomyces cerevisiae that results in a microsatellite instability phenotype. Biodiscovery. 2012;(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Denver DR, Feinberg S, Steding C, Durbin M, Lynch M. The relative roles of three DNA repair pathways in preventing Caenorhabditis elegans mutation accumulation. Genetics. 2006;174(1):57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Denver DR, Feinberg S, Estes S, Thomas WK, Lynch M. Mutation rates, spectra and hotspots in mismatch repair-deficient Caenorhabditis elegans. Genetics. 2005;170(1):107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bell JS, Harvey TI, Sims AM, McCulloch R. Characterization of components of the mismatch repair machinery in Trypanosoma brucei. Mol Microbiol. 2004;51(1):159–73.. [DOI] [PubMed] [Google Scholar]
- 22. Scherer SJ, Avdievich E, Edelmann W. Functional consequences of DNA mismatch repair missense mutations in murine models and their impact on cancer predisposition. Biochem Soc Trans. 2005;33(Pt 4):689–93. [DOI] [PubMed] [Google Scholar]
- 23. Bridge G, Rashid S, Martin SA. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers (Basel). 2014;6(3):1597–614. 10.3390/cancers6031597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iyama T, Wilson DM. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 2013;12(8):620–36. 10.1016/j.dnarep.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. [DOI] [PubMed] [Google Scholar]
- 26. de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82(2):321–30. [DOI] [PubMed] [Google Scholar]
- 27. Nowosielska A, Marinus MG. DNA mismatch repair-induced double-strand breaks. DNA Repair (Amst). 2008;7(1):48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240(4857):1302–9. [DOI] [PubMed] [Google Scholar]
- 29. Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313 (Pt 1):17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wyrzykowski J, Volkert MR. The Escherichia coli methyl-directed mismatch repair system repairs base pairs containing oxidative lesions. J Bacteriol. 2003;185(5):1701–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ni TT, Marsischky GT, Kolodner RD. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol Cell. 1999;4(3):439–44. [DOI] [PubMed] [Google Scholar]
- 32. Zlatanou A, Despras E, Braz-Petta T, Boubakour-Azzouz I, Pouvelle C, Stewart GS, et al. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and Pol η in response to oxidative DNA damage in human cells. Mol Cell. 2011;43(4):649–62. 10.1016/j.molcel.2011.06.023 [DOI] [PubMed] [Google Scholar]
- 33. Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309(5733):416–22. [DOI] [PubMed] [Google Scholar]
- 34. El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309(5733):409–15. [DOI] [PubMed] [Google Scholar]
- 35. Bell JS, McCulloch R. Mismatch repair regulates homologous recombination, but has little influence on antigenic variation, in Trypanosoma brucei. J Biol Chem. 2003;278(46):45182–8. [DOI] [PubMed] [Google Scholar]
- 36. Barnes RL, McCulloch R. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res. 2007;35(10):3478–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campos PC, Silva VG, Furtado C, Machado-Silva A, Darocha WD, Peloso EF, et al. Trypanosoma cruzi MSH2: Functional analyses on different parasite strains provide evidences for a role on the oxidative stress response. Mol Biochem Parasitol. 2011;176(1):8–16. 10.1016/j.molbiopara.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Machado-Silva A, Teixeira SM, Franco GR, Macedo AM, Pena SD, McCulloch R, et al. Mismatch repair in Trypanosoma brucei: heterologous expression of MSH2 from Trypanosoma cruzi provides new insights into the response to oxidative damage. Gene. 2008;411(1–2):19–26. 10.1016/j.gene.2008.01.010 [DOI] [PubMed] [Google Scholar]
- 39. Bringaud F, Rivière L, Coustou V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol. 2006;149(1):1–9. [DOI] [PubMed] [Google Scholar]
- 40. Paes LS, Suárez Mantilla B, Zimbres FM, Pral EM, Diogo de Melo P, Tahara EB, et al. Proline dehydrogenase regulates redox state and respiratory metabolism in Trypanosoma cruzi. PLoS One. 2013;8(7):e69419 10.1371/journal.pone.0069419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. CAMARGO EP. GROWTH AND DIFFERENTIATION IN TRYPANOSOMA CRUZI. I. ORIGIN OF METACYCLIC TRYPANOSOMES IN LIQUID MEDIA. Rev Inst Med Trop Sao Paulo. 1964;6:93–100. [PubMed] [Google Scholar]
- 42. DaRocha WD, Silva RA, Bartholomeu DC, Pires SF, Freitas JM, Macedo AM, et al. Expression of exogenous genes in Trypanosoma cruzi: improving vectors and electroporation protocols. Parasitology research. 2004;92(2):113–20. [DOI] [PubMed] [Google Scholar]
- 43. Wang Z, Morris JC, Drew ME, Englund PT. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J Biol Chem. 2000;275(51):40174–9. [DOI] [PubMed] [Google Scholar]
- 44. Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99(1):89–101. [DOI] [PubMed] [Google Scholar]
- 45. Teixeira SM, Kirchhoff LV, Donelson JE. Post-transcriptional elements regulating expression of mRNAs from the amastin/tuzin gene cluster of Trypanosoma cruzi. The Journal of biological chemistry. 1995;270(38):22586–94. [DOI] [PubMed] [Google Scholar]
- 46. Hope M, MacLeod A, Leech V, Melville S, Sasse J, Tait A, et al. Analysis of ploidy (in megabase chromosomes) in Trypanosoma brucei after genetic exchange. Mol Biochem Parasitol. 1999;104(1):1–9. [DOI] [PubMed] [Google Scholar]
- 47. Struthers L, Patel R, Clark J, Thomas S. Direct detection of 8-oxodeoxyguanosine and 8-oxoguanine by avidin and its analogues. Anal Biochem. 1998;255(1):20–31. [DOI] [PubMed] [Google Scholar]
- 48. Maynard S, de Souza-Pinto NC, Scheibye-Knudsen M, Bohr VA. Mitochondrial base excision repair assays. Methods. 2010;51(4):416–25. 10.1016/j.ymeth.2010.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Furtado C, Kunrath-Lima M, Rajão MA, Mendes IC, de Moura MB, Campos PC, et al. Functional characterization of 8-oxoguanine DNA glycosylase of Trypanosoma cruzi. PLoS One. 2012;7(8):e42484 10.1371/journal.pone.0042484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schamber-Reis BL, Nardelli S, Régis-Silva CG, Campos PC, Cerqueira PG, Lima SA, et al. DNA polymerase beta from Trypanosoma cruzi is involved in kinetoplast DNA replication and repair of oxidative lesions. Mol Biochem Parasitol. 2012;183(2):122–31. 10.1016/j.molbiopara.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 51. Alsford S, Horn D. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol Biochem Parasitol. 2008;161(1):76–9. 10.1016/j.molbiopara.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ben-Dov CP, Levin MJ, Vázquez MP. Analysis of the highly efficient pre-mRNA processing region HX1 of Trypanosoma cruzi. Mol Biochem Parasitol. 2005;140(1):97–105. [DOI] [PubMed] [Google Scholar]
- 53. Kaniak A, Dzierzbicki P, Rogowska AT, Malc E, Fikus M, Ciesla Z. Msh1p counteracts oxidative lesion-induced instability of mtDNA and stimulates mitochondrial recombination in Saccharomyces cerevisiae. DNA Repair (Amst). 2009;8(3):318–29. 10.1016/j.dnarep.2008.11.004 [DOI] [PubMed] [Google Scholar]
- 54. Pogorzala L, Mookerjee S, Sia EA. Evidence that msh1p plays multiple roles in mitochondrial base excision repair. Genetics. 2009;182(3):699–709. 10.1534/genetics.109.103796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19(12):1580–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Loon B, Markkanen E, Hübscher U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair (Amst). 2010;9(6):604–16. 10.1016/j.dnarep.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 57. Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22(4):501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Negureanu L, Salsbury FR. The molecular origin of the MMR-dependent apoptosis pathway from dynamics analysis of MutSα-DNA complexes. J Biomol Struct Dyn. 2012;30(3):347–61. 10.1080/07391102.2012.680034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Edelbrock MA, Kaliyaperumal S, Williams KJ. Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities. Mutat Res. 2013;743–744:53–66. 10.1016/j.mrfmmm.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gonçalves RL, Barreto RF, Polycarpo CR, Gadelha FR, Castro SL, Oliveira MF. A comparative assessment of mitochondrial function in epimastigotes and bloodstream trypomastigotes of Trypanosoma cruzi. J Bioenerg Biomembr. 2011;43(6):651–61. 10.1007/s10863-011-9398-8 [DOI] [PubMed] [Google Scholar]
- 61. Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166(12 Pt 2):S4–8. [DOI] [PubMed] [Google Scholar]
- 62. Paiva CN, Feijó DF, Dutra FF, Carneiro VC, Freitas GB, Alves LS, et al. Oxidative stress fuels Trypanosoma cruzi infection in mice. J Clin Invest. 2012;122(7):2531–42. 10.1172/JCI58525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Paiva CN, Bozza MT. Are reactive oxygen species always detrimental to pathogens? Antioxid Redox Signal. 2014;20(6):1000–37. 10.1089/ars.2013.5447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mittra B, Andrews NW. IRONy OF FATE: role of iron-mediated ROS in Leishmania differentiation. Trends in parasitology. 2013;29(10):489–96. Epub 2013/08/21. 10.1016/j.pt.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klungland A, Bjelland S. Oxidative damage to purines in DNA: role of mammalian Ogg1. DNA Repair (Amst). 2007;6(4):481–8. [DOI] [PubMed] [Google Scholar]
- 66. Sampath H. Oxidative DNA damage in disease—insights gained from base excision repair glycosylase-deficient mouse models. Environ Mol Mutagen. 2014;55(9):689–703. 10.1002/em.21886 [DOI] [PubMed] [Google Scholar]
- 67. Le Page F, Klungland A, Barnes DE, Sarasin A, Boiteux S. Transcription coupled repair of 8-oxoguanine in murine cells: the ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc Natl Acad Sci U S A. 2000;97(15):8397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Furtado C, Kunrath-Lima M, Rajao MA, Mendes IC, de Moura MB, Campos PC, et al. Functional characterization of 8-oxoguanine DNA glycosylase of Trypanosoma cruzi. PLoS One. 2012;7(8):e42484 10.1371/journal.pone.0042484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krauth-Siegel RL, Comini MA. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta. 2008;1780(11):1236–48. 10.1016/j.bbagen.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 70. Dufernez F, Yernaux C, Gerbod D, Noël C, Chauvenet M, Wintjens R, et al. The presence of four iron-containing superoxide dismutase isozymes in trypanosomatidae: characterization, subcellular localization, and phylogenetic origin in Trypanosoma brucei. Free Radic Biol Med. 2006;40(2):210–25. [DOI] [PubMed] [Google Scholar]
- 71. Nogueira FB, Krieger MA, Nirdé P, Goldenberg S, Romanha AJ, Murta SM. Increased expression of iron-containing superoxide dismutase-A (TcFeSOD-A) enzyme in Trypanosoma cruzi population with in vitro-induced resistance to benznidazole. Acta Trop. 2006;100(1–2):119–32. [DOI] [PubMed] [Google Scholar]
- 72. Wilkinson SR, Meyer DJ, Taylor MC, Bromley EV, Miles MA, Kelly JM. The Trypanosoma cruzi enzyme TcGPXI is a glycosomal peroxidase and can be linked to trypanothione reduction by glutathione or tryparedoxin. J Biol Chem. 2002;277(19):17062–71. [DOI] [PubMed] [Google Scholar]
- 73. Wilkinson SR, Temperton NJ, Mondragon A, Kelly JM. Distinct mitochondrial and cytosolic enzymes mediate trypanothione-dependent peroxide metabolism in Trypanosoma cruzi. J Biol Chem. 2000;275(11):8220–5. [DOI] [PubMed] [Google Scholar]
- 74. Wilkinson SR, Obado SO, Mauricio IL, Kelly JM. Trypanosoma cruzi expresses a plant-like ascorbate-dependent hemoperoxidase localized to the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2002;99(21):13453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Trenaman A, Hartley C, Prorocic M, Passos-Silva DG, van den Hoek M, Nechyporuk-Zloy V, et al. Trypanosoma brucei BRCA2 acts in a life cycle-specific genome stability process and dictates BRC repeat number-dependent RAD51 subnuclear dynamics. Nucleic Acids Res. 2013;41(2):943–60. 10.1093/nar/gks1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) T.brucei (Tb) MSH2 or MLH1 genes were deleted by replacement of the coding sequence (CDS) of the gene with a DNA construct that consisted of Puromycin (PURR) or Blasticidin (BSDR) resistance genes flanked by processing sequences (providing signals for splicing and poliadenylation) derived from the αβ tubulin intergenic regions. Homologous integration was guided by 5’ and 3’ UTR sequences of either gene that surrounded the antibiotic resistance cassettes. The two alleles of T.cruzi MSH2 were each deleted by homologous recombination using different DNA constructs. The first allele was deleted by using 5’ and 3’ UTR sequences to guide integration of the CDS for the Hygromycin resistance gene (HygroR); in this case, splicing and polyadenylation of the resistance gene is assumed to be derived from endogenous MSH2 processing signals. For the second allele, the CDS was not deleted in its entirety, but instead it is was partly deleted by a construction containing a Neomycin resistance gene (NeoR) flanked by splicing and polyadenylation signal sequences derived from HX1 and GAPDH intergenic regions containing signals for splicing and poliadenylation. Integration of the resistance cassette here is guided by 5’ and 3’ MSH2 CDS sequence. (B) A PCR screen to identify double knockout clones (-/-) in which both alleles of TcMSH2 have been targeted was conducted with specific primers annealing in the HygroR or NeoR resistance genes together with common primers that recognised sequences that flanked the MSH2 fragment included in the the constructs. An agarose gel is shown of the PCR products generated using genomic DNA from T. cruzi wild type cells (WT), first allele mutant clones (cl1 and cl2 +/-) in which HygroR has been integrated, or double allele mutants (cl1 and cl2 -/-) in which both HygroR and NeoR have integrated. c- indicates a control reaction without any input genomic DNA.
(TIF)
PCR amplifications were carried out to verify the knockout of both TbMSH2 (A) or TbMLH1 alleles (B) using specific primers annealing in the target gene or the PURR or BSDR resistance genes together with common primers that recognised sequences that flanked the MSH2 or MLH1 fragment included in the the constructs. Arrows shown in the above panels denote regions of the wild type or mutated loci complementary to the different primers. PCR products generated using genomic DNA from T. brucei wild type cells (WT), PUR mutant clones (Tbmsh2/Tbmlh1 +/-) in which PURR has been integrated, BSD mutant clones (Tbmsh2/Tbmlh1 +/-) in which BSDR has been integrated or double allele mutants (Tbmsh2/Tbmlh1-/-) in which both PURR and BSDR have integrated are shown after separation on agarose gels.
(TIF)
WT T. cruzi epimastigote and two clonal cell lines with one (Tcmsh2+/-) or both msh2 alleles disrupted (Tcmsh2-/-) were diluted in LIT medium to 1x107 parasites.mL-1 and grown for additional 3 days. Cell densities were measured at 24 hours intervals; bars indicate standard deviation.
(TIF)
PCR amplification of the microsatellite locus JS2 from PCF WT, Tbmsh2+/-, Tbmsh2-/-, Tbmlh1+/-, and Tbmlh1-/- grown in the absence (-) and presence (+) of 20μM H2O2 for 48 hours and then cloned by limiting dilution in 96 well culture dishes. The JS2 locus was amplified from 10 clones from each cell line using primers JS2A and JS2B [21]. PCR products were separated on 3% agarose gesl; note, the two alleles of this JS2 locus have distinct sizes. PCR product from one of the wild type sub clones was also added in the first and last lane parallel to the mutants, for size comparison. Clones that show a difference in size relative to WT are indicated by an arrow; size markers are shown.
(TIF)
T. brucei wild type (WT), msh2+/-, msh2-/-, mlh1+/- and mlh1-/- procyclic form cells were grown in culture medium with 0 μM, 10 μM or 20 μM H2O2. Cell density was measured after 24 and 72 hours and plotted as the percentage survival of the H2O2 treated cells relative to untreated; vertical lines show standard deviation. ***p<0.001, **p<0.001, difference between mutants relative to wild type.
(TIF)
The upper diagram shows that the gene knockout strategy deletes the entire TbMSH2 ORF and indicates the position of primers (‘ORF’) used to test for the presence or absence of MSH2 ORF in genomic DNA of wild type (WT) cells, Tbmsh2+/- and Tbmsh2-/- mutants, and in three PCF Tbmsh2-/-/+ clones (1–3) in which the MSH2 ORF was reintegrated into the endogenous locus of the-/- mutant using a construct described previously [21]. The lower diagram shows two agarose gels, the uppermost displaying PCR products generated using the ORF primers and the different cells, relative to a control PCR (lower gel) in which the unaltered MSH2 5’ UTR was PCR-amplified (‘UTR’); size markers are shown (kb).
(TIF)
WT, Tbmsh2+/-, Tbmsh2-/- and Tbmsh2-/-/+ cells were grown in the absence (-) or presence (+) of 20 μM H2O2 for 48 hours and then cloned by limiting dilution in 96 well culture dishes. The JS2 locus was amplified from 10 clones from each cell line using primers JS2A and JS2B [21] and PCR products were separated on 3% agarose gel; note, the two alleles of the JS2 locus are distinct sizes and are marked by a dashed red line (‘c’ indicates a control PCR reaction run without genomic DNA). PCR product from one of the untreated wild type sub clones (wt-) was added in the first and last lane of the gels showing the mutants, for size comparison. Clones that show a difference in size relative to WT are indicated by an arrow; size markers are shown (bp).
(TIF)
(A) Growth curve of T. brucei PCF cells in which RNAi against MSH2 was induced by the presence of tetracycline (Tet+) (2 μg.ml-1) Cell density was measured every 24 hours over a period of 216 hours and compared between the Tet+ cells and PCF cells grown in the absence of tetracycline (Tet-).; the cultures were diluted to their starting density every 72 hours. The graph shows the average cell density of two independent clones, repeated twice; vertical lines indicate standard error. P<0.001 (B) mRNA levels of MSH2 and MLH1 were determined in one cloned T. brucei PCF cell line before and after RNAi induction by quantitative reverse-transcriptase PCR. Total RNA extracted from RNAi-induced (Tet+) and non-induced (Tet-) T. brucei PCF cells was reverse transcribed and quantified using GPI8 mRNA as an endogenous control. Error bars are standard deviations for four replicates. ***P<0.001
(TIF)
T. brucei BSF Tbmsh2+/- were transfected to express MSH2 C-terminally fused to a c-myc tag (Tbmsh2:myc) and the MMR efficiency was compared in these cells to T. brucei wild type (WT), Tbmsh2+/- and Tbmsh2-/- cells after treatment with MNNG. Cells were grown at a starting density of 1 x 105 cells.ml-1 in the presence of increasing concentrations of MNNG (from 0.39 μM—400 μM) in fluorescence-readable 96 well plates. After 48 hours, Alamar Blue was added to each well and fluorescence was measured after a further 24 hours of growth. IC50 values were determined and are shown as the mean of three experiments, with standard deviations indicated by vertical bars.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.