

Abstract
Background: Classical homocystinuria due to cystathionine β-synthase (CBS) deficiency (OMIM 236200) is a recessively inherited condition caused by mutations in the CBS gene. The founder mutation p.R336C accounts for almost all CBS deficiency in Qatar, affecting approximately 1 in 1,800 births, making it the most prevalent monogenic disease among the Qatari population. Untreated patients can have severe intellectual disability (ID), devastating multisystem complications and premature death. Current treatment is based on pharmacology therapy and life-long methionine-restricted diet, which is difficult to maintain particularly in late diagnosed individuals. Data on the neurodevelopmental and psychological impact of the disease on outcomes among Qatari patients are generally lacking and have not been studied.
Objectives: To examine the cognitive, educational and psychological outcomes of classical homocystinuria on Qatari patients.
Subjects and Methods: Thirty-two cases with classical homocystinuria and 25 sibling controls were recruited to evaluate the neurodevelopmental and cognitive outcomes. We reviewed the subjects’ medical record and collected pertinent clinical and educational data from parents. Stanford–Binet Intelligence Test (Arabic translation – 4th ed.) was used for cognitive (IQ) testing.
Results: The mean age for the subjects was 11.2 years (range 0.6–29) with 56% males. The majority of cases (93%) carried the mutation (p.R336C), and parental consanguinity was 84%. There were no differences between the two groups in the fine motor, expressive language, behavioural and visual skills. However, cases have much lower total IQ particularly in the domains of short memory, quantitative reasoning and visual–spatial domains. A significant number of adolescents and adult cases had medical co-morbidities as well as behavioural and emotional problems.
Conclusion: Individuals with classical homocystinuria have many developmental and cognitive difficulties with significant number of cases having learning disability and lower IQs (cf. sibling controls) with adolescents and adults more affected. Those diagnosed by newborn screening have better developmental and cognitive outcomes compared to late diagnosed cases. Psychological and psychiatric referrals should be part of the standard of care for those cases
Keywords: Cognitive, Homocystinuria, Neurodevelopmental, Newborn screening, Outcomes, Qatar
Introduction
Classical homocystinuria (CHU) due to CBS deficiency (OMIM 236200) is caused by mutations in the CBS gene. Although this disease is the most frequent disorder of sulphur and methionine metabolism, it is still considered a rare inborn error of metabolism with estimated prevalence between 1/20,000 and 344,000. However, in Qatar, its prevalence is extremely high, the highest in the world, of approximately 1/1,800 births (Mudd et al. 1964; Mudd 1985; Naughten et al. 1998; El-Said et al. 2006; Zschocke et al. 2009; Gan-Schreier et al. 2010; Yap 2012). Clinically, CBS-deficient patients may present with ectopia lentis, osteoporosis, and skeletal deformities often associated with Marfanoid features, but most importantly with intellectual disability and life-threatening complications of the vascular system leading to premature death (Mudd 1985; Mudd et al. 1985; Yap 2012). Therapy of homocystinuria usually includes administration of high doses of pyridoxine, the cofactor of CBS; however, only less than 50% of affected subjects show a substantial plasma homocysteine reduction (Mudd 1985; Lindner et al. 2007; Yap 2012). The homocystinuria patients in Qatar are known to be pyridoxine nonresponsive with more severe phenotype and complications (El-Said et al. 2006; Zschocke et al. 2009; Gan-Schreier et al. 2010).
Human CBS is a pyridoxal 5′-phosphate (PLP)-containing enzyme (E.C. 4.2.1.22), and it is the enzyme involved in the first step of the methionine transsulphuration pathway, where Hcy is combined with serine forming cystathionine, which in turn is the precursor of cysteine. Consequently, an impaired CBS activity leads to hyperhomocysteinaemia and homocystinuria, hypermethioninaemia and hypocysteinaemia (Mudd 1985; Yap 2012). The human CBS gene, mapped to chromosome 21q21-3, encompasses 30 kb of genomic DNA, and a total of 23 exons have been reported (Lindner et al. 2007). However, only exons 1–14 and 16 encode the CBS protein. More than 140 different disease-causing mutations have been identified in the CBS gene, and although scattered along the entire gene length, mutations in exons 3, 8 and 10 are most prevalent (Schiff and Blom 2012). Most of these mutations are missense, and only three nonsense mutations have been reported. In addition, several splicing mutations as well as deletions and insertion mutations have been found (Kraus et al. 1999; Schiff and Blom 2012).
The founder mutation p.R336C (c.1006C>T) can cause a severe B6 nonresponsive phenotype. If untreated, homozygous patients for this mutation (the usual situation in Qatar) are clinically affected with severe intellectual disability as the most prominent feature, in addition to devastating multisystem complications and premature death (El-Said et al. 2006; Zschocke et al. 2009; Gan-Schreier et al. 2010). Furthermore, p.R336C mutation exhibited an activity lower than 4% of the wild-type protein (Urreizti et al. 2006). This disease imposes a huge clinical, financial and psychosocial overall burden on the population of Qatar (Gan-Schreier et al. 2010).
Treatment of patients with CHU whether diagnosed early by newborn screening or later is based on a combined life-long methionine-restricted diet and pharmacological therapy. Dietary treatment includes low methionine diet with cysteine enriched methionine free amino acid supplement. The use of dietary control and medications is an effective intervention to reduce the mean homocysteine and methionine levels. However, dietary control is rather difficult to maintain, in particular for late diagnosed patients and during puberty, adolescence and adulthood. Pharmacological therapy includes betaine, in addition to vitamin B6, vitamin B12 and folate supplementation. In our cohort of cases with CHU, the overall disease control (measured by homocysteine and methionine levels) is significantly better among those diagnosed through newborn screening than those diagnosed clinically. This is attributed mainly to poor compliance with both diet and medications (Table 1).
Table 1.
Mean homocysteine and methionine pre- and posttreatment levels among CHU cases diagnosed by NBS and those of late diagnosis
| Diagnosed by NBS | Late diagnosis | |
|---|---|---|
| Mean homocysteine level (umol/L) | ||
| Pretreatment | 141.6 | 130.5 |
| Posttreatment | 40.8 | 115.2 |
| Mean methionine level (umol/L) | ||
| Pretreatment | 468.9 | 770.6 |
| Posttreatment | 256.1 | 928.2 |
In this study, we prospectively assessed the neurodevelopmental and cognitive outcomes in 32 children and adults with CHU and compared them with 25 sibling controls using standardized assessments.
Methodology
Between April 2011 and March 2013, a total of 32 cases with CHU who attended the Metabolic Clinic at Hamad Medical Corporation (HMC), Doha, Qatar, were invited to participate in the study. We also recruited one (the immediate older or younger) unaffected sibling of the same sex to serve as control for the subject. Sibling controls were used as the neurodevelopmental and cognitive outcomes assessed in this study may be affected by family circumstances and genetic makeup.
The diagnosis was achieved in late diagnosis/symptomatic patients by measuring plasma total homocysteine. The diagnosis is further supported by simultaneously high or borderline high plasma methionine. Confirmation of the diagnosis is by mutation analysis of the CBS gene (common Qatari mutations). For newborn babies (identified by NBS), we measure total homocysteine in dried blood spots by LC/MS/MS. Then, in positive cases, confirmation of the diagnosis is achieved by measuring plasma total homocysteine, plasma methionine and mutation analysis of the CBS gene.
A detailed medical assessment including history, physical examination and medical records review was conducted by the study investigators to collect pertinent data on demographic factors, medical, laboratory, neurodevelopmental, psychosocial and educational attainment. Direct questioning of parents and or adult cases was used to collect pertinent data that was not available in the medical records.
Cognitive assessment was conducted using Stanford–Binet Intelligence Scale – 4th ed. (Arabic translation) by a trained clinical psychologist. The test yields a total mental processing composite based on subtests that measure verbal reasoning, quantitative reasoning, visual–spatial processing, working memory and total IQ. For children under 3 years of age at testing, cognitive assessment was not done.
T-test or Fisher’s exact test was used for normally distributed data, and for non-normally distributed data, non-parametric tests were used. Matched and unmatched analysis for all variables was used to adjust for the use of sibling controls.
Differences in baseline characteristics were assessed using McNemar’s test for comparing dichotomous variables and chi-square and Fisher’s test for higher-order categorical variables. Two-tailed significance test was used with a p-value of <0.05 as significance level. Statistical analyses were conducted in STATA 13.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
Results
Thirty-two cases and 25 sibling controls were available for evaluation. The mean age for the study’s subjects (cases and controls) was 11.2 years (range 0.6–29) and 32 (56%) were males. The mean age at diagnosis of all cases was 66 months (range 1–240), and 9 (28%) were diagnosed in the first month of life through the national expanded newborn screening programme that was established in early 2006. The mean age of the 14 (44%) cases that were diagnosed prior to 2006 was 92 months (range 14–240) compared with a mean age of 100 months (range 56–142) for those diagnosed on or after 2006 excluding cases diagnosed by newborn screening (p = NS).
Parental consanguinity was positive in 27 (84%) of the cases. Interestingly, all cases carry the founder mutation p.R336C, except one child who had the G347S mutation.
Developmental Domains
In 25 (78%) of cases, the parents reported one or more concerns about their children’s development. Table 2 shows developmental difficulties among cases and controls. Parents reported more difficulties in the domains of fine motor skills and coordination, and expressive language, but only the vision and behavioural/emotional problems were statistically significant when compared with their sibling controls. All cases were able to walk independently; however, three cases needed help to climb stairs or to run. None of them had cerebrovascular insult; however, one child had white matter changes in his brain MRI. There were no significant differences in developmental domains among boys and girls in the study.
Table 2.
Developmental difficulties among cases and controls as reported by parents
| Domain | Cases (n = 32) | Controls (n = 25) | p-value |
|---|---|---|---|
| Gross motor skills | 3 (9%) | 0 (0%) | NS |
| Fine motor skills + coordination | 4 (12.5%) | 0 (0%) | NS |
| Receptive language | 7 (22%) | 3 (12%) | NS |
| Expressive language | 11 (34%) | 3 (12%) | NS |
| Self-care skills | 2 (6.2%) | 0 (0%) | NS |
| Behavioural/emotional | 13 (40.6%) | 1 (4%) | 0.002 |
| Vision problems | 18 (56%) | 3 (12%) | < 0.001 |
| Hearing problems | 0 (0%) | 1 (4%) | NS |
NS not significant
There were no parental concerns regarding daily living activities (eating, personal care skills and independent use of toilet), but four cases aged 10, 13, 22 and 29 years have major coordination and handwriting difficulty skills.
Seven cases had both receptive and expressive language difficulties, and in four cases expressive language difficulties were only reported. At the time of enrolment, two children were getting private speech therapy sessions.
A significant number (13/32) of cases were reported by their parents to have behavioural and emotional difficulties. The mean age for those cases was 14.6 years (range 9–29) and six were females. Eight (61%) of the cases had a mean total IQ < 70. According to the families, none of their children had a formal psychiatric assessment or an explicit diagnosis.
Over half of the cases reported to regularly attending the ophthalmology clinics because of their visual difficulties and two needed eye operation. Four cases were labelled as visually impaired and attend a special school for children with visual impairment.
Education Attainment
Table 3 shows the educational attainment history as reported by parents. At the time of enrolment, a significant number of cases were attending or previously attended special needs school or needing extra help in the classroom to access the mainstream education curriculum. A number of parents reported that their child’s school performance was average or below what is expected especially in arithmetic and language. Overall, cases and controls had good school attendance record.
Table 3.
Education variables among cases and controls as reported by parents
| Variable | Cases (n = 27) | Controls (n = 21) | p-value |
|---|---|---|---|
| Attends education | 26 (96.3%) | 20 (95.2%) | NS |
| Attends special needs school or centre | 6 (22%) | 0 (0%) | 0.028 |
| School performance rated by parents as average or below | 16 (59%) | 9 (43%) | NS |
| Extra help in class | 10 (37%) | 3 (14.3%) | NS |
| > 3 days off school in 3 months | 4 (15%) | 2 (9.5%) | NS |
NS not significant
Cognitive Function
The cognitive data was obtained from 28 cases and 24 controls. Cognitive testing was not available in the remainder of cases and controls due to young age and refusal to conduct the test. Total IQ score as well as three of the four subset scales (quantitative reasoning, visual–spatial processing, working memory) for the Stanford–Binet Intelligence Scale – 4th ed (Arabic translation) were significantly lower among cases compared with controls (P < 0.001) (Fig. 1).
Fig. 1.
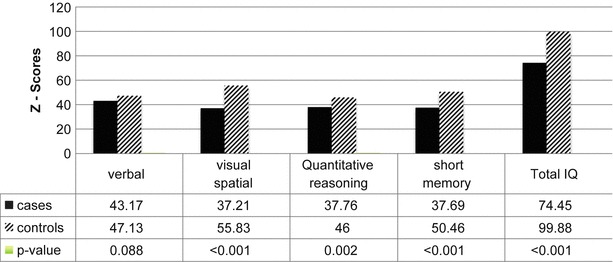
Total and subscale IQ scores among cases and controls
Figure 2 shows the classification of cognitive levels according to the total IQ score. Unlike the controls whom the majority (19/24) were classified to have average or high average cognitive level, only 9 (32%) cases had total IQ in the average or high average (p < 0.001). Moreover, more than a third of the cases (10/28) had total IQ < 70 and were classified as mental retardation but none of the controls (p = 0.001). There were no significant differences in IQ among boys and girls in the study.
Fig. 2.
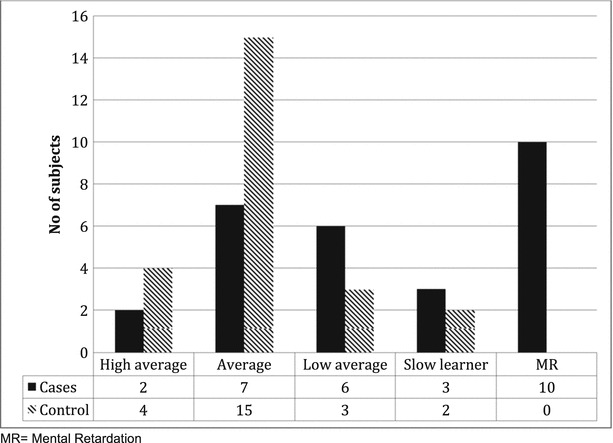
Classification of IQ scores among cases and controls
Newborn Screening and Outcomes
Table 4 shows the developmental and cognitive outcomes among cases stratified by age at diagnosis. Nine cases were diagnosed within the first month of life by newborn screening, whereas the remainder of the cases were diagnosed at different ages according to their presentation. None of the nine cases had visual problems compared with 18 (78%) in the late diagnosed group. This is a significant difference between the two groups. One child in the newborn screening group has receptive and expressive language difficulties at the time of enrolment; however, his total IQ was 116 (high average).
Table 4.
Age at diagnosis and developmental, educational and cognitive outcomes among the cases as reported by parents
| Variable | Newborn screening (n = 9) | Late detection (n = 23) | p-value |
|---|---|---|---|
| Mean (SD) age at enrolment | 4.2 (1.7) | 14.0 (6.2) | <0.001 |
| Vision problems | 0 | 18 | <0.001 |
| Receptive language difficulties | 1 | 6 | NS |
| Expressive language difficulties | 1 | 10 | NS |
| Special school | 0 (n = 4) | 6 | NS |
| Extra help in class | 1 | 9 | NS |
| Mean (SD) total IQ | 101 (14.2) | 66 (19.8) | 0.002 |
NS not significant, SD standard deviation
The differences in language domains, attendance at special school and access to extra support in class were not statistically significant between those diagnosed soon after birth and cases detected/diagnosed later in life. Nonetheless, it is apparent that the “late detection” group had more reported difficulties. In addition, the late diagnosed group total mean IQ was significantly lower when compared with the newborn screening group (p < 0.001) (Table 4).
Discussion
We conducted a case–control study to report on the neurodevelopmental, educational and cognitive outcomes of cases with CHU. To our knowledge, only a limited number of studies have reported on these outcomes in subjects with CHU. We used siblings as control to adjust for genetic, environmental (protective or risk) factors and socioeconomic status that may affect the outcomes in question especially the IQ which could be highly heritable (Bailey and Revelle 1991; Devlin et al. 1997; Dickens and Flynn 2001; Turkheimer et al. 2003). The use of sibling controls in this study is not novel, as a number of previous studies have used siblings as controls possibly for the same reasons mentioned above (Koch et al. 1984; Eldridge et al. 1989; Yap et al. 2001).
In 2003, Qatar has established an expanded newborn screening (NBS) programme for a number of metabolic and endocrine conditions including CHU (by using methionine) (Lindner et al. 2007; Zschocke et al. 2009; Gan-Schreier et al. 2010). In 2006, NBS for CHU performed by measuring total homocysteine. The introduction of the screening programme has clearly helped in the early detection of newborn cases. All cases bar one were homozygous for the founder p.R336C mutation, and considering the high consanguinity rate among the families, this explains the high prevalence rate of CHU in Qatar (El-Said et al. 2006; Zschocke et al. 2009; Gan-Schreier et al. 2010). It is interesting that cases reported from a neighbouring country (Saudi Arabia) had different common mutations for CHU (Al-Essa et al. 1998).
Our study showed a large number of families had raised concerns about one or more domains of their child’s development, and clearly there was a trend of difficulties among the cases; nonetheless, the data did not show statistical differences between cases and controls in the gross or fine motor skills or the language domains. This could be attributed to the small sample size.
Forty percent of our cases were reported by their parents to have behavioural and emotional difficulties. Our study concurs with previous studies that reported similar or even higher rates of psychiatric/behavioural disorders among cases of homocystinuria (Abbott et al. 1987). Another significant finding is that all those cases bar one had total IQs that are low average or below 70. The association between psychiatric/behavioural problems and low IQ in CHU has been reported previously (Abbott et al. 1987). However, it is not clear from our study if those cases have behavioural/emotional issues secondary to their low cognitive abilities or they have primary behavioural/emotional problems that are independent of their IQ; particularly the parents have denied any formal diagnosis or follow-up of psychiatric illness among the cases. This may be explained by cultural factors or more family support particularly among female patients. A recent systematic review has reported difficulty in assigning causality association of metabolic disorders with psychiatric illnesses in adults (Bonnot et al. 2014).
Visual problems were very prevalent among our cases and clearly represent a long-term morbidity. However, it was reassuring that the majority of cases are regularly followed up and closely monitored in the Ophthalmology Clinic. A significant number of cases have low cognitive abilities as highlighted by their low total and subset IQs. The high prevalence of visual problems may have contributed further to their lower score. Low IQs in the subset of short-term memory and reasoning negatively impact on the child’s learning ability.
Our study is susceptible to biases common to case–control studies. It was not possible to blind the study team to the subject’s status. Another limitation is the relatively small sample size which could be the reason why many of the differences between cases and controls did not achieve statistical significance; however, CHU is still a rare condition and most of the studies that addressed the neurodevelopmental and cognitive outcomes have published similar number of subjects or even less (Yap et al. 2001; Weisfeld-Adams et al. 2013). Another important limitation is that we relied on parent/subject report and recall on some of the developmental and educational domains. It is not clear if those reports have over- or underestimated the prevalence of difficulties in those domains. Further studies with larger sample size and using assessment tools that have been translated and validated into Arabic language may help to control this limitation.
Conclusion
It is clear that current treatment for CHU in early-treated children, particularly in those detected by NBS, has eliminated severe intellectual and cognitive impairment. More support is needed for cases attending mainstream schools especially those with low cognitive abilities and those with visual problems as they may need extra support in class or individual educational plans that take into account their overall difficulties. Future research assessing intellectual and neurocognitive outcome in CHU will enhance the development of new treatment strategies. Long-term follow-up studies are needed to explore the behavioural/emotional difficulties especially among adolescents and adults with this condition. Psychological and/or psychiatric referrals should be part of continuing care of those cases especially for those with low cognitive (IQ) abilities who may be at greater risk.
Compliance with Ethics Guidelines
Haitham El Bashir, Lubna Dekair, Yasmeen Mahmoud and Tawfeg Ben-Omran declare that they have no conflict of interest. The study was approved by the Hamad Medical Corporation, Medical Research Center Ethics Committee (# 10263/10).
Haitham El Bashir: conceived the idea, wrote the study protocol and ethical approval, study design, data analysis, wrote the first draft of manuscript, study guarantor
Lubna Dekair: study design, recruitment, data collection, developmental assessment, critical review of manuscript
Yasmeen Mahmoud: study design, recruitment, data collection, cognitive assessment, critical review of manuscript
Tawfeg Ben-Omran: study design, recruitment of cases, data collection, critical review of manuscript
Funding
The study was funded by a research grant from the Medical Research Center, Hamad Medical Corporation, Doha, Qatar. The funding body had no role in the content of this article.
Footnotes
Competing interests: None declared
Contributor Information
Haitham El Bashir, Email: helbashir@hmc.org.qa.
Collaborators: Johannes Zschocke
References
- Abbott MH, Folstein SE, Abbey H, Pyeritz RE. Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am J Med Genet. 1987;26(4):959–969. doi: 10.1002/ajmg.1320260427. [DOI] [PubMed] [Google Scholar]
- Al-Essa M, Rashed M, Ozand PT. Saudi experience with classic homocystinuria. Ann Saudi Med. 1998;18(3):230–233. doi: 10.5144/0256-4947.1998.230. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Revelle W. Increased heritability for lower IQ levels? Behav Genet. 1991;21(4):397–404. doi: 10.1007/BF01065975. [DOI] [PubMed] [Google Scholar]
- Bonnot O, Klunemann HH, Sedel F, Tordjman S, Cohen D, Walterfang M. Diagnostic and treatment implications of psychosis secondary to treatable metabolic disorders in adults: a systematic review. Orphanet J Rare Dis. 2014;9:65. doi: 10.1186/1750-1172-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Daniels M, Roeder K. The heritability of IQ. Nature. 1997;388(6641):468–471. doi: 10.1038/41319. [DOI] [PubMed] [Google Scholar]
- Dickens WT, Flynn JR. Heritability estimates versus large environmental effects: the IQ paradox resolved. Psychol Rev. 2001;108(2):346–369. doi: 10.1037/0033-295X.108.2.346. [DOI] [PubMed] [Google Scholar]
- Eldridge R, Denckla MB, Bien E, Myers S, Kaiser-Kupfer MI, Pikus A, Schlesinger SL, Parry DM, Dambrosia JM, Zasloff MA, et al. Neurofibromatosis type 1 (Recklinghausen’s disease). Neurologic and cognitive assessment with sibling controls. Am J Dis Child. 1989;143(7):833–837. doi: 10.1001/archpedi.1989.02150190083027. [DOI] [PubMed] [Google Scholar]
- El-Said MF, Badii R, Bessisso MS, Shahbek N, El-Ali MG, El-Marikhie M, El-Zyoid M, Salem MS, Bener A, Hoffmann GF, Zschocke J. A common mutation in the CBS gene explains a high incidence of homocystinuria in the Qatari population. Hum Mutat. 2006;27(7):719. doi: 10.1002/humu.9436. [DOI] [PubMed] [Google Scholar]
- Gan-Schreier H, Kebbewar M, Fang-Hoffmann J, Wilrich J, Abdoh G, Ben-Omran T, Shahbek N, Bener A, Al Rifai H, Al Khal AL, Lindner M, Zschocke J, Hoffmann GF. Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. J Pediatr. 2010;156(3):427–432. doi: 10.1016/j.jpeds.2009.09.054. [DOI] [PubMed] [Google Scholar]
- Koch R, Azen C, Friedman EG, Williamson ML. Paired comparisons between early treated PKU children and their matched sibling controls on intelligence and school achievement test results at eight years of age. J Inherit Metab Dis. 1984;7(2):86–90. doi: 10.1007/BF01805813. [DOI] [PubMed] [Google Scholar]
- Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de Franchis R, Andria G, Kluijtmans LA, Blom H, Boers GH, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, Gaustadnes M. Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat. 1999;13(5):362–375. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Lindner M, Abdoh G, Fang-Hoffmann J, Shabeck N, Al-Sayrafi M, Al-Janahi M, Ho S, Abdelrahman MO, Ben-Omran T, Bener A, Schulze A, Al-Rifai H, Al-Thani G, Hoffmann GF. Implementation of extended neonatal screening and a metabolic unit in the State of Qatar: developing and optimizing strategies in cooperation with the Neonatal Screening Center in Heidelberg. J Inherit Metab Dis. 2007;30(4):522–529. doi: 10.1007/s10545-007-0553-7. [DOI] [PubMed] [Google Scholar]
- Mudd SH. Vascular disease and homocysteine metabolism. N Engl J Med. 1985;313(12):751–753. doi: 10.1056/NEJM198509193131210. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Finkelstein JD, Irreverre F, Laster L. Homocystinuria: an enzymatic defect. Science. 1964;143(3613):1443–1445. doi: 10.1126/science.143.3613.1443. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, Andria G, Boers GH, Bromberg IL, Cerone R, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985;37(1):1–31. [PMC free article] [PubMed] [Google Scholar]
- Naughten ER, Yap S, Mayne PD. Newborn screening for homocystinuria: Irish and world experience. Eur J Pediatr. 1998;157(Suppl 2):S84–S87. doi: 10.1007/PL00014310. [DOI] [PubMed] [Google Scholar]
- Schiff M, Blom HJ. Treatment of inherited homocystinurias. Neuropediatrics. 2012;43(6):295–304. doi: 10.1055/s-0032-1329883. [DOI] [PubMed] [Google Scholar]
- Turkheimer E, Haley A, Waldron M, D’Onofrio B, Gottesman II. Socioeconomic status modifies heritability of IQ in young children. Psychol Sci. 2003;14(6):623–628. doi: 10.1046/j.0956-7976.2003.psci_1475.x. [DOI] [PubMed] [Google Scholar]
- Urreizti R, Asteggiano C, Cozar M, Frank N, Vilaseca MA, Grinberg D, Balcells S. Functional assays testing pathogenicity of 14 cystathionine-beta synthase mutations. Hum Mutat. 2006;27(2):211. doi: 10.1002/humu.9395. [DOI] [PubMed] [Google Scholar]
- Weisfeld-Adams JD, Bender HA, Miley-Akerstedt A, Frempong T, Schrager NL, Patel K, Naidich TP, Stein V, Spat J, Towns S, Wasserstein MP, Peter I, Frank Y, Diaz GA. Neurologic and neurodevelopmental phenotypes in young children with early-treated combined methylmalonic acidemia and homocystinuria, cobalamin C type. Mol Genet Metab. 2013;110(3):241–247. doi: 10.1016/j.ymgme.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Yap S (2012) Classical homocystinuria: newborn screening with early treatment effectively prevents complications. Hamdan Med J 5(3): 351–362
- Yap S, Rushe H, Howard PM, Naughten ER. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2001;24(4):437–447. doi: 10.1023/A:1010525528842. [DOI] [PubMed] [Google Scholar]
- Zschocke J, Kebbewar M, Gan-Schreier H, Fischer C, Fang-Hoffmann J, Wilrich J, Abdoh G, Ben-Omran T, Shahbek N, Lindner M, Al Rifai H, Al Khal AL, Hoffmann GF. Molecular neonatal screening for homocystinuria in the Qatari population. Hum Mutat. 2009;30(6):1021–1022. doi: 10.1002/humu.20994. [DOI] [PubMed] [Google Scholar]