

Abstract
RING E3 ligases catalyze the transfer of ubiquitin (Ub) from E2 ubiquitin-conjugating enzyme thioesterified with Ub (E2~Ub) to substrate. For RING E3 dimers, the RING domain of one subunit and tail of the second cooperate to prime Ub, but how this is accomplished by monomeric RING E3s in the absence of a tail-like component is unknown. Here, we present a crystal structure of a monomeric RING E3, Tyr363-phosphorylated human CBL-B, bound to a stabilized Ub-linked E2, revealing a similar mechanism in activating E2~Ub. Both pTyr363 and the pTyr363-induced element interact directly with Ub’s Ile36 surface, improving the catalytic efficiency of Ub transfer by ~200-fold. Hence, interactions outside the canonical RING domain are crucial for optimizing Ub transfer in both monomeric and dimeric RING E3s. We propose that an additional non-RING Ub-priming element may be a common RING E3 feature.
Keywords: CBL, RING E3, E2, ubiquitin, phosphorylation, monomeric
Posttranslational modification by Ub regulates protein functions by altering their stability, activity, localization and protein-protein interaction network and is therefore essential for diverse biological processes. Ub conjugation requires the consecutive actions of three classes of enzymes: Ub activating enzyme (E1), E2 and E3 (Ref. 1). Initially, an E1 activates and transfers Ub to an E2, where a covalent thioester is formed between E2’s catalytic cysteine and the C-terminal glycine on the tail of Ub. Subsequently, an E3 recruits substrate and E2~Ub to catalyze the formation of an isopeptide bond between the C-terminal glycine of Ub and an amino group of a lysine side chain on a protein substrate. E3s confer specificity to ubiquitination by promoting Ub transfer to select substrates; defects in E3 function are associated with many major disease pathologies including cancer, neurodegenerative disorders and metabolic disease2,3.
Over 600 E3s have been identified in mammals and most belong to the RING family of E3 ligases (reviewed in 4,5). RING E3s act as scaffolds, simultaneously recruiting both substrate and E2~Ub via different domains to promote Ub transfer in the absence of a covalent E3-Ub intermediate. The RING finger domain is essential for the recruitment of E2~Ub and comprises a defined motif of cysteine and histidine residues that bind two zinc ions in a “cross-brace” arrangement. Several types of RING E3s are recognized, depending on the oligomeric state required for function. Regardless of type, the RING–E2 interface is always distal to the E2 active site in RING E3–E2 complex structures6-9 and biochemical and recent structural studies suggest all types promote Ub transfer through an allosteric mechanism where multiple interactions between the E3, E2 and Ub lock the conformation of the tail of Ub, thereby activating the thioester bond for nucleophilic attack10-13. E2-mediated interactions with the Ile44 surface of Ub are an essential component of this mechanism14,15, and for dimeric RING E3s, recent structural studies of RING–E2–Ub complexes have shown that E3-mediated interactions with the Ile36 surface of Ub are also crucial12,13. These Ile36 surface interactions are facilitated by a cross-dimer arrangement where one subunit of the RING dimer binds E2 and Ub’s Ile36 surface and the tail of the second subunit also binds this Ub surface. Both RING- and dimer tail-mediated interactions with Ub are required for optimal Ub transfer. In monomeric RING E3s, the RING domain can mediate interactions with the Ile36 surface of Ub, but whether this is sufficient for optimal Ub transfer or requires an additional Ub-interacting structural component like the tail in RING E3 dimers is unclear.
The monomeric family of CBL RING E3 ligases (c-CBL, CBL-B, and CBL-C) attenuate non-receptor and receptor tyrosine kinase (RTK) signaling by ubiquitinating and thereby directing these kinases for degradation via the endocytic or proteasomal pathway (reviewed in 16). Members of the CBL family share a highly conserved N-terminus comprised of a tyrosine kinase-binding domain (TKBD), a linker helix region (LHR) and a RING domain (~435 amino acids). The C-terminus is more variable, comprising a proline-rich region followed by an extension. The TKBD contains a phosphotyrosine recognition motif that functions as a substrate-binding site, where phosphorylated RTKs and non-RTKs like epidermal growth factor receptor (EGFR) and ZAP-70, respectively, are recruited for ubiquitination17-19. Phosphorylation of a strictly conserved Tyr within the LHR (Tyr363 in CBL-B, Tyr371 in c-CBL and Tyr341 in CBL-C) enhances ligase activity and is required for CBL-mediated ubiquitination of RTKs17,20,21. To investigate how monomeric RING E3s promote Ub transfer, we used CBL as a model system and determined the structure of a fragment of pTyr363-CBL-B encompassing the TKBD, LHR and RING domain (residues 36–427, hereby referred to as CBL-B, Fig. 1a) bound to a ZAP-70 substrate peptide and the E2 UbcH5B linked to Ub via an isopeptide bond (UbcH5B–Ub). E3 binding promotes numerous Ub contacts required to lock the position of Ub’s tail for transfer and activate the E2~Ub thioester. The RING–Ub and E2–Ub interactions are almost identical to those observed in the dimeric RING E3–E2-Ub complexes. Notably, pTyr363 directly contacts Ub’s Ile36 surface in a manner similar to the tail of the RING dimers. Although previous studies have shown that phosphorylation of this LHR Tyr activates the ligase by inducing conformational changes that eliminate autoinhibition and bridge the gap between the RING domain and substrate-binding site22,23, this structure and accompanying biochemical and NMR data demonstrate that the phosphorylation-induced structural element is also required for positioning Ub for catalysis. We propose that monomeric RING E3s require an additional non-RING component to mediate Ub interactions for optimal activity. This is the first structure-based elucidation of the mechanism of monomeric RING-mediated Ub transfer.
Figure 1.

Structure of pCBL-B–UbcH5B-Ub–ZAP-70 peptide. (a) Diagram of the crystallized CBL-B fragment: TKBD in gray, LHR in yellow and RING domain in orange. Residues encompassing each domain are indicated. (b) Cartoon representation of the complex. Left and right panels are related by 120° rotation about the y-axis. pCBL-B domains are colored as in a. Ub is colored green; UbcH5B, light blue; ZAP-70 peptide, pink; pTyr363 side chain is shown in stick and phosphorus is colored in cyan. Zn2+ atoms are depicted as purple spheres. (c) Detailed interactions between Ub’s Ile36 surface and pCBL-B. (d) Close-up view of Ub Ile36 surface–BIRC7 dimer interactions (PDB 4AUQ13). One subunit of the BIRC7 RING dimer, RINGA, is in the same orientation as the pCBL-B RING domain in c and colored orange. The second subunit of the RING dimer, RINGB, is colored yellow and Ub and UbcH5B are colored as in b. (e) ClustalW sequence alignment of RING and U-box domains. The four highly conserved Ub-binding residues (H(Φ/T)CR) on the C-terminal Zn2+-binding loop are colored in orange. Φ indicates hydrophobic residue. Asterisks indicate monomeric RING or U-box E3s.
Results
Structure of pCBL-B–E2-Ub–ZAP-70 peptide complex
We made several protein modifications to generate suitable UbcH5B–Ub and pTyr363-CBL-B for structural and biochemical studies. We mutated UbcH5B’s Cys85 to lysine to form an isopeptide bond with the terminal glycine of Ub and Ser22 to arginine to prevent backside binding with Ub12,24. In CBL-B, both Tyr360 and Tyr363 (the phosphorylation activation site) are located on the linker helix and accessible for modification, so we mutated Tyr360 to Phe to generate homogeneously Tyr363-phosphorylated CBL-B (hereby referred to as pCBL-B). Neither the activity nor the structure is affected when the equivalent site is mutated (Y368F) in Tyr371-phosphorylated c-CBL22, and, when compared to the previously elucidated solution-based structure of the LHR and RING domain of wild type pTyr363-CBL-B23, no considerable differences are evident in the corresponding region of our pCBL-B structure (r.m.s. deviation 1.52 Å for Cα atoms of residues 353–426).
To investigate the mechanism of monomeric RING E3-mediated Ub transfer, we determined the structure of pCBL-B bound to UbcH5B–Ub and a ZAP-70 substrate peptide (Fig. 1 and Table 1). The peptide comprises the amino acid sequence from ZAP-70 (residues 286–297) recognized by the phosphotyrosine-binding motif of CBL-B and c-CBL but contains no lysines and thus cannot function as a substrate. The heterotrimeric protein-peptide complex crystallized in space group P1211 and there are four copies in the asymmetric unit (r.m.s. deviation ranging from 0.41-0.57 Å for Cα atoms); our analysis focuses on the subunit comprising chains E-H, which had the best quality of density at the Ub–RING interface. The structures of the individual protein domains are similar to the models used for molecular replacement (See Methods) and the substrate peptide-binding mode is comparable to other CBL TKBD-peptide complexes25,26.
Table 1. Data collection and refinement statistics.
| pCBL-B–UbcH5B–Ub–ZAP-70 peptide | |
|---|---|
| Data collection | |
| Space group | P1211 |
| Cell dimensions | |
| a, b, c (Å) | 95.0, 131.8, 122.0 |
| αβγ(°) | 90.0, 91.9, 90.0 |
| Resolution (Å) | 30.54–2.21(2.27–2.21)a |
| Rmerge | 0.058(0.688) |
| I/σI | 14.2(2.1) |
| Completeness (%) | 99.6(99.9) |
| Redundancy | 3.4(3.5) |
| Refinement | |
| Resolution (Å) | 30.5–2.21 |
| No. reflections | 149230 |
| Rwork/Rfree | 0.175/0.211 |
| No. atoms | |
| Protein | 19758 |
| Ligand/ion | 12 |
| Water | 776 |
| B factors | |
| Protein | 48.1 |
| Ligand/ion | 44.1 |
| Water | 41.0 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.811 |
Values in parentheses are for highest-resolution shell.
pCBL-B adopts an active configuration where pTyr363 directly contacts the RING domain, thereby placing the linker helix adjacent to the RING domain and extending the E2-binding surface as observed previously23 (Fig. 1b). The RING domain contacts the substrate-binding face of the TKBD and orients the UbcH5B–Ub linkage toward the substrate-binding site. This pCBL-B-E2–ZAP-70 peptide conformation is nearly identical to the one observed in the complex structure of Tyr371-phosphorylated c-CBL bound to UbcH5B and ZAP-70 peptide22 (r.m.s. deviation 1.43 Å for Cα atoms of pTyr371-c-CBL–UbcH5B–ZAP-70 peptide, Supplementary Fig. 1a–c). Despite the presence of Ub in the pCBL-B complex, the UbcH5B-RING interactions are comparable in the two phosphorylated CBL complexes and resemble other E2–RING interfaces6,7.
pTyr363 directly contacts Ub
pCBL-B’s pTyr363 directly interacts with Ub’s Ile36 surface (Fig. 1c and Supplementary Fig. 1d). The side chain of Ub’s Thr9 forms a hydrogen bond with the phosphate moiety from pCBL-B’s pTyr363, and Ub’s Lys11 forms hydrophobic interactions with pCBL-B’s Leu362, pTyr363 and Met366. Like the deubiquitinating enzyme DUBA, the single phosphate appears to precisely position Ub for catalysis27. To investigate whether this pTyr363–Ub interaction affects enzymatic activity, we compared Ub transfer mediated by unphosphorylated and phosphorylated CBL-B. Phosphorylation of CBL’s linker helix contributes to enzyme activity enhancement by abolishing RING–TKBD-mediated autoinhibition and positioning the RING domain adjacent to the substrate-binding site on the TKBD22,23; to eliminate these contributions so that the effect of pTyr363 on E2~Ub transfer could be assessed, we used a fragment of CBL-B comprising only the LHR and RING domain (CBL-BLRR, residues 346–427). This fragment lacks the TKBD and therefore cannot adopt an autoinhibited conformation but is defective in both auto- and substrate-ubiquitination assays; hence, we investigated Ub transfer using single turnover lysine discharge assays. Notably, pCBL-BLRR promoted discharge more quickly than the corresponding unphosphorylated fragment (Fig. 2a).
Figure 2.

Validation of pTyr363-Ub interactions. (a) Non-reduced pulse-chase lysine discharge reactions showing the disappearance of UbcH5B~Ub in the presence of CBL-BLRR or pCBL-BLRR over time. (b-f) 31P-NMR chemical shift perturbation data for pCBL-BLRR (b) alone, (c) with UbcH5B, (d) with UbcH5B–Ub, (e) with UbcH5B–Ub T9A, and (f) with UbcH5B–Ub K11A.
Furthermore, to determine whether pTyr363 contacts Ub in solution, we performed 31P-NMR on pCBL-BLRR with several UbcH5B–Ub variants under saturating conditions. Given that there is only one phosphate group in pCBL-BLRR, the chemical shift observed corresponds to the phosphate environment of pTyr363. Addition of UbcH5B alone induced a small chemical shift in pTyr363 (Δ0.02 p.p.m.). This shift substantially increased (Δ0.58 p.p.m.) when Ub was linked to UbcH5B (Fig. 2b–d). Furthermore, when Ub’s Thr9 or Lys11 were mutated to alanine, the effects of adding UbcH5B–Ub diminished, more so for Ub T9A than K11A (Fig. 2e-f). These data are consistent with our structural observations.
pTyr363-induced structural element is required for activity
Ub appears poised for transfer in the complex. In addition to the Ub–linker helix interactions, extensive contacts between Ub’s Ile36 surface and pCBL-B’s RING domain, Ub’s Ile44 surface and UbcH5B’s α3, and Ub’s tail and UbcH5B’s α2 along with residues 112–117 (Supplementary Fig. 2a–c) bury ~55% of Ub’s accessible surface area. These Ub interactions are nearly identical to those observed in the UbcH5–Ub complexes with the dimeric RING E3s BIRC7 and RNF4 (Refs. 12,13); superposition of the RING–E2–Ub portion of the pCBL-B complex onto the BIRC7 and RNF4 complexes reveals r.m.s deviations of 0.87 Å and 0.77 Å, respectively, for Cα atoms (Supplementary Fig. 2d). For the dimeric RING E3s, multiple interactions between the RING, E2 and Ub lock the tail of Ub into a conformation where the thioester is optimally oriented for nucleophilic attack12; activating the thioester not only requires these Ub–E2 and Ub–RING interactions but also depends on Ub interactions with the tail of the second dimer12,13,28,29. Remarkably, the phosphorylated linker helix in pCBL-B seemingly replaces the dimer tail in BIRC7 and RNF4 – both structural elements contact the Ile36 surface of Ub though the interaction is predominantly mediated by an aromatic residue in the dimer and the phosphate moiety in pCBL-B (Fig. 1c,d).
The parallels between pCBL-B and these two dimeric RING E3s suggest they use similar mechanisms to promote Ub transfer. To investigate pCBL-B’s mechanism, we mutated key residues in the Ub–UbcH5B and Ub–pCBL-B interfaces and tested them in single-turnover lysine discharge assays. All of these mutants were defective in Ub transfer, including those in the pTyr363-linker helix–Ub interface, thus the observed Ub interactions are crucial for Ub transfer (Supplementary Fig. 2e–g). For the dimeric RING E3s, mutations within any of the Ub–E2 or Ub–E3 interfaces increase Km and decrease kcat in steady-state analyses because stabilization of Ub is a key requirement for optimizing the conformation of the E2~Ub thioester bond for transfer13. We postulate that if the dimer tail and pTyr363-linker helix have similar functions, then removing the phosphate, perturbing the phosphate-binding site, or disrupting the pTyr363–Ub interface will likewise affect both Km and kcat. We tested these three hypotheses using CBL-BLRR, pCBL-BLRR K381A and Ub T9A, respectively, in di-Ub formation assays and found that, indeed, all three variants detrimentally affected Km and kcat when compared to the reaction with pCBL-BLRR and wild type Ub (Fig. 3 and Supplementary Fig. 3). Together, our structural and biochemical data demonstrate that the monomeric RING E3 pCBL-B uses an Ub-tail locking mechanism to promote Ub transfer like the RING E3 dimers BIRC7 and RNF4.
Figure 3.

Effect of pTyr363 on the kinetics of Ub transfer. (a) Kinetics of diUb formation catalyzed by CBL-BLRR. The rate of diUb formation was plotted against UbcH5B concentration for wild type [32P]Ub. (b) As in a but performed with pCBL-BLRR. (c) As in b but with [32P]Ub T9A (d) As in b but with pCBL-BLRR K381A. Kinetic parameters were determined from three independent datasets (n = 3). Error bars, s.d.
Discussion
Our present work provides the first structural evidence elucidating the mechanism of Ub transfer by a monomeric RING E3. Like other RING and U-box E3s, CBL-B uses an allosteric mechanism where E3–E2, E3–Ub and E2–Ub interactions restrain Ub into a closed, “folded-back” conformation and position Ub’s tail for transfer10-15. Here, we show that CBL-B positions Ub’s C-terminal tail along UbcH5B’s active site cleft in the same manner as the dimeric RING E3s BIRC7 and RNF4 (Refs. 12,13) and mirrors SUMO’s C-terminal tail as observed in the complex structure of the non-RING E3, RanBP2, bound to Ubc9 and SUMO-modified substrate30. UbcH5B’s α2 residues and the loop encompassing residues 112–117 cooperate to lock Ub’s tail into a conformation that activates the E2~Ub thioester; UbcH5B’s Asn77 stabilizes Ub’s Gly76 carbonyl oxygen via a hydrogen bond and UbcH5B’s Asp117 is poised to activate the incoming lysine as described previously12,31 (Supplementary Fig. 2b).
It is noteworthy that stabilization of the globular Ub body is a critical component for optimal positioning of Ub’s tail and hence Ub transfer12,13. This is mediated by direct interactions between the E3 and Ub’s Ile36 surface and between Ub’s Ile44 surface and UbcH5B’s α3. CBL-B adopts a strategy similar to the dimeric RING E3s BIRC7 and RNF4 in stabilizing the Ile36 surface of Ub. Two E3 components mediate these stabilizing interactions, the RING itself and an additional structural element outside the canonical RING domain (Fig. 1c,d and Fig. 4a,b). In the RING E3 dimers, the tail acts as this additional Ub-binding component, and in CBL-B, phosphorylation of Tyr363 generates a structural element adjacent to the RING domain that functionally mimics the dimer tail and enhances catalytic efficiency by ~200-fold (Fig. 3). We postulate that optimal enzymatic activity is essential in cells, where ubiquitination is highly dynamic and requires efficiency. Indeed, mutations that disrupt the linker helix pTyr-binding interface in c-CBL reduce catalytic efficiency and compromise EGFR ubiquitination in cells22. Likewise, dimeric RING E3 tail mutations also hamper substrate ubiquitination in cells8,32,33.
Figure 4.
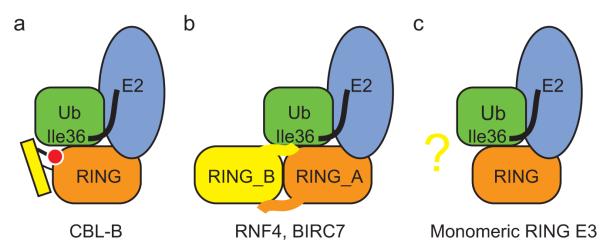
Requirements for optimal Ub transfer by RING E3s. (a-c) An additional component outside the canonical RING domain is required for stabilizing Ub by the monomeric RING E3, CBL-B (a), and the dimeric RING E3s, RNF4 and BIRC7 (b). Whether other monomeric RING E3s harbor an additional Ub-binding component requires further investigation (c). RING domain is in orange, Ub is in green with Ile36 surface indicated, E2 is in light blue, additional Ub-binding component is in yellow and phosphorylated tyrosine is in red.
Interestingly, a similar allosteric Ub-priming mechanism is used to promote transfer in SUMO E3 ligases30,34. For the SUMO RING E3 Siz1, modeling and biochemical assays suggest an acidic patch on an α-helix outside of the RING domain directly contacts the conserved basic patch on the backside of SUMO to position E2~SUMO for transfer34 (Supplementary Fig. 4). Notably, this basic patch on SUMO does not correspond to Ub’s Ile36 surface. Likewise, in the SUMO E3 RanBP2, SUMO is positioned for transfer by contacts with the E2-binding domain and an N-terminal extension that forms an anti-parallel β sheet with the adjacent SUMO surface, which is not the Ub Ile36 surface equivalent30 (Supplementary Fig. 4). Thus, additional Ub surfaces aside from Ile36 may be used to stabilize E3-mediated Ub transfer.
Our findings raise the question of whether other monomeric RING E3s require an additional Ub-binding element to stabilize Ub for transfer (Fig. 4). In all three RING E3–UbcH5–Ub complexes, the bulk of the RING-mediated Ub Ile36 surface interactions involve four highly conserved residues on the C-terminal Zn2+-binding loop of the RING domain (Fig. 1c–e). This Ub-binding RING quadrad is shared by several dimeric and monomeric RING E3s (Fig. 1e). Given that this Ub-binding quadrad alone is not sufficient for optimal activity in CBL-B but depends upon a second non-RING Ub-binding element, it seems likely that other monomeric E3s harboring this RING motif will likewise require an additional Ub-binding component to stabilize Ub for transfer. Unfortunately, no such elements are readily identifiable in other monomeric RING E3s publicly available in the Protein Data Bank. Notably, in most cases, only the canonical RING domain of the E3 ligase has been structurally elucidated.
Several monomeric RING and U-box E3s do not share this Ub-binding RING quadrad (Fig. 1e). Two examples include RBX1, a RING finger protein from a multi-subunit Cullin-RING ligase (CRL) and the U-box E4B. The structures of full length Ufd2p (a yeast homolog of E4B)35 and RBX1 in both unmodified and NEDD8-modified CRL complexes36-38 do not reveal any obvious additional Ub-binding surface outside the canonical RING or U-box domain similar to pCBL-B’s phosphorylated linker helix or the dimeric RING E3 tail. Despite this, RBX1 binds CDC34~Ub with ~50-fold higher affinity than unconjugated CDC34 (Ref. 39), and, in the case of E4B, weak U-box–Ub contacts are sufficient to drive E2~Ub toward a catalytically active closed conformation11. Variations in the Ub–E3 binding interface may preclude the necessity for additional Ub-binding components in these E3s. Alternatively, full-length protein may undergo conformational changes or require post-translational modifications to expose such an element. Likewise, another binding partner might assume this role. It is clear that further studies are required to investigate how other monomeric RING and U-box E3s optimize Ub for catalysis. Identification of additional Ub-binding elements, if present, may open new avenues for therapeutic targeting of RING or U-box E3s.
Online Methods
Protein preparation
CBL-B variants were cloned into pGEX4T1 (GE Healthcare) which contains an N-terminal glutathione S-transferase (GST) tag followed by a TEV protease or thrombin cleavage site. Proteins were expressed in E. coli BL21 (DE3) Gold or BL21 (DE3) RIL (Stratgene). For crystallization, pCBL-B was generated and purified using the same protocol as described for c-CBL22. For lysine discharge and di-Ub kinetics assays, CBL-BLRR and pCBL-BLRR variants were expressed and purified as described for crystallization except cleavage was performed on glutathione sepharose beads and the pass-back step omitted. For lysine discharge and di-Ub kinetic assays, mouse Uba1, UbcH5B and [32P]Ub were prepared as described previously13. His-tag Ub lacking Gly75 and Gly76 (His-Ub ΔGG) was expressed in pRSF_1b vector and purified by Ni-NTA affinity and size exclusion chromatography. Protein concentrations were determined by Bio-RAD protein assay using BSA as a standard and Ub concentration was determined as described previously40. Proteins were stored in 25 mM Tris-HCl (pH 7.6), 0.15 M NaCl and 1 mM DTT or 25 mM HEPES (pH 7.0), 0.15 M NaCl and 1 mM DTT at −80 °C. ZAP-70 phosphotyrosine peptide (sequence TLNSDG(p)YTPEPA) was purchased from Alta Bioscience.
Generation of UbcH5B–Ub for crystallization and NMR
Ub variants with a C-terminal Gly-Gly motif were cloned into a modified pGEX4T1 vector, containing an N-terminal His-GST-tag followed by a TEV cleavage site and a Gly-Gly-Ser linker. His-GST-Ub variants were purified by glutathione affinity chromatography and dialyzed into 25 mM Tris-HCl (pH 8.0) and 0.15 M NaCl. Untagged Arabidopsis thaliana Uba1 was expressed and purified as described previously13. Untagged UbcH5B S22R C85K was expressed from pRSF_1b and purified by SP-sepharose chromatography and dialyzed into 25 mM Tris-HCl (pH 8.0) and 0.15 M NaCl. Isopeptide-linked UbcH5B-Ub variants were formed by mixing Uba1, UbcH5B S22R C85K, and His-GST-Ub variants in 50 mM Tris-HCl 9.0, 0.2 M NaCl, 10 mM MgCl2 and 10 mM ATP at 30°C for 1 d. These UbcH5B–His-GST-Ub variants were purified by Ni-NTA affinity chromatography and His-GST tag was removed by TEV protease. Then, untagged UbcH5B–Ub variants were purified by cation exchange and SD75 1660 gel filtration chromatography and stored in 25 mM HEPES, pH 7.5, 0.2 M NaCl and 1 mM DTT at −80 °C.
Crystallization
Crystals were obtained by mixing the protein with an equal volume of reservoir solution and were grown by hanging-drop vapor diffusion at 4 °C. pCBL-B (3.5 mg ml−1) was mixed with UbcH5B–Ub (20 mg ml−1) and ZAP-70 peptide (10 mM) at a 1:1:1 molar ratio, and crystals were grown in conditions containing 0.1 M bicine, pH 9.0, 8-11% (w/v) PEG 3350 and 0.1 M sodium formate. The crystals were flash frozen in 0.1 M bicine, pH 9.0, 13% (w/v) PEG 3350 and 0.1 M sodium formate and 30% (v/v) ethylene glycol. Data were collected at beamlines I04 and I24 at DLS.
Structural determination
The data were integrated with automated XDS41 and scaled using the CCP4 program suite42. pCBL-B–UbcH5B-Ub–ZAP70 peptide complex crystals belong to space group P1211 with four molecules in the asymmetric unit. Initial phases were obtained by molecular replacement with PHASER43 using CBL-B TKBD from PDB 3PFV, a model of phosphorylated CBL-B LHR-RING bound to UbcH5B generated from PDBs 2LDR and 4A49, and Ub from PDB 4AUQ as the search models. The r.m.s. deviations for Cα atoms between the molecular replacement models and the four copies in the final model ranged from 0.55–0.59 Å, 1.20–1.22 Å, and 0.41–0.46 Å for the TKBD, pCBL-B LHR-RING–UbcH5B, and Ub, respectively. All models were built in COOT44 and refined using PHENIX45.
The complex structure (Chains A–P) was refined at a resolution of 2.21 Å and the final model contained four copies of pCBL-B (Chains A, E, M residues 38–427 and Chain I residues 41–426), ZAP-70 peptide (Chains B and N residues 4–12 and Chains F and J residues 5–12), UbcH5B (Chains C, G, K and O residues 2–147) and Ub (Chains D, H, L and P residues 1–76). Atoms from residue side chains with poor electron density were omitted. Details of the refinement statistics are shown in Table 1. All figure models were generated using PYMOL (Schrödinger).
Single-turnover lysine discharge assays
UbcH5B variants (7 μM) were charged with mouse Uba1 (0.4 μM) and Ub variants (40 μM) for 15 min at 23 °C as described previously13. Charging was stopped by incubating the reaction with 0.25 U apyrase (Sigma) and 30 mM EDTA for 5 min at 23 °C. The lysine discharge reactions were then initiated by the addition of a mixture of CBL-BLRR or pCBL-BLRR (120 nM) and L-lysine (150 mM). The final E3 and L-lysine concentrations are in parenthesis and the final UbcH5B~Ub variant concentrations were ~1.8 μM for all reactions. Reactions were quenched with 4× SDS loading buffer at the indicated time, resolved by SDS-PAGE and stained with InstantBlue (Expedeon).
Di-Ub formation assay
Concentrations of UbcH5B specified in Fig. 3 were charged independently with mouse Uba1 (1.8 μM) and [32P]Ub variants (70 μM) for 15 min at 23 °C in a buffer containing 50 mM HEPES (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 5 mM ATP, 1 mM DTT, 0.3 U ml−1 inorganic pyrophosphatase, 0.3 U ml−1 creatine kinase and 5 mM creatine phosphate. UbcH5B~[32P]Ub variants were then added to a reaction containing a mixture of His-Ub ΔGG (1 mM) and CBL-BLRR or pCBL-BLRR variants in 50 mM HEPES (pH 7.5), 50 mM NaCl. Final E3 concentrations are indicated in Supplementary Fig. 3. After 2 min, reactions were quenched with 2X SDS loading buffer containing 500 mM DTT, resolved by SDS-PAGE, dried and exposed to a phosphorimager. Under these conditions, UbcH5B~[32P]Ub variants are continuously regenerated by E1 and less than 0.2% of His-Ub ΔGG is modified by [32P]Ub at the highest UbcH5B concentration, thus representing an initial rate of reaction. In addition there was no observable autoubiquitination of CBL-BLRR or pCBL-BLRR variants. Di-Ub bands were quantified using ImageQuant (GE Healthcare). Control reactions lacking E3 for each UbcH5B~[32P]Ub variant concentration were performed for background subtraction during quantification. All reported kinetic parameters were determined by fitting at least three independent datasets to the Michaelis-Menten equation using SigmaPlot 8.0 (Systat Software Inc.).
NMR spectroscopy
Data were recorded at 25 °C on a Varian 400-MR spectrometer using a 5-mm OneNMR probe operating at 161.86 MHz for phosphorus-31. All protein samples were dialyzed into buffer containing 25 mM HEPES (pH 7.0), 0.15 M NaCl and 10% D2O. 10 μM deuterated phosphoric acid was added as an internal reference to normalize the chemical shifts from different spectra. 31P spectra were recorded for pCBL-BLRR (150 μM) alone and together with UbcH5B or UbcH5B–Ub variants (150 μM) in an equimolar ratio. Due to peak broadening in the pCBL-BLRR–UbcH5B–Ub complex spectra, higher protein mixture concentrations (225 μM for pCBL-BLRR and 300 μM for UbcH5B–Ub) were used to enhance the signal.
Supplementary Material
Acknowledgements
We would like to thank A. Schuettelkopf for discussion. W. Clark and A. Keith for in-house DNA sequencing; Diamond Light Source (DLS) for access to beamlines I04 and I24 beamlines (mx6683) that contributed to the results presented here. This work was supported by Cancer Research UK.
Footnotes
Accession codes
Coordinates and structure factors for pCBL-B-UbcH5B–Ub–ZAP-70 peptide complex have been deposited in Protein Data Bank under accession code 3zni.
References
- 1.Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 2.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–44. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 3.Petroski MD. The ubiquitin system, disease, and drug discovery. BMC Biochem. 2008;9(Suppl 1):S7. doi: 10.1186/1471-2091-9-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 5.Budhidarmo R, Nakatani Y, Day CL. RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012;37:58–65. doi: 10.1016/j.tibs.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–9. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 7.Dominguez C, Bonvin AM, Winkler GS, van Schaik FM, Timmers HT, Boelens R. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure. 2004;12:633–44. doi: 10.1016/j.str.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Mace PD, et al. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 2008;283:31633–40. doi: 10.1074/jbc.M804753200. [DOI] [PubMed] [Google Scholar]
- 9.Yin Q, et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 2009;16:658–66. doi: 10.1038/nsmb.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozkan E, Yu H, Deisenhofer J. Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc. Natl. Acad. Sci. USA. 2005;102:18890–5. doi: 10.1073/pnas.0509418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pruneda JN, et al. Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell. 2012;47:933–42. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489:115–20. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 2012;19:876–83. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saha A, Lewis S, Kleiger G, Kuhlman B, Deshaies RJ. Essential role for ubiquitin-ubiquitin-conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol. Cell. 2011;42:75–83. doi: 10.1016/j.molcel.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011;144:769–81. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohapatra B, et al. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta. 2013;1833:122–39. doi: 10.1016/j.bbamcr.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levkowitz G, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell. 1999;4:1029–40. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- 18.Lupher ML, Jr., Songyang Z, Shoelson SE, Cantley LC, Band H. The Cbl phosphotyrosine-binding domain selects a D(N/D)XpY motif and binds to the Tyr292 negative regulatory phosphorylation site of ZAP-70. J. Biol. Chem. 1997;272:33140–4. doi: 10.1074/jbc.272.52.33140. [DOI] [PubMed] [Google Scholar]
- 19.Rao N, Lupher ML, Jr., Ota S, Reedquist KA, Druker BJ, Band H. The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J. Immunol. 2000;164:4616–26. doi: 10.4049/jimmunol.164.9.4616. [DOI] [PubMed] [Google Scholar]
- 20.Kassenbrock CK, Anderson SM. Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J. Biol. Chem. 2004;279:28017–27. doi: 10.1074/jbc.M404114200. [DOI] [PubMed] [Google Scholar]
- 21.Ryan PE, Sivadasan-Nair N, Nau MM, Nicholas S, Lipkowitz S. The N terminus of Cbl-c regulates ubiquitin ligase activity by modulating affinity for the ubiquitin-conjugating enzyme. J. Biol. Chem. 2010;285:23687–98. doi: 10.1074/jbc.M109.091157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dou H, Buetow L, Hock A, Sibbet GJ, Vousden KH, Huang DT. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat. Struct. Mol. Biol. 2012;19:184–92. doi: 10.1038/nsmb.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobashigawa Y, Tomitaka A, Kumeta H, Noda NN, Yamaguchi M, Inagaki F. Autoinhibition and phosphorylation-induced activation mechanisms of human cancer and autoimmune disease-related E3 protein Cbl-b. Proc. Natl. Acad. Sci. USA. 2011 doi: 10.1073/pnas.1110712108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE. A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell. 2006;21:873–80. doi: 10.1016/j.molcel.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 26.Ng C, Jackson RA, Buschdorf JP, Sun Q, Guy GR, Sivaraman J. Structural basis for a novel intrapeptidyl H-bond and reverse binding of c-Cbl-TKB domain substrates. EMBO J. 2008;27:804–16. doi: 10.1038/emboj.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang OW, et al. Phosphorylation-dependent activity of the deubiquitinase DUBA. Nat. Struct. Mol. Biol. 2012;19:171–5. doi: 10.1038/nsmb.2206. [DOI] [PubMed] [Google Scholar]
- 28.Plechanovova A, et al. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 2011;18:1052–9. doi: 10.1038/nsmb.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakatani Y, Kleffmann T, Linke K, Condon SM, Hinds MG, Day CL. Regulation of ubiquitin transfer by XIAP, a dimeric RING E3 ligase. Biochem. J. 2012 doi: 10.1042/BJ20121702. [DOI] [PubMed] [Google Scholar]
- 30.Reverter D, Lima CD. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005;435:687–92. doi: 10.1038/nature03588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yunus AA, Lima CD. Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat. Struct. Mol. Biol. 2006;13:491–9. doi: 10.1038/nsmb1104. [DOI] [PubMed] [Google Scholar]
- 32.Feltham R, et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 2011;286:17015–28. doi: 10.1074/jbc.M111.222919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26:102–12. doi: 10.1038/sj.emboj.7601469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yunus AA, Lima CD. Structure of the Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO modification of PCNA. Mol. Cell. 2009;35:669–82. doi: 10.1016/j.molcel.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu D, Li W, Ye Y, Brunger AT. Structure and function of the yeast U-box-containing ubiquitin ligase Ufd2p. Proc. Natl. Acad. Sci. USA. 2007;104:15599–606. doi: 10.1073/pnas.0701369104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng N, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 37.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calabrese MF, et al. A RING E3-substrate complex poised for ubiquitin-like protein transfer: structural insights into cullin-RING ligases. Nat. Struct. Mol. Biol. 2011;18:947–9. doi: 10.1038/nsmb.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spratt DE, Wu K, Kovacev J, Pan ZQ, Shaw GS. Selective recruitment of an E2~ubiquitin complex by an E3 ubiquitin ligase. J. Biol. Chem. 2012;287:17374–85. doi: 10.1074/jbc.M112.353748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 40.Bohnsack RN, Haas AL. Conservation in the mechanism of Nedd8 activation by the human AppBp1-Uba3 heterodimer. J. Biol. Chem. 2003;278:26823–30. doi: 10.1074/jbc.M303177200. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. Xds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collaborative Computational Project, N The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 43.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:432–8. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.