

Abstract
Defects in the mitochondrial respiratory chain can induce a heterogeneous range of clinical and biochemical manifestations. Hepatic involvement includes acute fulminant hepatic failure, microvesicular steatosis, neonatal non-alloimmune haemochromatosis and cirrhosis. Recently pathogenic mutations in tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase (TRMU) gene (OMIM 610230) have been demonstrated to cause transient infantile liver failure (OMIM 613070). The human TRMU gene encodes a mitochondrial protein, 5-methylaminomethyl-2-thiouridylate methyltransferase, whose molecular function is that of mitochondrial tRNA modification.
We report an infant who presented with acute liver failure, in whom we observed hepatic copper intoxication and cirrhosis on liver biopsy. We postulate that the hepatic copper intoxication observed in our patient is most likely a secondary event associated with cholangiopathy. Periportal copper accumulation has been implicated in causing secondary mitochondrial dysfunction; the impact of copper accumulation in patients with TRMU mutations is unclear and warrants long-term clinical follow-up.
Introduction
The mitochondrial respiratory chain (MRC) is an essential component of normal cellular activity. Mitochondria are double-membraned intracellular organelles whose main function is to generate the high-energy phosphate molecule ATP via the process of oxidative phosphorylation (OXPHOS) (Lee and Sokol 2007). In order to achieve OXPHOS, the MRC is constructed into five separate complexes in which the subcomponents are encoded by both nuclear and mitochondrial DNA (mtDNA), with Complex II being the only complex without mtDNA contributions to its structure or maintenance.
The clinical features of a mitochondrial disorder are protean, in which “high-energy requirement” organs are more likely to produce a measurable disease burden. Hepatic involvement can occur as part of a single-organ or multi-organ clinical phenotype. The hepatic involvement of MRC disorders is common and demonstrates heterogeneity in clinical presentations and age of onset and is often fatal especially in the neonatal-infantile period. The hepatic features most commonly associated with MRC diseases include cholestasis, coagulopathy, cirrhosis, non-alloimmune neonatal haemochromatosis and acute fulminant hepatic failure (Lee and Sokol 2007; Fellman and Kotarsky 2011). Liver biopsy most commonly demonstrates hepatic steatosis.
In recent years nuclear-encoded genes that play a role in the maintenance and translational efficiency of mtDNA have been linked with hepatic phenotypes due to mitochondrial depletion. Nuclear-encoded genes associated with mitochondrial depletion syndromes include POLG (OMIM 174736), MPV17 (OMIM 137960), SUCLG1 (OMIM 611224) and DGUOK (OMIM 601465) (Fellman and Kotarsky 2011). Mutations in the tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase (TRMU) gene (OMIM 610230) have recently emerged as a cause of infantile-onset acute hepatic failure (Zeharia et al. 2009). The TRMU gene product functions as a mitochondrial tRNA modification and thus mitochondrial translation (Sasarman et al. 2011). An encouraging feature of this clinical presentation is that the hepatic failure can be reversible with appropriate supportive treatment. However, long-term neurological and hepatic outcomes are yet to be defined. Our case differs to the reported cases with TRMU mutations in that periportal hepatocyte copper loading was evident on liver biopsy.
Case Report
Our previously well female infant presented in extremis in hypovolaemic shock with acute hepatic failure at 4.5 months of age. There was no obvious precipitating infective illness. She had a gastrointestinal haemorrhage secondary to severe coagulopathy and was found to be profoundly hypoglycaemic and acidotic from a high lactate. She ultimately made a full systemic, hepatic and neurological recovery with intensive respiratory, circulatory and liver supportive therapy (N-acetylcysteine, carnitine and coenzyme Q10).
A primary mitochondrial defect was considered as part of the differential diagnosis in view of the abruptness and severity of clinical and biochemical presentation. Persistent mild abnormalities in liver function combined with the absence of an alternative diagnosis prompted the collection of liver and muscle biopsies at 8 months of age.
Significant abnormalities were detected on histological examination of the liver biopsy, most notably copper loading. Copper staining with rhodanine was performed revealing conspicuous copper and copper binding protein deposition in hepatocytes in a periportal distribution with no evidence of Mallory hyaline deposits (Fig. 1). Other prominent histological findings included portal tract fibrosis, cholangiopathy and small droplet steatosis (Figs. 2 and 3). Electron microscopy of the liver tissue revealed excessive numbers of mitochondria per cell, providing ultrastructural morphology correlation of oncocytosis noted on light microscopy with microvesicular steatosis and normal mitochondrial morphology.
Fig. 1.
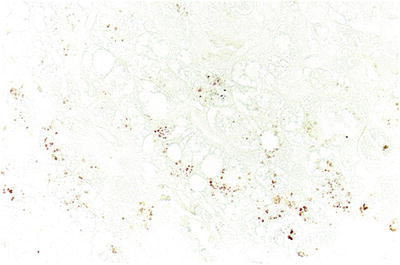
Rhodanine staining revealing conspicuous copper and copper binding protein deposition in hepatocytes in a periportal distribution with no evidence of Mallory hyaline deposits
Fig. 2.
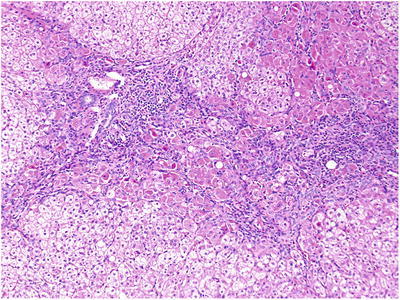
Portal tract fibrosis, cholangiopathy and small droplet steatosis
Fig. 3.
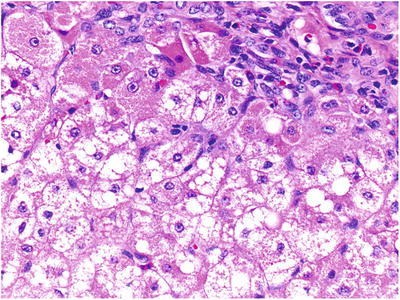
Portal tract fibrosis, cholangiopathy and small droplet steatosis
The patient had no dietary history to suggest excess copper ingestion and no wider clinical features associated with known genetic defects in copper transportation. Serum copper levels (16 μmol/L, reference range 13–26) and ceruloplasmin levels (1.75 μmol/L, reference range 1.9–3.41) were within normal limits.
Mitochondrial respiratory chain (MRC) enzyme analysis occurred via previously published methods (Bernier et al. 2002) with normal results in the muscle. Liver MRC chain activity levels of Complexes I, III and IV were low compared to citrate synthase, and Complex IV activity was low compared to Complex II activity (see Table 1). Mutation analysis for the common mitochondrial DNA mutations and the three common POLG mutations yielded normal results. The hepatic mtDNA/nDNA ratio was 1.02. TRMU sequencing proceeded via previously published methods (Zeharia et al. 2009) identifying compound heterozygosity for two pathogenic mutations c.835G>A and c.1037-1040 del TCAA.
Table 1.
Mitochondrial respiratory chain enzymes – liver
| Unit (nmol/min/mg) | Activity (ref range) | % Activity | % CS ratio | % CII ratio |
|---|---|---|---|---|
| Complex I | 12 (8–11) | 126 | 29 | 58 |
| Complex II | 134 (54–73) | 220 | 49 | |
| Complex III | 9.5(5.2–10.3) | 125 | 28 | 57 |
| Complex IV | 0.45 (0.5–0.9) | 63 | 14 | 29 |
| Citrate synthase | 122 (26–31) | 436 |
Having identified periportal copper staining and hepatic fibrosis, we instituted a hepatocellular carcinoma surveillance programme involving yearly liver ultrasounds and plasma alpha-fetoprotein levels. In the absence of a formal diagnosis of Wilson’s disease, specific copper chelation was avoided, but it was thought prudent to commence zinc acetate to reduce enterocyte absorption of copper. Now 4 years later our patient continues to demonstrate normal neurological developmental and hepatic function. Liver synthetic function and transaminases, copper studies and regular hepatobiliary ultrasound scanning are normal.
Discussion
The aetiology of acute infantile hepatic failure is heterogeneous in nature, and mitochondrial respiratory chain defects are potentially under-recognised. Diagnosis is confounded by non-specific clinical phenotypes, technical difficulties in obtaining and interpreting mitochondrial respiratory chain studies and molecular heterogeneity of mitochondrial defects. An accurate and timely diagnosis for the hepatic failure and of the molecular aetiology of mitochondrial respiratory chain defects is of significant practical importance for the hepatologist in terms of liver transplantation suitability. The reversible hepatic failure seen in patients with TRMU mutations underscores the importance of making this diagnosis and the value of aggressive supportive care. The clinical presentation and recovery of our infant are similar to reported cases associated with TRMU mutations (Gaignard et al. 2013; Schara et al. 2011; Uusimaa et al. 2011; Zeharia et al. 2009). Our infant presented with acute hepatic failure, had mild deficiency of MRC Complex IV on liver samples and has made a full and sustained clinical recovery. This case is unique in that the histopathological findings revealed excessive periportal copper deposition in the liver biopsy.
Copper is an essential trace element, and both excess and deficiency are associated with clinically relevant pathology. Once dietary copper is absorbed from the enterocyte, it is transported to the liver via the portal venous system bound to amino acids like histidine and serum proteins like albumin. Delivery of copper into the hepatocyte occurs via the copper transport protein, and once into the hepatocyte, most copper is bound to caeruloplasmin, the major transport protein for copper. Within the liver, there are four potential routes for copper distribution, (1) joining the copper/metallothionein pool, (2) binding to copper chaperone protein for delivery to zinc superoxide mutase, (3) attaching to cox17 for trafficking into the mitochondria for MRC Complex IV assembly and (4) trafficking to the trans-Golgi network (TGN) via human atox-1 homologue (Shim and Harris 2003). Copper plays an essential role in numerous crucial enzymatic pathways including neurotransmitter production, lysyl oxidase cross-linkage of collagen, free radical savaging via superoxide dismutase and metallothioneins and formation of MRC Complex IV and clotting factors (Corkins 2011). Genetic diseases of copper excretion and intracellular transport are associated with varied clinical phenotypes. These genetic diseases are associated with the TGN transportation of copper and are Menkes disease (MD) (OMIM 309400) and Wilson’s disease (WD) (OMIM 277900).
Under normal physiologic conditions 80% of copper is excreted via the biliary system attached to bile acids and 20% via the urine (Corkins 2011). Diseases associated with copper excretion are caused by a primary genetic defect in the excretion pathway, such as those associated with the TGN or by the process of biliary obstruction (Corkins 2011; Elmes et al. 1989; Evans et al. 1980; Goldfischer et al. 1980; Prohaska 2008). The periportal location of copper staining in our patient is more in keeping with being secondary to altered biliary excretion rather than from dietary copper excess as seen in Indian childhood cirrhosis (Pankit and Bhave 2002), endemic Tyrolean infantile cirrhosis (Pankit and Bhave 2002) or a TGN-related copper transport defect in which the copper distribution tends to be pan-lobar (Goldfischer et al. 1980).
Our case raises unanswered questions regarding the role of copper in the pathogenicity of liver failure secondary to patients with TRMU mutations. The most likely reason for the periportal copper staining in our patient is secondary to abrupt biliary cholestasis secondary to severe mitochondrial impairment. To our knowledge, the only reported functions of the TRMU gene product have been of mitochondrial 2-thiolation of the wobble U in tRNAlys, tRNAGlu and tRNAGln (Sasarman et al. 2011). A cytosolic role, especially one associated with intracellular copper trafficking, has not been postulated. MRC Complex IV is a critical component of the OXPHOS pathway in which the catalytic core contains three copper atoms (Horn and Barrientos 2008; Mehta et al. 2006). The TRMU gene product has not been postulated to have a role as a cytosolic or an intra-mitochondrial copper chaperone molecule or in that of MRC Complex IV assembly. However, MRC Complex IV deficiency has been described in patients with TRMU mutations including our case (Gaignard et al. 2013; Schara et al. 2011; Uusimaa et al. 2011; Zeharia et al. 2009). Hepatic copper deposition does not equate to systemic copper excess, as demonstrated in our patient. Cases of symptomatic copper deficiency have been described in patients with chronic cholestasis when copper has been removed from total parenteral nutrition supplements (Corkins 2011), highlighting the importance of copper for normal cellular function.
Oxidative stress is implicated in the pathogenesis and progression of specific liver diseases including biliary cirrhosis (Sastre et al. 2007; Tiao et al. 2009). In chronic cholestasis liver mitochondria have demonstrated increased H2O2 production and GSH depletion and oxidation (Sastre et al. 2007). Mitochondrial oxidative stress is a precursor to apoptosis. Copper excretion from the TGN involves interaction between MURR1/COMMD1 and the X-linked inhibitor of apoptosis protein (XIAP), which is a potent suppressor of apoptosis that directly inhibits specific members of the caspase family of cysteine proteases (Burstein et al. 2004). XIAP levels are greatly reduced by intracellular copper accumulation in Wilson’s disease and other copper toxicosis disorders (Mufti et al. 2006, 2007). Elevated copper levels result in a profound, reversible conformational change in XIAP, which accelerates degradation and significantly decreases the ability of XIAP to inhibit caspase-3 (Mufti et al. 2006, 2007). The observation of periportal copper accumulation in our patient is likely to be secondary to cholestasis; however, copper accumulation has been associated with the initiation of apoptosis via XIAP and mitochondrial oxidative stress. The TRMU gene product functions in the mitochondrial matrix; what is unclear from our case is if the copper accumulation will induce long-term secondary mitochondrial oxidative stress in an already abnormal mitochondrial system.
In conclusion, this case highlights the importance of recognising TRMU mutations as a cause of reversible, transient liver failure in infants and provides some insight into the potential interaction of mitochondrial disorders and intrahepatic copper accumulation. As the long-term outcome from liver failure in infants with TRMU mutations, in particular those with copper accumulation, has yet to be defined, our case also highlights the need for long-term follow-up of these patients.
Acknowledgements
The authors thank Professor Alex Kinsley (King’s College Hospital, London) for kindly reviewing the histopathological slides and Professor David Thorburn (MCRI, Melbourne) for performing the MRC studies.
Compliance with Ethics Guidelines
Conflict of Interest
Zubin Grover, Pete Lewindon, Andrew Clousten, Avraham Shaag, Orly Elpeleg and David Coman declare that they have no conflicts of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Author Contributions
Dr. Zubin Grover was a paediatric gastroenterologist involved in patient care and the manuscript development.
A/Prof. Lewindon was a paediatric gastroenterologist involved in patient care and the manuscript development.
Dr. Andrew Clousten is a histopathologist who reports the liver biopsy histology and has been involved in the manuscript development.
Drs. Orly Elpeleg and Avrahm Shaag performed the TRMU gene sequencing and assisted in the manuscript development.
A/Prof. David Coman is a metabolic physician and the patient’s primary care giver and has coordinated the manuscript development and design as the senior author.
Footnotes
Competing interests: None declared
Contributor Information
D. Coman, Email: david.coman@health.qld.gov.au
Collaborators: Johannes Zschocke
References
- Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59(9):1406–1411. doi: 10.1212/01.WNL.0000033795.17156.00. [DOI] [PubMed] [Google Scholar]
- Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, Wijmenga C, Brewer GJ, Nabel GJ, Duckett CS. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004;23(1):244–254. doi: 10.1038/sj.emboj.7600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkins MR. Copper metabolism and pediatric cholestasis. Curr Opin Clin Nutr Metab Care. 2011;14(6):642–646. doi: 10.1097/MCO.0b013e32834b2b1b. [DOI] [PubMed] [Google Scholar]
- Elmes ME, Clarkson JP, Mahy NJ, Jasani B. Metallothionein and copper in liver disease with copper retention–a histopathological study. J Pathol. 1989;158(2):131–137. doi: 10.1002/path.1711580208. [DOI] [PubMed] [Google Scholar]
- Evans J, Newman SP, Sherlock S. Observations on copper associated protein in childhood liver disease. Gut. 1980;21(11):970–976. doi: 10.1136/gut.21.11.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellman V, Kotarsky H. Mitochondrial hepatopathies in the newborn period. Semin Fetal Neonatal Med. 2011;16(4):222–228. doi: 10.1016/j.siny.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Gaignard P, Gonzales E, Ackermann O, Labrune P, Correia I, Therond P, Jacquemin E, Slama A. Mitochondrial infantile liver disease due to TRMU gene mutations: three new cases. JIMD Rep. 2013;11:117–123. doi: 10.1007/8904_2013_230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfischer S, Popper H, Sternlieb I. The significance of variations in the distribution of copper in liver disease. Am J Pathol. 1980;99(3):715–730. [PMC free article] [PubMed] [Google Scholar]
- Horn D, Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life. 2008;60(7):421–429. doi: 10.1002/iub.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, Sokol RJ. Liver disease in mitochondrial disorders. Semin Liver Dis. 2007;27(3):259–273. doi: 10.1055/s-2007-985071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Templeton DM, O’Brien PJ. Mitochondrial involvement in genetically determined transition metal toxicity II. Copper toxicity. Chem Biol Interact. 2006;163(1–2):77–85. doi: 10.1016/j.cbi.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Mufti AR, Burstein E, Csomos RA, Graf PC, Wilkinson JC, Dick RD, Challa M, Son JK, Bratton SB, Su GL, et al. XIAP Is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol Cell. 2006;21(6):775–785. doi: 10.1016/j.molcel.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Mufti AR, Burstein E, Duckett CS. XIAP: cell death regulation meets copper homeostasis. Arch Biochem Biophys. 2007;463(2):168–174. doi: 10.1016/j.abb.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankit AN, Bhave SA. Copper metabolic defects and liver disease: environmental aspects. J Gastroenterol Hepatol. 2002;17(Suppl 3):S403–S407. doi: 10.1046/j.1440-1746.17.s3.35.x. [DOI] [PubMed] [Google Scholar]
- Prohaska JR. Role of copper transporters in copper homeostasis. Am J Clin Nutr. 2008;88(3):826S–829S. doi: 10.1093/ajcn/88.3.826S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F, Antonicka H, Horvath R, Shoubridge EA. The 2-thiouridylase function of the human MTU1 (TRMU) enzyme is dispensable for mitochondrial translation. Hum Mol Genet. 2011;20(23):4634–4643. doi: 10.1093/hmg/ddr397. [DOI] [PubMed] [Google Scholar]
- Sastre J, Serviddio G, Pereda J, Minana JB, Arduini A, Vendemiale G, Poli G, Pallardo FV, Vina J. Mitochondrial function in liver disease. Front Biosci. 2007;12:1200–1209. doi: 10.2741/2138. [DOI] [PubMed] [Google Scholar]
- Schara U, von Kleist-Retzow JC, Lainka E, Gerner P, Pyle A, Smith PM, Lochmuller H, Czermin B, Abicht A, Holinski-Feder E, et al. Acute liver failure with subsequent cirrhosis as the primary manifestation of TRMU mutations. J Inherit Metab Dis. 2011;34(1):197–201. doi: 10.1007/s10545-010-9250-z. [DOI] [PubMed] [Google Scholar]
- Shim H, Harris ZL. Genetic defects in copper metabolism. J Nutr. 2003;133(5 Suppl 1):1527S–1531S. doi: 10.1093/jn/133.5.1527S. [DOI] [PubMed] [Google Scholar]
- Tiao MM, Lin TK, Wang PW, Chen JB, Liou CW. The role of mitochondria in cholestatic liver injury. Chang Gung Med J. 2009;32(4):346–353. [PubMed] [Google Scholar]
- Uusimaa J, Jungbluth H, Fratter C, Crisponi G, Feng L, Zeviani M, Hughes I, Treacy EP, Birks J, Brown GK, et al. Reversible infantile respiratory chain deficiency is a unique, genetically heterogenous mitochondrial disease. J Med Genet. 2011;48(10):660–668. doi: 10.1136/jmg.2011.089995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeharia A, Shaag A, Pappo O, Mager-Heckel AM, Saada A, Beinat M, Karicheva O, Mandel H, Ofek N, Segel R, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009;85(3):401–407. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]