

Abstract
Glutaric acidemia I (GA I, #231670) is one of the treatable, autosomal recessively inherited metabolic disorders. Macrocephaly, acute encephalitis-like crises, dystonia and characteristic frontotemporal atrophy are the hallmarks of this disease. In this communication, we present the clinical, biochemical and molecular profile of seventeen GA I patients from 15 unrelated families from India and report seven novel mutations in GCDH gene (c.281G>A (p.Arg94Gln), c.401A>G (p.Asp134Gly), c.662T>C (p.Leu221Pro), c.881G>C (p.Arg294Pro), c.1173dupG (p.Asn392Glufs*5), c.1238A>G (p.Tyr413Cys) and c.1241A>C (p.Glu414Ala)). Out of these, c.662T>C (p.Leu221Pro) in exon 8 and c.281G>A (p.Arg94Gln) allele in exon 4 were low excretor alleles, whereas c.1241A>C (p.Glu414Ala), c.1173dupG and c.1207C>T (p.His403Tyr) in exon 11 were high excretor alleles. We conclude that c.1204C>T (p.Arg402Trp) is probably the most common mutant allele. Exons 11 and 8 are the hot spot regions of GCDH gene in Indian patients with GA I. An early diagnosis and timely intervention can improve the underlying prognosis. Molecular confirmation is helpful in providing genetic counselling and prenatal diagnosis in subsequent pregnancy.
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2014_377) contains supplementary material, which is available to authorized users.
Introduction
Glutaric acidurias are a group of autosomal recessively inherited metabolic disorders that are characterized by abnormal excretion of glutaric acid due to a defect in amino acid or fatty acid metabolism pathways. Glutaric acidemia type 1 or glutaric aciduria type 1 (GA I) occurs due to the deficiency of glutaryl-CoA dehydrogenase (GCDH) enzyme (EC 1.3.8.6, old number EC 1.3.99.7), a member of the acyl-CoA dehydrogenase family and a key enzyme in the catabolic pathways of the amino acids tryptophan, lysine and hydroxylysine. Deficiency of GCDH causes increased organic acid excretion of glutaric acid, 3-hydroxyglutaric acid and glutaconic acid in urine and elevated glutarylcarnitine (C5DC) in plasma. The estimated worldwide frequency of GA I is 1 in 100,000 newborns (Lindner et al. 2004), but increased frequency has been reported in the inbred Old-Order Amish community of Pennsylvania (Morton et al. 1991) and the Ojibway Indians in Manitoba (Haworth et al. 1991). About ninety percent of the affected children present classically between 2 and 37 months of age with extrapyramidal symptoms, predominantly dystonia superimposed on axial hypotonia after an acute encephalopathic crisis precipitated by intercurrent febrile illness, infection, fasting or immunization (Hoffmann et al. 1991; Kolker et al. 2006; Strauss et al. 2003). About 75% of the patients have macrocephaly with soft neurological signs such as head lag, irritability and feeding difficulties. Extrapyramidal symptoms are due to bilateral striatal injury during the acute episode. Early diagnosis and prompt initiation of treatment can prevent the long-term complications and mortality which have led to its inclusion in conservative newborn screening programs (Heringer et al. 2010; Kolker et al. 2007). GCDH enzyme is encoded by GCDH gene, which is located on the chromosome 19p13.2 spanning about ~7 kb and contains 12 exons (Transcript ID ENST00000222214) and encodes 438 amino acid-long precursor protein. It is a genetically heterogeneous condition and is caused by different types of mutations such as missense, nonsense and intronic variations in GCDH gene. Till date, 163 mutations have been reported from different ethnic groups in HGMD (Goodman et al. 1998; Busquets et al. 2000a; Zschocke et al. 2000). Although there is some correlation between genotype and the urinary excretion of glutaric acid, the correlation between genotype and phenotype has been elusive (Kolker et al. 2006). There is no published data from India on the mutation spectrum of GA I. This retrospective study is aimed to present clinical and molecular profile of Indian patients and report seven novel mutations. In silico analysis was performed to predict the role of these novel mutations on protein function. The effects of novel missense mutations on the structure of gene product have been analyzed by computational modelling. Prenatal diagnosis was offered in one family.
Materials and Methods
Patient Enrolment
Over a period of 5 years (June 2008 till June 2013), 17 patients of varying age from 15 unrelated families were recruited from the Genetic Clinic and Pediatric Neurology Clinic. The study was approved by the institutional ethics committee, and written informed consent was taken from all parents. Clinical details were filled in a predesigned proforma for neurometabolic disorders. Diagnosis was made based upon the clinical presentation, neuroimaging, urinary organic acids and tandem mass spectrometry results. Wherever possible, clinical outcome was assessed through follow-up either clinically and by review of records or through parents' interview where the child was not alive. During follow-up assessment, patients were evaluated mainly clinically and not biochemically for amino acids due to lack of availability of amino acids and affordability. Age-dependent developmental assessment tools were used for the developmental delay. It included calculation of developmental age and quotient using the Developmental Assessment Scale for Indian Infants (DASII) for 0–30 months, Developmental Profile 3 (DP 3) for 0–12 years and Malin’s Intelligence Scale for Indian Children (MISIC) for 6–15 years. No formal scoring for the severity of movement disorder or morbidity was used.
GCDH Gene Analysis
Peripheral blood was collected in EDTA vacutainer, and genomic DNA was isolated by salt method (Miller et al. 1988). Primers were designed by primer 3 software (bioinfo.ut.ee/primer 3–0.4.0) (Supplementary Table 1), and coding sequences were amplified in eight different fragments by polymerase chain reaction (PCR). Amplified PCR products were purified by enzyme treatment (ExoI & SAP) and sequenced by ABI 3130 Genetic Analyzer (Applied Biosystem, USA). Sequence analysis was performed by using ChromasPro software (http://technelysium.com.au) and NCBI blast. Nomenclature of novel mutations was done according to the guidelines of Human Genome Variation Society (http://www.hgvs.org/mutnomen). Novel mutations were checked in the 1000 Genome Projects database (http://www.1000genomes.org/), dbSNP database (http://www.ncbi.nlm.nih.gov/snp/) and HGMD (Human Gene Mutation Database, http://www.hgmd.org/).
In Silico Analysis of Novel Mutations
Multiple sequence alignments were performed to check the conservation of amino acid across some other species. PolyPhen2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org) programs were used to assess the qualitative effect of missense changes on the protein function. Both these programs predict the effect of amino acid replacement due to mutation on the function of protein. PolyPhen2 predicts the effect based on physicochemical properties of residue and comparative analysis, while SIFT predicts on the basis of degree of conservation of residues through PSI-BLAST sequence alignment.
Three-dimensional structure of the proteins is required to understand the role of mutations on their structure and function at atomic level. Crystal structure of wild-type human GCDH complexed with coenzyme FAD (PDB: 1SIQ) (Fu et al. 2004) is available in PDB. Model structure of protein with novel missense mutation was built with the help of crystal structure of wild-type protein and “Build Mutant” protocol incorporated in MODELLER 8.2 program (Sali and Blundell 1993) in the modelling environment of Discovery Studio (DS 2.0) (Discovery Studio 2006, Accelrys Inc., San Diego, CA, USA). The “Build Mutant” protocol changes the selected residue into the desired residue and optimizes the conformation of mutated residue along with neighbouring residues. Side chains of the residues in the models were further refined to correct their conformation and orientation using the modelling program “Chi Rotor” (Spassov et al. 2007). The stereochemical quality of the mutant models was verified by PROCHECK (Laskowski et al. 1993). The generated models were energy minimized to remove any steric clash between atoms, followed by short MD simulations for the final refinement using CHARMm force field simulation engine (Brooks et al. 1983; Momany and Rone 1992).
Results
A total of fifteen unrelated families and seventeen patients were recruited during the study period. The male/female ratio was 1.4:1. The median age at onset of symptoms and final diagnosis was 6 months (2−12 months) and 18 months (3–192 months), respectively. Out of 15 families, 3 had positive family history. Consanguinity was seen in 8/15 (53%) families with different religious backgrounds (5 Muslim, 1 South Indian, 2 Hindu). Twelve patients (70%) had precipitating illness like fever associated with seizure (4/12), fever associated with/without gastroenteritis or pneumonia (4/12), seizure alone (3/12) or after immunization (1/12). The main presenting features were macrocephaly, developmental regression mainly motor due to development of dystonia, seizures or a combination of these. Table 1 shows the distribution of clinical, biochemical and radiological abnormalities across various families. Patients were divided into four developmental categories: patients with (1) normal development, (2) developmental regression, (3) developmental delay only and (4) developmental delay with regression category. “Regression” in GA I essentially refers to loss of intentional motor control due to development of dystonia. Six patients (35.2%) had normal initial development followed by regression and dystonia, two had normal development without regression or dystonia but had associated seizures and one had normal development with dystonia. The rest of the eight patients (47%) had developmental delay with or without regression with associated extrapyramidal symptoms in all except one. Based upon the developmental assessment by DASII, DP3 and MISIC at different ages, all patients except F2, F10a, F12 and F15 had delay in the physical and adaptive domain primarily because of the development of dystonia. Patients F7, F9 and F15 had mild global developmental delay, whereas patients F1, F3, F4, F8 and F13 had severe to profound delay.
Table 1.
Distribution of clinical, biochemical and radiological abnormalities across various families
| Case number | Gender | Age at onset (months) | Age at diagnosis (months) | Precipitating illness | Developmental category (n = 17) | Macrocephaly | Extrapyramidal symptoms | Seizures | Abnormal neuro radiological changesa | Elevated glutaryl carnitine (C5 DC) | Elevated urine elevated glutaric acid and its metabolites | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F2 | M | 2 | 3 | Fever, acute gastroenteritis, seizures | Normal development (3) | + | − | + | + | + | + | Alive |
| F10a | F | 6 | 132 | Seizure, vomiting | + | − | + | + | + | + | Alive | |
| F6 | M | 4.6 | 5 | Seizures | − | + | + | + | + | + | Alive | |
| F5 | F | 6 | 18 | Fever, seizures | Regressionb (6) | + | + | + | + | + | + | Died |
| F14 | F | 6 | 7 | Fever, seizures | + | + | + | + | + | + | Alive | |
| F10b | M | 5 | 24 | Fever, gastroenteritis | + | + | − | + | + | + | Alive | |
| F11a | F | 7 | 24 | Fever | − | + | − | + | + | − | Alive | |
| F11b | M | 6 | 12 | Seizures once only | − | + | − | + | − | − | Alive | |
| F12 | M | 7.6 | 10 | No | + | − | +c | + | + | + | Alive | |
| F9 | F | 12 | 192 | No | Developmental delay (4) | + | +d | +c | + | + | Alive | |
| F1 | M | 10 | 54 | No | + | + | − | + | + | + | Alive | |
| F4 | M | 6 | 10 | Pneumonia | + | − | − | + | + | + | Alive | |
| F15 | M | 6 | 30 | No | + | − | − | + | + | + | Alive | |
| F7 | M | 7 | 60 | Fever, seizures | Developmental delay and regressionb (4) | + | + | + | + | + | + | Alive |
| F8 | F | 6 | 36 | Injection | − | + | − | + | + | + | Alive | |
| F3 | F | 6 | 6 | Fever, seizures | + | + | − | + | + | + | Died | |
| F13 | M | 8 | 10 | No | − | + | +c | + | + | + | Alive |
a Extrastriatal neuroradiological abnormalities
Temporal hypoplasia/frontotemporal atrophy, wide temporal and sylvian CSF spaces (bat-wing appearance) in F2, F4, F6, F7, F8, F9, F10b, F12, F15
Subdural hygroma F2, F4, F10a, F12
Hyperintensities in globus pallidus (GP)] and thalamus (T) – F4 (GP), F8 (T), F13 (GP)
Striatal neuroradiological abnormalities – hyperintensities in caudate (C), putamen (P), F1 (P), F2 (C,P), F3 (C,P), F4 (P), F5 (P,C), F6 (P), F8 (P), F10b (C,P), F11a (P), F15 (C,P)
bLoss of intentional motor control
cMyoclonic seizures
dChorieform movements
Macrocephaly, dystonia, choreoathetoid movements and seizures were present in 70.5% (12/17), 64.7% (11/17), 0.05% (1/17) and 52.9% (9/17), respectively. Patients with generalized tonic–clonic seizures had initial one or two episodes followed by complete disappearance. Three patients had myoclonic seizures. Both striatal (hyperintensities in caudate and putamen) and extrastriatal (subdural hygroma, wide sylvian fissure, frontotemporal atrophy, hyperintensities in globus pallidus and thalamus) abnormalities on neuroimaging were seen in all the patients. Majority of the patients had feeding difficulties especially in swallowing. Soon after diagnosis, all the patients were kept on indigenous lysine- and tryptophan-restricted diet (predominantly a vegetarian Indian (native/local) diet in natural form) and carnitine supplementation (50–100 mg/kg/day) due to nonavailability and affordability of GA I-specific diet. Lysine restriction was achieved by natural and locally available diet. The amount of lysine varied depending upon the different age groups with an average daily intake of 90–120 mg/kg body weight/day, the highest being in infants. The total protein intake was 1–2 g protein/kg actual body weight/day. All patients were also supplemented with riboflavin (100–300 mg/day), calcium and other micronutrients soon after the diagnosis, and none of them reported to have any major deficiencies. These patients were on naturally achieved lysine-restricted vegetarian diet, and lysine-free, tryptophan-reduced amino acid supplements were not given separately due to nonavailability of these amino acid supplements. Baclofen (10–20 mg/day) was used for dystonia. Two patients (F3 and F5) died at the age of 1.6 and 2.6 years, respectively. Both these children had severe feeding difficulties and were malnourished. They died at home probably due to aspiration pneumonia.
GCDH Gene Mutations
A total of 15 different mutations were identified in GCDH gene in different exons in 17 patients of 15 unrelated families. Seven novel (c.281G>A (p.Arg94Gln), c.401A>G (p.Asp134Gly), c.662T>C (p.Leu221Pro), c.881G>C (p.Arg294Pro), c.1173dupG (p.Asn392Glufs*5), c.1238A>G (p.Tyr413Cys) and c.1241A>C (p.Glu414Ala)) and eight reported (c.650C>T (p.Pro217Leu), c.769C>T (p.Arg257Trp), c.764C>T (p.Ser255Leu), c.770G>A (p.Arg257Gln), c.937C>T (p.Arg313Trp), 1173delG (p.Asn392Metfs*9), c.1204C>T (p.Arg402Trp) and c.1207C>T (p.His403Tyr)) mutations (Fig. 1, Table 2) were found only in five different exons of GCDH gene. Mutation c.1207C>T (p.His403Tyr) was initially thought to be novel in nature, but recently it has been reported (Wang et al. 2013). Amongst the seven novel mutations, one is frameshift and six are missense mutations. Prenatal diagnosis was performed for one family (F1). Upon counselling, pregnancy was terminated as the foetus was affected.
Fig. 1.
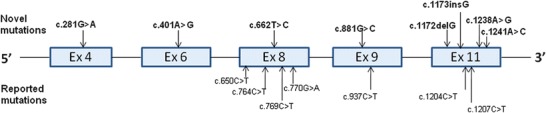
Mutation distribution across GCDH gene
Table 2.
Mutations detected in 17 Indian patients with glutaric aciduria type 1
| Case no. | Exon | Mutation at nucleotide level | Mutation at protein level | Frequency (alleles) | Type of mutations | Reported/novel | PolyPhen2/SIFT prediction | References |
|---|---|---|---|---|---|---|---|---|
| F11a | 4 | c.281G>A | p.Arg94Gln | 2/34 | Missense | Novel | Probably damaging/damaging | This study |
| F11b | ||||||||
| F10a | 6 | c.401A>G | p.Asp134Gly | 4/34 | Missense | Novel | Probably damaging/damaging | This study |
| F10b | ||||||||
| F12 | 8 | c.650C>T | p.Pro217Leu | 1/34 | Missense | Reported | – | Zschocke et al. (2000) |
| F11a | 8 | c.662T>C | p.Leu221Pro | 2/34 | Missense | Novel | Probably damaging /damaging | This study |
| F11b | ||||||||
| F3 | 8 | c.764C>T | p.Ser255Leu | 1/34 | Missense | Reported | – | Busquets et al. (2000a) |
| F8 | 8 | c.769C>T | p.Arg257Trp | 2/34 | Missense | Reported | – | Schwartz et al. (1998) |
| F9 | 8 | c.770G>A | p.Arg257Gln | 2/34 | Missense | Reported | – | Schwartz et al. (1998) |
| F3 | 9 | c.881G>C | p.Arg294Pro | 1/34 | Missense | Novel | Probably damaging/damaging | This study |
| F15 | 9 | c.937C>T | p.Arg313Trp | 1/34 | Missense | Reported | – | Goodman et al. (1998) |
| F15 | 11 | c.1173delG | p.Asn392Metfs*9 | 1/34 | Frameshift | Reported | Truncated protein | Anikster et al. (1996) |
| F12 | 11 | c.1173dupG | p.Asn392Glufs*5 | 1/34 | Frameshift | Novel | Truncated protein | This study |
| F4, F5, F6, F7 | 11 | c.1204C>T | p.Arg402Trp | 8/34 | Missense | Reported | – | Biery et al. (1996) |
| F13, F14 | 11 | c.1207C>T | p.His403Tyr | 4/34 | Missense | Reported | – | Wang et al. (2013) |
| F1 | 11 | c.1238A>G | p.Tyr413Cys | 2/34 | Missense | Novel | Probably damaging/damaging | This study |
| F2 | 11 | c.1241A>C | p.Glu414Ala | 2/34 | Missense | Novel | Probably damaging/damaging | This study |
In Silico Analysis
All the novel mutations were found in the highly conserved region of the protein. Both PolyPhen2 and SIFT programs predict the effect of observed novel mutation in this study to have deleterious effect on the function of protein. Stereochemical qualities of mutant models are good, and substituted residues lie in the most favoured region of Ramachandran’s plot. In silico study through the three-dimensional structure of the model of the mutant proteins helps to understand the effect of mutation on the structure of protein.
Genotype and Biochemical Phenotype
Patients were divided into high excretors and low excretors based upon the criteria by Baric et al. (1999) and Kolker et al. (2006). High excretors were defined as glutaric acid excretion of >100 mmol GA/mol creatinine and low excretors as <100 mmol GA/mol creatinine. Levels of glutaric acids were available only in six patients with five novel alleles. In our study, out of seven novel alleles, two alleles, c.662T>C (p.Leu221Pro) and c.281G>A (p.Arg94Gln), had almost no glutaric aciduria (F11a and b) and hence were low excretors; however, c.1241A>C (p.Glu414Ala) (F2), c.1173dupG (F12) and c.1207C>T (p.His403Tyr) in Ex-11 (F13, F14) were found to be high excretor alleles.
Discussion
There have been few reports of GA I in Indian patients, the majority being on neuroimaging (Kamate et al. 2012; Sen and Pillay 2011). This is the first report on clinical, biochemical and molecular profile of Indian patients.
Clinical Spectrum
Glutaric aciduria type I is probably the second common organic acidemia in India, the commonest being methylmalonic acidemia (unpublished data, personal communication). These patients have a variable course of illness, the majority of patients presenting with loss of intentional motor control due to development of dystonia precipitated by an acute febrile illness, macrocephaly and extrapyramidal symptoms. In our cohort, the median age at the onset of symptoms was 6 months. The age at diagnosis was quite variable and there was a gap of nearly 12 months between the onset and diagnosis probably because of the variable clinical spectrum of the disorder or due to the delayed diagnosis. Twelve out of 17 (70%) had some precipitating factor at around 6 months. Out of these three had normal development, while others had developmental delay and/or regression. In accordance with other studies (Wang et al. 2013; Mushimoto et al. 2011), a combination of macrocephaly, developmental regression, dystonia and seizures emerged as the predominant presenting feature. Three out of 17 had normal development (F2, F10a, F6) despite having the precipitating event and radiological abnormalities. Case F2 was diagnosed at 3 months and kept on modified diet and carnitine supplementation. He has good compliance and is developmentally normal. All patients with or without crises had MRI abnormalities (Harting et al. 2009). Eighty two percent of the patients (14/17) were already neurologically compromised (categories 2–4) at the time of presentation. Category 2 patients had mainly motor regression due to incapacitating dystonia. A correlation between MRI findings and dystonia has been studied in detail earlier by Harting et al. (2009) and Garbade et al. (2014). These authors clearly showed that the severity of striatal lesions on MRI did correlate with the severity of the movement disorder. In contrast, extrastriatal MRI finding did not correlate with the motor dysfunction. In our study, 5/17 patients did not develop dystonia at a mean age of 37 months (3–132 months). Two of these patients (F10a and F12) had only extrastriatal features, whereas F2, F4 and F15 had both extrastriatal and striatal involvement. In our study, we did not perform any follow-up MRI. It has been shown (Heringer et al. 2010) that prompt vigorous intervention during acute episodes with the provision of glucose, fluids and electrolytes along with carnitine supplementation can prevent striatal degeneration and has improved outcome. Most of our patients had acute crises prior to the diagnosis. They were diagnosed after routine evaluation for either developmental delay, seizures or macrocephaly. Hence no emergency regimen was given at the time of acute precipitating factors. However, after initial diagnosis, patients were explained about emergency home treatment (Kolker et al. 2006, 2007, 2011).
An individualized long-term dietary treatment (Kolker et al. 2011; Viau et al. 2012; Lee et al. 2013) includes a low lysine diet through natural protein with or without lysine-free and tryptophan-reduced amino acid supplements along with L-carnitine supplementation for adequate growth (Boy et al. 2013). Due to lack of availability of lysine-free and tryptophan-reduced therapeutic formulae, the metabolic maintenance treatment was tried mainly through locally available protein-restricted diet with age-specific minimum requirement of lysine and tryptophan, micronutrients, riboflavin and carnitine supplementation. Baclofen was given for dystonia.
Mutation Spectrum
GA I mutations are classified either as severe or mild depending upon the residual enzyme activity and the urinary excretion of glutaric acid. p.Arg402Trp mutation in exon 11 is described as one of the severe and common mutations that account for about 20% of the mutations in Caucasians. Most of the patients are reported as compound heterozygotes, but in our cohort, 80% of the family mutations were in homozygous state including Arg402Trp mutation.
Figure 1 shows the distribution of 15 different mutations in five different exons of GCDH gene, in which seven of them (c.1238A>G, c.1241A>C, c.1173dupG, c.881G>C, c.401A>G, c.662T>C, c.281G>A) were novel and found in seven unrelated patients. Three families were homozygous for novel mutations, whereas five were compound heterozygous. Mutation prediction software PolyPhen2 and SIFT (Table 2) depicted that these missense mutations are probably damaging/damaging the protein. Frameshift mutations c.1173dupG (p.Asn392Glufs*5) and c.1173delG (p.Asn392Metfs*9) were generated truncated proteins. Family 11 had two novel mutations in compound heterozygous state, the father being a carrier of c.281G>A (p.Arg94Gln) and the mother a carrier of c.662T>C (p.Leu221Pro). However, these mutations appear as leaky or mild as both these sibs did not show urinary excretion of glutaric acid.
In Silico Analysis of Effect of Mutation on Their Structure
GCDH is a member of acyl-CoA dehydrogenase family and catalyses the oxidative decarboxylation of glutaryl-CoA using flavoprotein as its electron acceptor. Structural analysis reveals that the proper positioning of cofactor FAD (flavin adenine dinucleotide) and glutaryl-CoA is essential for decarboxylation reaction followed by dehydrogenation (Fu et al. 2004). Sequence analysis shows that residues Arg294, Tyr413 and Glu414 are highly conserved amongst the dehydrogenase family. Residue Glu414 is an active-site residue and functions as catalytic base. It is responsible for proton abstraction. Hence, the position of carboxylate group of Glu414 is extremely important and is stabilized by two hydrogen bonds with side chain of Arg294 (Fig. 2a). Therefore, Glu414Ala mutation will abolish the catalytic function of proton abstraction. The natural variant of Glu414 leads to loss of its enzymatic activity (Keyser et al. 2008). In addition, replacement of Glu414 by Ala also leads to a change in side chain conformation of crucial residues Arg294 and Tyr413 (Fig. 2b). Hence, this substitution will hamper its function severely. It clearly indicates that this mutation has a direct and crucial role in the pathogenesis of GA I.
Fig. 2.
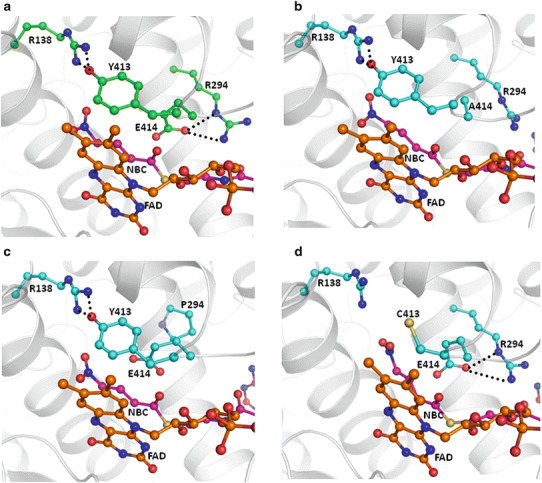
Cartoon representation of (a) crystal structure of wild-type human mitochondrial GCDH and model structure of mutants (b) Glu414Ala, (c) Tyr413Cys and (d) Arg294Pro. Side chain of important residues are shown (ball and stick in green for wild type and cyan for mutants), glutaryl-Co-A (ball and stick in magenta) and cofactor FAD (ball and stick in orange) and hydrogen-bonded interactions in black dotted lines. Glutaryl-CoA and FAD are shown partially
Tyr413 is essential for the stabilization of FAD position through π–π aromatic interaction with phenyl ring of flavin (Fig. 2a). It is present on the flexible loop region, and conformation as well as the position of its aromatic side chain is stabilized by hydrogen-bonded interactions with another crucial residue Arg138. Arg138 plays an important role in enzymatic function as its replacement by Gln or Gly leads to manyfold increase in Km value for glutaryl-CoA and decrease in its catalytic activity (Dwyer et al. 2001). The conformation of Arg138 side chain is known to be crucial for the enzymatic function as it acts as a hydrogen bond donor to γ-carboxylate of substrate glutaryl-CoA and electrostatic catalyst to stabilize the negative charges during catalysis (Keyser et al. 2008). Positively charged guanidinium group of Arg138 in wild-type model complex was in close proximity (3.0 Å) of carboxylate moiety of glutaryl-CoA. Substitution of Tyr413 by Cys will lead to loss of a crucial edge to face π–π interaction between Tyr and flavin; this might lead to destabilization of FAD position (Fig. 2c). Simultaneously this also alters the conformation of Arg138 as it is stabilized by hydrogen bond with Tyr413, and important guanidinium group in mutant model complex moves away from carboxylate group of glutaryl-CoA. Therefore, Tyr413Cys mutation causes the double adverse impact and might lead to a significant decrease in the enzymatic activity of GCDH.
The substitution of Arg294 by Pro causes loss of crucial hydrogen bonds with Glu414, and this leads to a change in the conformation of active-site residue Glu414 required for proton abstraction (Fig. 2d). This also changes the loop conformation on which another crucial residue Tyr413 resides. Moreover, residue 294 lies in the helical region, and Pro is known to destabilize the helical conformation. Hence, this mutation might also adversely affect its enzymatic activity.
The rest of the three novel missense mutations (Arg94Gln, Asp134Gly and Leu221Pro) lie away from the active site. Residue Arg94 is positively charged and surface exposed. In mutant, Arg94 is replaced by Gln which is neutral in nature. This change in nature of residue can affect the interactions with the interacting partner. The degree of effect of this mutation on its function will depend on the importance of the interacting partner. Recently two important proteins, dihydrolipoamide S-succinyltransferase involved in glutaryl-CoA synthesis and β subunit of electron transfer flavoprotein acting as electron acceptor, have been identified as directly binding partners of GCDH (Schmiesing et al. 2014). Another mutation site residue Leu221 lies on the loop region and away from the active site. It is nonpolar in nature and its side chain is buried. Its substitution by Pro does not affect the conformation of the protein significantly. Hence, this mutation does not appear to affect the enzymatic function of GCDH. Similarly Asp134 is pointing away from the active site; however, OD1 and OD2 of its side chain form hydrogen bonds with residues present on the neighbouring helices and might be assisting in the proper arrangement of those helices. Its substitution by Gly which lacks any side chain will not be able to form hydrogen bonds with residues on the neighbouring helices and might be decreasing the stability of helical arrangement. Though the role of this mutation on the catalytic function cannot be directly linked, this may lead to decreased enzymatic activity.
Biochemical, Genotype and Phenotype Correlation
The genotype and phenotype correlation has been elusive in GA I; however, genotype often predicts the biochemical phenotype in GA I. Severe mutations tend to have no residual enzyme activity and have a typical urinary metabolite pattern. Milder mutations have significant residual enzyme activity, hence low or normal urinary excretion of glutaric acids. Some of the mutations correlate well with the excretion of glutaric acid and 3 hydroxyglutaric acids. Arg402Trp and Ala293Thr correspond to high excretor group (Busquets et al. 2000a), whereas Arg227Pro and Val400Met account for low excretor group (Christensen et al. 1997). In our study, out of seven novel alleles, two (c.662T>C (p.Leu221Pro) and c.281G>A (p.Arg94Gln)) in exons 8 and 4, respectively, were low excretor alleles, whereas c.1241A>C (p.Glu414Ala), c.1173dupG and c.1207C>T (p.His403Tyr) in exon 11 were high excretor alleles.
In accordance with previous studies, we also did not observe any genotype and phenotype correlation (Christensen et al. 2004; Mushimoto et al. 2011). Interestingly, in our cohort, Arg402Trp (allele frequency 8/34, 23.5%) mutation emerged as one of the common mutations in four unrelated Muslim families. Two of these families had consanguinity. Haplotype analysis could not be done on these families to examine the founder effect. This mutation constituted about 23.5% of all the mutant alleles followed by His403Tyr (alleles frequency 4/34, 11.76%). Both these mutations were seen in unrelated families and did not have a similar phenotypic effect, suggesting a poor genotype–phenotype correlation and interfamilial variability in GA I. Intrafamilial variability as previously reported by Gregersen et al. (1977) and Kolker et al. (2007) was also observed in one of our families; the siblings in F10 and F11 (F10 showed presence of isolated macrocephaly and seizure in one sib and severe dystonia with regression in another sib) with the same mutation and genetic background had different phenotypes, suggesting intrafamilial variability in GA I. This intrafamilial and interfamilial variability can probably be explained by the epigenetic factors or complex interactions at tissue or metabolic levels and the extent of neurological damage caused by the various precipitating factors (Wang et al. 2013). In our cohort, exon 11 (in 53%; 18/34 alleles) followed by exon 8 (in 23.5%; 8/34 alleles) appear to harbour most of the mutations (Fig. 1). Hence, we recommend that molecular testing for exon 11 should be done first followed by exon 8 in resource-constrained settings. The combined diagnostic yield for these two exons appears to be as high as 76.5%.
Although the clinical, radiological and biochemical analyses are helpful in arriving at the diagnosis, molecular confirmation of the diagnosis undoubtedly gives an opportunity to the family for prenatal diagnosis in the subsequent pregnancies. Prenatal diagnosis could be offered in one family, and the pregnancy was terminated as the foetus was found to be affected.
The limitations in our study include qualitative results of urinary organic acids, because of which the exact correlation between biochemical phenotype and genotype could not be established. Secondly, after initiating the modified indigenous diet, the effectiveness of the diet could not be evaluated as the subsequent follow-ups for growth, biochemical monitoring for various amino acids and repeated follow-up MRI scans were not available.
In India, due to lack of uniformly available newborn screening, the diagnosis of GA I is delayed. Other factors for delayed diagnosis and poor final outcome could be lack of awareness amongst physicians, distance to the appropriate medical facility, lack of home management and poor patient compliance due to illiteracy and socioeconomic status. Excellent outcomes after early diagnosis and early dietary intervention for favourable neurological outcome have led to the inclusion of GA I in newborn screening panel across various countries. In countries like India, where a national newborn screening program is yet to start, authors would like to recommend its inclusion in the expanded panel. Till that time it would be plausible to have a high index of suspicion for early diagnosis and timely dietary intervention and parent education to prevent further progress of neurological damage and birth of another affected child. Although no formal biochemical follow-up evaluation has been done for the low-cost, culturally acceptable and locally available Indian indigenous diets, it was observed that with good patient compliance, patients on these diets did not further deteriorated neurologically. Biochemical evaluation with lysine and tryptophan levels and evaluation of growth parameters of Indian patients with GA I on indigenous diet are warranted to examine the effectiveness of these diets before drawing any conclusions.
Conclusion
Recognition of modifiable inherited metabolic disorders such as GA I requires high index of suspicion and timely referral for further management to an appropriate centre. Early diagnosis and prompt management for acute illness and initiation of indigenous diet and carnitine are likely to have a beneficial effect on the neurological outcome. Further studies and appropriate follow-up are required to evaluate the effectiveness of Indian indigenous diets in the growth and long-term outcome of patients with GA. In accordance with the previous reports, our results also showed that there is a lot of intrafamilial and interfamilial variability even with the same mutation and there is no genotype–phenotype correlation. Molecular confirmation of the diagnosis is particularly helpful in prenatal diagnosis in the next pregnancy. p.Arg402Trp mutation emerged as a common mutation in Muslim families. Exon 11 and exon 8 are two hot spot exons and should be analyzed first in Indian patients for molecular diagnosis. Confirmation by molecular diagnosis aids in providing genetic counselling and prenatal diagnosis in future pregnancy.
Electronic Supplementary Material
Acknowledgement
Indian Council of Medical Research for funding
Dr Rita Christopher, NIMHANS Bangalore, Sandor proteomics, Hyderabad
Take-Home Message
Exons 11 and 8 of the GCDH gene seem to be the mutational hot spot regions in Indian patients with GA I.
Compliance with Ethics Guidelines
Conflict of Interest
Neerja Gupta, Pawan Kumar Singh, Manoj Kumar, Shivaram Shastri, Sheffali Gulati, Atin Kumar, Anuja Agarwala, Seema Kapoor, Mohandas Nair, Savita Sapra, Sudhisha Dubey, Ankur Singh, Punit Kaur and Madhulika Kabra declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
Planning: Neerja Gupta, Pawan Kumar Singh, Sudhisha Dubey, Madhulika Kabra
Conducting: Neerja Gupta, Pawan Kumar Singh, Shivaram Shastri, Sheffali, Gulati, Atin Kumar, Anuja Agarwala, Seema Kapoor, Ankur Singh, Mohandas Nair, Savita Sapra, Sudhisha Dubey, Manoj Kumar, Punit Kaur and Madhulika Kabra
Reporting: Neerja Gupta, Pawan Kumar Singh, Shivaram Shastri, Sheffali Gulati, Seema Kapoor, Manoj Kumar and Madhulika Kabra
Neerja Gupta and Pawan Kumar Singh participated equally and should share “first authorship”.
Footnotes
Competing interests: None declared
Contributor Information
Madhulika Kabra, Email: madhulikakabra@hotmail.com.
Collaborators: Johannes Zschocke
References
- Anikster Y, Shaag A, Joseph A, Mandel H, Ben-Zeev B, Christensen E, Elpeleg ON. Glutaric aciduria type I in the Arab and Jewish communities in Israel. Am J Hum Genet. 1996;59:1012–1018. [PMC free article] [PubMed] [Google Scholar]
- Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann GF, et al. Sensitivity and specificity of free and total glutaric and 3-hydroxyglutaric acid measurements by stable- isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis. 1999;22:867–881. doi: 10.1023/A:1005683222187. [DOI] [PubMed] [Google Scholar]
- Biery BJ, Stein DE, Morton DH, Goodman SI. Gene structure and mutations of glutaryl-coenzyme A dehydrogenase: impaired association of enzyme subunits that is due to an A421V substitution causes glutaric acidemia type I in the Amish. Am J Hum Genet. 1996;59:1006–1011. [PMC free article] [PubMed] [Google Scholar]
- Boy N, Haege G, Heringer J, et al. Low lysine diet in glutaric aciduria type I- effect on anthropometric and biochemical follow-up parameters. J Inherit Metab Dis. 2013;36:525–533. doi: 10.1007/s10545-012-9517-7. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Busquets C, Merinero B, Christensen E, et al. Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically and biochemically distinct. Pediatr Res. 2000;48:315–322. doi: 10.1203/00006450-200009000-00009. [DOI] [PubMed] [Google Scholar]
- Christensen E, Ribes A, Busquets C, et al. Compound heterozygosity in the glutaryl-CoA dehydrogenase gene with R227P mutation in one allele is associated with no or very low free glutarate excretion. J Inherit Metab Dis. 1997;20:383–386. doi: 10.1023/A:1005390214391. [DOI] [PubMed] [Google Scholar]
- Christensen E, Ribes A, Merinero B, Zschocke J. Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:861–868. doi: 10.1023/B:BOLI.0000045770.93429.3c. [DOI] [PubMed] [Google Scholar]
- Discovery Studio 2.0 (2006) Molecular modeling program package. Accelrys Software Inc, San Diego
- Dwyer TM, Rao KS, Westover JB, Kim JJ, Frerman FE. The function of Arg-94 in the oxidation and decarboxylation of glutaryl-CoA by human glutaryl-CoA dehydrogenase. J Biol Chem. 2001;276:133–138. doi: 10.1074/jbc.M007672200. [DOI] [PubMed] [Google Scholar]
- Fu Z, Wang M, Paschke R, Rao KS, Freman FE, Kim JJ. Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: structural bases of dehydrogenation and decarboxylation reactions. Biochemistry. 2004;43:9674–9684. doi: 10.1021/bi049290c. [DOI] [PubMed] [Google Scholar]
- Garbade SF, Greenberg CR, Demirkol M, et al. Unravelling the complex MRI pattern in glutaric aciduria type I using statistical models-a cohort study in 180patients. J Inherit Metab Dis. 2014;37:763–773. doi: 10.1007/s10545-014-9676-9. [DOI] [PubMed] [Google Scholar]
- Goodman SI, Stein DE, Schlesinger S, et al. Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): review and report of thirty novel mutations. Hum Mutat. 1998;12:141–144. doi: 10.1002/(SICI)1098-1004(1998)12:3<141::AID-HUMU1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Gregersen N, Brandt NJ, Christensen E, Gron I, Rasmussen K, Brandt S. Glutaric aciduria: clinical and laboratory findings in two brothers. J Pediatr. 1977;90:740–745. doi: 10.1016/S0022-3476(77)81239-0. [DOI] [PubMed] [Google Scholar]
- Harting I, Neumaier-Probst E, Seitz A, et al. Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain. 2009;132:1764–1782. doi: 10.1093/brain/awp112. [DOI] [PubMed] [Google Scholar]
- Haworth JC, Dilling LA, Seargeant LE, et al. Increased prevalence of hereditary metabolic diseases among native Indians in Manitoba and northwestern Ontario. CMAJ. 1991;145:123–129. [PMC free article] [PubMed] [Google Scholar]
- Heringer J, Boy SP, Ensenauer R, et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol. 2010;68:743–752. doi: 10.1002/ana.22095. [DOI] [PubMed] [Google Scholar]
- Hoffmann GF, Trefz FK, Barth PG, et al. Glutaryl-coenzyme A dehydrogenase deficiency: a distinct encephalopathy. Pediatrics. 1991;88:1194–1203. [PubMed] [Google Scholar]
- Kamate M, Patil V, Chetal V, Darak P, Hattiholi V. Glutaric aciduria type I: a treatable neurometabolic disorder. Ann Indian Acad Neurol. 2012;15:31–34. doi: 10.4103/0972-2327.93273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyser B, Muehlhausen C, Dickmanns A, Christensen E, Muschol N, Ullrich K. Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH) Hum Mol Genet. 2008;17:3854–3863. doi: 10.1093/hmg/ddn284. [DOI] [PubMed] [Google Scholar]
- Kolker S, Garbade S, Greenberg CR, et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res. 2006;59:840–847. doi: 10.1203/01.pdr.0000219387.79887.86. [DOI] [PubMed] [Google Scholar]
- Kolker S, Garbade SF, Boy N, et al. Decline of acute encephalopathic crises in children with glutaryl-CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr Res. 2007;62:357–363. doi: 10.1203/PDR.0b013e318137a124. [DOI] [PubMed] [Google Scholar]
- Kolker S, Christensen E, Leonard JV, Greenberg CR, et al. Diagnosis and management of glutaric aciduria type I–revised recommendations. J Inherit Metab Dis. 2011;34:677–694. doi: 10.1007/s10545-011-9289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Lee CS, Chien YH, Peng SF, et al. Promising outcomes in glutaric aciduria type I patients detected by newborn screening. Metab Brain Dis. 2013;28:61–67. doi: 10.1007/s11011-012-9349-z. [DOI] [PubMed] [Google Scholar]
- Lindner M, Kölker S, Schulze A, Christensen E, Greenberg CR, Hoffmann GF. Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:851–859. doi: 10.1023/B:BOLI.0000045769.96657.af. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momany FA, Rone R. Validation of the general purpose QUANTA ®3.2/CHARMm® force field. J Comput Chem. 1992;13:888–900. doi: 10.1002/jcc.540130714. [DOI] [Google Scholar]
- Morton DH, Bennett MJ, Seargeant LE, Nichter CA, Kelley RI. Glutaric aciduria type I: a common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, Pennsylvania. Am J Med Genet. 1991;41:89–95. doi: 10.1002/ajmg.1320410122. [DOI] [PubMed] [Google Scholar]
- Mushimoto Y, Fukuda S, Hasegawa Y, et al. Clinical and molecular investigation of 19 Japanese cases of glutaric acidemia type 1. Mol Genet Metab. 2011;102:343–348. doi: 10.1016/j.ymgme.2010.11.159. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schmiesing J, Schluter H, Ullrich K, Braulke T, Muhlhausen C. Interaction of glutaric aciduria type 1-related glutaryl-CoA dehydrogenase with mitochondrial matrix proteins. PLoS One. 2014;9(2):e87715. doi: 10.1371/journal.pone.0087715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Christensen E, Superti-Furga A, Brandt NJ. The human glutaryl-CoA dehydrogenase gene: report of intronic sequences and of 13 novel mutations causing glutaric aciduria type I. Hum Genet. 1998;102:452–458. doi: 10.1007/s004390050720. [DOI] [PubMed] [Google Scholar]
- Sen A, Pillay RS. Striatal necrosis in type 1 glutaric aciduria: different stages in two siblings. J Pediatr Neurosci. 2011;6:146–148. doi: 10.4103/1817-1745.92845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov VZ, Yan L, Flook PK. The dominant role of side-chain backbone interactions in structural realization of amino acid-code. ChiRotor: a side-chain prediction algorithm based on side-chain backbone interactions. Protein Sci. 2007;16:494–506. doi: 10.1110/ps.062447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Robinson DL, et al. Type I glutaric aciduria, part 1: natural history of 77 patients. Am J Med Genet. 2003;121C:38–52. doi: 10.1002/ajmg.c.20007. [DOI] [PubMed] [Google Scholar]
- Viau K, Ernst SL, Vanzo RJ, et al. Glutaric acidemia type 1: outcomes before and after expanded newborn screening. Mol Genet Metab. 2012;106:430–438. doi: 10.1016/j.ymgme.2012.05.024. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li X, Ding Y et al (2013) Clinical and mutational spectra of 23 Chinese patients with glutaric aciduria type 1. Brain Dev pii: S0387-7604(13)00309-4 [DOI] [PubMed]
- Zschocke J, Quak E, Guldberg P, Hoffmann GF. Mutation analysis in glutaric aciduria type I. J Med Genet. 2000;37:177–181. doi: 10.1136/jmg.37.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.