

Abstract
The movement of ions across specific channels embedded on the membrane of individual cardiomyocytes is crucial for the generation and propagation of the cardiac electrical impulse. Emerging evidence over the last 20 years strongly suggests that the normal electrical function of the heart is the result of dynamic interactions of membrane ion channels working in an orchestrated fashion as part of complex molecular networks. Such networks work together with exquisite temporal precision to generate each action potential and contraction. Macromolecular complexes play crucial roles in transcription, translation, oligomerization, trafficking, membrane retention, glycosylation, posttranslational modification, turnover, function and degradation of all cardiac ion channels known to date. In addition, the accurate timing of each cardiac beat and contraction demands, a comparable precision on the assembly and organizations of sodium, calcium and potassium channel complexes within specific subcellular microdomains, where physical proximity allows for prompt and efficient interaction. This review article, part of the Compendium on Sudden Cardiac Death, discusses the major issues related to the role of ion channel macromolecular assemblies in normal cardiac electrical function and the mechanisms of arrhythmias leading to sudden cardiac death. It provides an idea of how these issues are being addressed in the laboratory and in the clinic, which important questions remain unanswered, and what future research will be needed to improve knowledge and advance therapy.
Keywords: Ion channels, multiprotein complexes, genetics, arrhythmias, sudden cardiac death
1. Introduction
Fundamental research conducted over the last 20 years has led to an explosion of knowledge on the genetic and molecular mechanisms that regulate the function of cardiac ion channels. One of the most important outcomes of such new understanding has been the realization that the traditional reductionist view that ionic currents are the expression of distinct proteins that are fixed and function independently expressed on an intracellular or surface membrane is no longer tenable. An ion channel protein may encounter and interact with hundreds of other proteins during its lifespan, from biosynthesis until degradation. Such a complex regulation over time and space suggests an important plasticity for these protein complexes which is a major determinant of cardiomyocyte function, including excitability, excitation-contraction (e-c) coupling, intercellular communication and the pathogenesis of arrhythmias. This article is part of the Circulation Research Compendium on Sudden Cardiac Death. It reviews research on many of the currently known multicomponent assemblies formed by the main cardiac ion channels with their protein partners. It looks also at the possible role that such assemblies may have in the molecular underpinnings of the normal electrical function of the cardiomyocyte and the mechanisms of complex cardiac arrhythmias and sudden cardiac death (SCD). We are focusing on cis-interacting proteins, i.e. within the same cell. While there is emerging evidence for important roles of proteins such as the β-subunits of the voltage-gated sodium channels in trans-interactions as cell-adhesion molecules,1 this aspect is not addressed in this review.
a. Ion channel macromolecular complexes
Macromolecular complexes consist of a handful to several thousand individual components, including proteins, nucleic acids, carbohydrates and lipids, and perform a wide array of vital tasks in the cell.2 As such, they are essential to the proper functioning of all cellular processes, including metabolism, cell signaling, gene expression, trafficking, cell cycle regulation and the formation of subcellular structures.3, 4 In the cardiac myocyte, macromolecular complexes also play crucial roles in converting energy, generating and propagating electrical signals and mediating contractility, as well intercellular communication.5, 6 To achieve these functions, complex molecular networks work together with exquisite temporal precision to generate each AP and contraction. The accurate timing of the molecular events demands, in addition, a comparable precision on the location of each molecule within the cell. Indeed, molecular networks assemble and organize within specific subcellular microdomains, where physical proximity allows for prompt and efficient interaction.7 For example, Petitprez et al8 described two separate pools of sodium channels in ventricular cardiomyocytes. One subpopulation localizes at the lateral membrane of the myocytes, while the other localizes at the intercalated disc (ID), and a recent study has shown that NaV1.5 and Kir2.1 co-localize at both the ID and the lateral membrane, which is important for mutual regulation and the control of cardiac excitability.9
b. Genetic cardiac channelopathies
Genetic cardiac channelopathies were identified over 20 years ago.10 As of today, more than 35 distinct genes encoding ion channel subunits or regulatory proteins are known to be linked to arrhythmogenic syndromes.11 The estimated prevalence of cardiac channelopathies in the general population remains however difficult to assess.12 Cardiac channelopathies are likely responsible for about half of sudden arrhythmic cardiac death cases.13 The most prevalent genetic disorder is the congenital long QT syndrome (LQTS). LQTS is caused by mutation-induced decrease in repolarizing currents or by increase in depolarizing currents. The second most frequent cardiac channelopathy is Brugada syndrome (BrS).14 The molecular mechanisms underlying BrS are still matter of controversy.15 Other important but more recently described forms of inherited arrhythmias caused by channel dysfunction include catecholaminergic polymorphic ventricular tachycardia (CPVT),16 congenital short QT syndrome (SQTS)17, 18 and mixed phenotypes.19
2. Sodium channel macromolecular complexes
The main voltage-gated sodium channel expressed in cardiac myocytes is NaV1.5;20 it is encoded by the human gene SCN5A. NaV1.5 is a large pore-forming protein, also called α-subunit, with 2016 amino acids and of a molecular weight of ~220 kDa (Fig. 1). The NaV1.5 protein has been shown to assemble with small (~30–40 kDa), single transmembrane segment proteins called β-subunits.21 Four of these β-subunits have been described in the human genome.21 The exact stoichiometry between the α and β-subunits of the cardiac Na+ channels is not known. However, the brain α-subunit of the Na+ channels was co-purified with one β1 and one β2 subunit suggesting a possible1:2 α to β stoichiometry.22 Several hundreds of mutations in SCN5A have been linked to cardiac arrhythmic disorders, such as the congenital and acquired LQTS, BrS, conduction slowing, sick sinus syndrome, atrial fibrillation, and dilated cardiomyopathy.23 This impressive list of allelic disorders underlines the crucial role of NaV1.5 in physiology and diseases.
Figure 1.
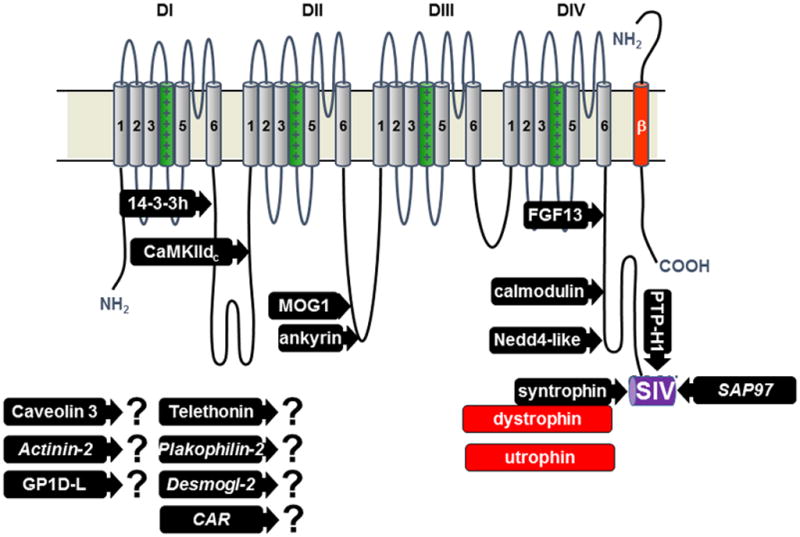
Topography of NaV1.5 channel and its interacting proteins. The proteins for which a binding site has been mapped are represented: 14-3-3 protein _-isoform, calmodulin-dependent protein kinase II delta-c, MOG1, ankyrin-g, fibroblast growth factor like 13, calmodulin, Nedd4-2 like ubiquitin ligases, syntrophin proteins adapting either dystrophin or utrophin, protein tyrosine phosphatase-H1, synapse associated protein-97. The proteins with question marks were found to interact with NaV1.5 but the sites of interaction are not yet known (CAR is coxsackie and adenovirus receptor, Desmogl-2 is desmoglein-2). Only one of the four beta subunits is represented (in red).
a. NaV1.5 interacting proteins
NaV1.5 interacts with and is regulated by a myriad of proteins6 hence forming macromolecular complexes (Fig. 1). These different interacting proteins reside in specific subcellular regions of the cardiac myocytes, such as the lateral membrane domains or the ID, thus defining distinct pools of NaV1.5 channels co-existing in cardiac cells.24 Importantly, mutations in the genes coding for some of these partner proteins were found in patients with genetic cardiac channelopathies, e.g. congenital LQTS and BrS.17 The proteins interacting with NaV1.5 may have different functions such as anchoring/adaptor proteins involved in trafficking, targeting, and anchoring of the channel protein to specific membrane compartments; as enzymes interacting with and modifying the channel structure via post-translational modifications; and as proteins modulating the biophysical properties of NaV1.5 upon binding. For further details see the recent review article.24
Among the proteins that have been proposed to be involved in targeting the Nav1.5 channel proteins to specific compartments, α1-syntrophin (Fig. 2A) and the MAGUK protein SAP97 play crucial roles (Fig. 1). Both proteins have PDZ (post synaptic density protein [PSD95], Drosophila disc large tumor suppressor [Dlg1], and zonula occludens-1 protein [zo-1]) protein-protein interacting domains allowing the direct interaction with the three last C-terminal residues of NaV1.5 (serine– isoleucine-valine or SIV-motif). Recent studies using genetically-modified mouse models indicated a role of the syntrophin-dystrophin macromolecular complex and the key role of the SIV-motif in determining the density of NaV1.5 channels at the lateral membranes of myocytes (Fig. 3).25, 26 While the role of the SIV-motif and SAP97 at the ID remains to be clarified, neither truncated channels (ΔSIV), nor the cardiac ablation of SAP97 were sufficient to perturb the expression of NaV1.5 at the ID of mouse cardiac cells (Fig. 2A and 2B).25, 26 Two other distinct protein-protein interacting domains are well-recognized in the C-terminal sequence of NaV1.5 (Fig. 1):27 the IQ-motif allowing specific interaction with calmodulin and the PY-motif, a domain found in membrane proteins permitting the binding of ubiquitin-ligases of the Nedd4/Nedd4-like family (reviewed in28). While the structural details and roles of the interaction of calmodulin and NaV1.5 have been controversial, a recent study29 suggested a model where calmodulin may be an essential molecular player in the transitions between the different channel states.29
Figure 2.
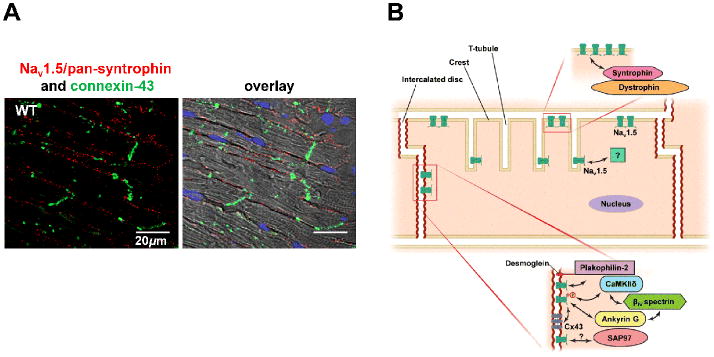
(A) Proximity ligation assay staining using antibodies for NaV1.5 and pan-syntrophin demonstrating the specific location of the interaction between these two proteins at the lateral membranes of mouse cardiac cells (red dots). In green, immunofluorescence staining demonstrating the present of connexin-43 at the IDs (modified with permission from Shy et al. 2014). (B) Depending on the partner proteins they interact with, NaV1.5 is found either at the ID region, or at the lateral membrane (composed of crest regions and T-tubules) of cardiomyocytes. Along the crests, functional sodium channels do not distribute homogenously, but segregate in densely-populated clusters, coexisting with areas devoid of functional channels.
Figure 3.
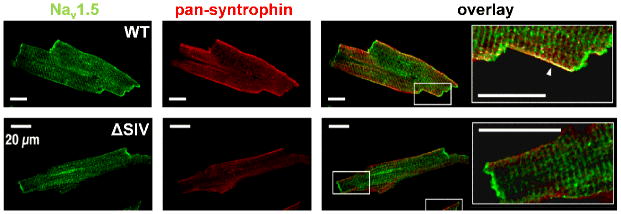
(Upper panels) Isolated mouse ventricular myocyte with double immunofluorescence staining (imaged with confocal microscopy). NaV1.5 (green) is expressed at the IDs, lateral membrane. The punctate staining most-likely represents the expression at the t-tubules. Syntrophin is only expressed at the lateral membrane where it co-localize with NaV1.5 (see arrow in merge showing the yellow region of co-localization). (Lower panels) Stainings of myocytes from genetically-modified mice (truncation of the last three residues of NaV1.5 interacting with syntrophins and SAP97, ΔSIV) illustrating the reduction of Nav1.5 expression exclusively at the lateral membrane, (modified with permission from Shy et al. 2014).
b. Mutations in genes coding for NaV1.5 channel interacting proteins and associated disorders
Among the long list of proteins interacting with Nav1.5 (Fig. 1),30 mutations in the genes coding for 6 of them were reported in patients with altered electrical function that may lead to SCD. Also important, more than 20 naturally-occurring mutations have been described in the genes coding for the 4 β-voltage-gated sodium channel subunits.31 These mutations were found in patients with SCD phenotypes such as BrS, sudden infant death syndrome (SIDS), sudden unexpected nocturnal death syndrome (SUNDS), and idiopathic VF. The molecular mechanisms underlying the observed phenotypes were diverse, but the majority of these β-subunit mutations reduced the NaV1.5-mediated INa.31
We review here briefly the evidence demonstrating that mutations of the proteins of the NaV1.5 macromolecular complexes cause severe electrical disturbances.
α1-syntrophin
NaV1.5 is part of the dystrophin multi-protein complex; Gavillet et al32 demonstrated that NaV1.5 interacts with dystrophin via adaptor syntrophin proteins (see also Fig. 2A).32 Similar to the binding with SAP97, this interaction is dependent on the last three residues (SIV) of the NaV1.5 protein. Two missense mutations in SNTA1, encoding α1-syntrophin, have been described in patients with congenital LQTS.33 The SNTA1 mutation, p.A390V was reported to disrupt a macromolecular complex comprising neuronal nitric oxide synthase (nNOS), plasma membrane Ca-ATPase type 4b and NaV1.5 with syntrophin.33 The mutant syntrophin protein increased the late Na+ current, a finding that is consistent with the LQTS phenotype. Increased nitrosylation of NaV1.5, when the mutant syntrophin was co-expressed in HEK293 cells was observed. Further, the mutation SNTA1 p.A257G was found in 3 unrelated LQTS probands.34 While no increase in the late INa was observed with this variant, significant increase in peak INa and slowed fast inactivation resulted from the co-expression of this mutant syntrophin. The gene SNTA1 was also found to be mutated in 8 cases of SIDS patients and these variants caused an increase of the NaV1.5-mediated late INa which was inhibited by nNOS inhibitors.34
Caveolin-3
Caveolin proteins are important components of caveolae, which are cholesterol-rich plasma membrane invaginations where signaling molecules and ion channels are enriched. Caveolin-3 is encoded by the gene CAV3; it is the predominant caveolin isoform expressed in cardiac cells. CAV3 was found to be mutated in patients with congenital LQTS and SIDS.35, 36 Caveolin-3 was co-immunoprecipitated with NaV1.5 in rat cardiac tissue and HEK293 cells.35, 37 Immunofluorescence stainings showed that the two proteins are co-localized at the lateral membrane of the cardiomyocytes.35 The co-expression of NaV1.5 and the LQTS and SIDS mutants of caveolin-3 in HEK293 cells were also shown to increase the late Na+ inward current.35, 36 It has been proposed that both CAV3 and SNTA1 mutations share a common mechanism in releasing inhibition of nNOS, leading to an increase in NaV1.5 S-nitrosylation and, as a result, augmented late INa.30
Glycerol-3-phosphate dehydrogenase 1 like protein
Glycerol-3-phosphate dehydrogenase like protein (GPD1L) is an enzyme homolog to glycerol-3-phosphate dehydrogenase 1, a key enzyme of the respiratory chain. In 2002, a locus on chromosome 3 in a family with BrS was detected, and later a missense mutation in the gene coding for GPD1L was observed.38, 39 Co-expression experiments showed that mutant GPD1L reduced the NaV1.5-mediated INa. Three other GPD1L mutations have been described in babies that died of SIDS.40 Expression of these SIDS variants in neonatal mouse cardiomyocytes also decreased the INa, demonstrating that SIDS patients may have decreased INa similarly to BrS. The mechanisms by which the mutations of GPD1L reduce the INa have been investigated in expression systems.41 It is proposed that Ser-1503 of NaV1.5 is phosphorylated by protein kinase C (PKC) and that this reduces the INa. It has been shown that the activity of PKC depends on GPD1L function, and that the mutant GPD1L variants lead to a further decrease in the INa following a diacylglycerol-dependent stimulation of PKC.41 Another possible mechanism is that the GPD1L mutant increases nicotinamide adenine dinucleotide phosphate and via PKC effects on mitochondria, and this decreases reactive oxygen species, which then reduce the INa by yet unknown mechanisms.42
MOG1
Multi-copy suppressor of gsp1 (MOG1) is a 29-kd protein encoded by the RANGRF gene. MOG1 interacts with the intracellular loop of NaV1.5 between domains II and III.43 The two proteins also co-localize at the IDs in mouse ventricular cells. MOG1 co-expression in HEK293 cells increased the NaV1.5-mediated current without altering its biophysical properties, suggesting that MOG1 is a co-factor for optimal channel expression at the cell membrane. A human study described one BrS variant of MOG1 that reduced the expression of NaV1.5 at the cell membrane of rat atrial cardiomyocytes and decreased the INa.44 The details about how MOG1 regulates the expression of NaV1.5 are still to be investigated.
Plakophillin-2
Plakophilin-2 (PKP2) is found at the IDs of cardiomyocytes. Delmar’s group demonstrated that NaV1.5 interacts not only with PKP2 at the IDs, but also in a complex with ankyrin-G and connexin-43.45, 46 Whether the interactions between these different proteins of the IDs are direct or indirect, and the site of interaction with NaV1.5 remains to be determined. In a recent study with 200 BrS patients, 5 missense mutations in the gene PKP2 were demonstrated.47 The INa and the density of Nav1.5 channels at the ID were reduced when PKP2 mutant proteins were co-expressed.48 This experimental evidence strongly supports a role for PKP2 in targeting and regulating the density of NaV1.5 at the IDs, and also its implication in BrS.
Fibroblast growth factor homologous factors (FGFs)
FGFs are cytosolic proteins that can modulate both cardiac Na+ and Ca2+ channels. 49 The proximal part of the C-terminal domain of Nav1.5 has been shown to bind to murine FGF13 - and human FGF12 (Fig. 1).50, 51 Knockdown of FGF13 in murine ventricular myocytes decreased INa and channel availability.50 Interestingly, a genetic variant of the gene coding for human FGF12 (p.Q7R) was identified in one BrS patient.51 When expressed in rat myocytes, this variant reduced INa and channel availability, hence leading to a loss of function which is consistent with the BrS phenotype.
Synapse Associated Protein 97
SAP97 regulates the targeting, localization and function of cardiac K+ and sodium channels via their PDZ-domain binding motifs located in the C-termini. The interaction between SAP97 and NaV1.5 has been demonstrated independently by the Abriel and the Jalife labs.8, 9, 25 While there is strong experimental evidence for a direct interaction between these two proteins via the NaV1.5-SIV motif and a SAP97 PDZ-domain, the exact role of SAP97 on the regulation of NaV1.5 function remains to be clarified.26 A mutation found in one patient with BrS modified the valine of the SIV motif of NaV1.5 into a methionine. This mutation was shown to specifically reduce the interaction with SAP97, but not α1-syntrophin. In parallel the mutation decreased the number of NaV1.5 channels and INa.25 It seems likely that genetic variants in DLG1, the gene coding for SAP97, will be found in patients with inherited channelopathies associated to SCD.
3. Calcium channel macromolecular complexes
The voltage-gated L-type calcium channel, CaV1.2, is the main pathway for the entry of calcium into cardiac cells.52 The pore-forming CaVα1 subunit carries the main biophysical and pharmacological properties of the channel that plays a key role in e–c coupling and AP duration. The CaVα1 subunit is modulated by interactions with different accessory subunits (Fig. 4). It is associated with 4 different β-isoforms (CaVβ1 to 4) and 4 different α2-δ-isoforms (CaVα2-δ1 to 4). Both Cavβ and Cavα2-δ have been shown to play dual roles in regulating both the biophysical properties and trafficking of CaV channels. In addition to these regulatory subunits, the γ subunits (8 isoforms) have been described as a third class of accessory subunits.53 In cardiomyocytes, the fully functional CaV1.2 channel which is composed of at least CaVα1, CaVβ, and CaVα1-δ subunits (Fig. 4) can be considered as the main core macromolecular calcium channel complex.
Figure 4.

Cav1.2 channels subunits (Cavα1, Cavβ, Cavα2-δ, and Cavγ) and their major interacting proteins. Ahnak1, Nedd4-1 (Neural precursor cell Expressed, Developmentally Down-regulated 4-1), RGK (Rem, Rem2, Rad, and Gem/Kir), Cav-3 (caveolin-3), PKA (protein kinase A), CaMKII (Ca2+/calmodulin-dependent protein kinase II), and USP2-45 (Ubiquitin carboxyl-terminal hydrolase 2 isoform 45). ). Illustration credit: Ben Smith.
a. CaV1.2 interacting proteins
All four CaVβs increase the Ca2+ current when they are co-expressed in heterologous expression systems along with a CaVα1 subunit. CaVβs also alter the voltage dependence and kinetics of activation and inactivation. Furthermore, CaVβ subunits either regulate or are needed for the modulation of CaVα1 by protein kinases, G proteins, ubiquitin ligases of the Nedd4 family, and small RGK proteins (Fig. 4).54, 55 CaVβ proteins have also been shown to interact with Ahnak1,56 a protein involved in the protein kinase A (PKA)-mediated control of the CaV1.2 channel. Altogether, these data demonstrate that CaVβ subunits play a pivotal role in the localization and regulation of cardiac calcium channels.
The CaVα1-δ auxiliary subunits are the product of a single gene that is post-translationally cleaved into α2 and δ peptides and remain associated via disulfide bonds.57 Only CaVα1-δ1 and CaVα1-δ2 proteins have been found to be expressed in mouse heart.58 Co-expression of the CaVα1-δ subunit, along with CaVα1 and Cavβ subunits, accelerated activation and deactivation kinetics and significantly increased ICa.58 Animals lacking the CaVα1-δ1 subunit demonstrated reduced basal myocardial contractility and relaxation and decreased L-type Ca2+ peak current amplitude.59 CaVα1-δ1 has been recently shown to be essential in the regulation of the CaV1.2 channel cell surface density mediated by the deubiquitylase USP2-45.60
The CaVγ proteins consist of 4 transmembrane domains with intracellular N- and C-terminal ends. In the human heart, only CaVγ4, 6, 7, and 8 have been found to be expressed53 The CaVγ subunits differentially modulate calcium channel function when co-expressed with the CaVβ1b and CaVα1-δ1 subunits, altering both activation and inactivation properties.53 The effects of Cavγ on Cav1.2 function are dependent on the subtype of Cavβ subunit.53
In cardiac myocytes, Cav1.2 channels are mainly localized in the t-tubular system (Fig. 5).61 L-type calcium channels are _ 3 - 9 - fold enriched in the t-tubule membrane than on the extra-tubular surface sarcolemma. Within the t-tubule, studies have estimated that ~75% of the L-type calcium channels are localized in the dyad domains (Fig. 5), thus constituting the main Cav1.2 macromolecular complex in cardiac cells. The dyad represents a small area where Cav1.2 channels, situated on the cytoplasmic side of the plasma membrane, are in opposition to the type-2 ryanodine receptors (RyR2) that are situated on the membrane of the sarcoplasmic reticulum (SR). An essential component of the dyadic cleft is junctophilin 2 (JPH2) (Fig. 5). JPH2 is a cleft protein that anchors the T-tubular membrane to the SR membrane; doing so it plays a key role in maintenance and function of that space.62 In JPH2 knock-down mice, reduction in the number of dyads was observed63 suggesting that JPH2 is responsible for maintaining the dyadic structure A calcium-binding protein, sorcin, expressed in cardiac cells, has also been shown to interact with both Cav1.2 and RyR2.64 Finally, PKA and Ca2+/CaM-dependent protein kinase II (CaMKII), which are known to mediate the regulation of Cav1.2 channel activity via their interactions with the C-terminal of the CaVα1C subunit, have also been shown to be part of the dyad.65
Figure 5.

Scheme showing the protein composition of the three CaV1.2-macromolecular complexes (dyad, extra-dyad, and extra t-tubule). (1) The extra-t-tubule; CaV1.2 channels and CaV-3 (caveolin-3) (2) The extra-dyad: CaV1.2 channels, Bin1 (bridging Integrator 1/amphiphysin 2), dysferlin, β2-AR (β2-adrenergic receptor), ahnak1, and calcineurin, and (3) The dyad: CaV1.2 channels, RyR2 (type 2 ryanodine receptor), sorcin, and JPH2 (junctophilin 2). Illustration credit: Ben Smith.
Recently, other partner proteins have been described to be important to regulate CaV1.2 expression at the t-tubule. Amphiphysin 2, also called Bridging Integrator 1 (BIN1), belongs to the BAR domain proteins superfamily and is involved in membrane invagination.66 Hong and colleagues have shown that BIN1 is expressed at the t-tubules,66 initiates t-tubule genesis and delivers CaV1.2 to t-tubules.66 Ahnak1 is indirectly associated with the L-type Ca2+ channel via its β2-subunit and has been shown to be located at the sarcolemma and t-tubules of cardiomyocytes.56 Similar to BIN1, the exact localization within the t-tubule system is not known. Nevertheless, its implication in the regulation of ICa via the β-adrenergic pathway suggests the presence of Ahnak1 and CaV1.2 channels in extra-dyad complexes. Dysferlin, a member of the ferlin family, has recently been shown to be expressed mainly at the ID of cardiomyocytes and is also present at the t-tubules.67 These observations suggest that other CaV1.2 macromolecular complexes, which may be caveolin-3–dependent, also exist in the extra-dyadic compartment (Fig. 5). Recently, a subpopulation of CaV1.2 channels that is located in the caveolae has been found to be part of a macromolecular signaling complex including β2-adrenergic receptor, adenylate cyclase, protein phosphatase 2A, and PKA.68 Calcineurin, another interacting/regulating protein of CaV1.2 channels, has also been shown to be present in caveolae and the t-tubule system.69 Via its association with the adapter protein AKAP5, calcineurin interacts with Cav-3.70 Altogether, these findings suggest the presence of t-tubular distinct CaV1.2 macromolecular complexes that are also present in extra-dyadic compartments (Fig. 5).
In parallel, other groups have demonstrated that a subpopulation of CaV1.2 channels is localized to caveolae in the extra-T-tubular lateral membrane of ventricular cardiomyocytes (Fig. 5), thus suggesting that at least a third CaV1.2 macromolecular complex exists.68
b. Mutations in genes coding for calcium channel interacting proteins and associated disorders
Mutations in genes coding for calcium channel accessory subunits have been linked to BrS and short QT syndrome type-6.71, 72 A loss-of-function mutation (p.S481L) of the CACNB2 gene, encoding the Cavβ2 subunit, was found in a BrS patient.71 Despite the large ICa mutation-induced decrease in heterologous expression systems, no reduction of CaV1.2 channel number has been observed at the plasma membrane. This suggests another mechanism of Cav regulation than the traffic defect that is generally observed in BrS. Templin et al. reported a mutation (p.S755T) in CACNA2D1, the gene encoding the CaVα2-δ1 subunit, in a SCD patient with SQTS.72 An important decrease of the ICa was observed with the expression of the mutant variant without any modification of the protein expression, thus suggesting that the single channel biophysical properties of the L-type channel were altered.
The p.I5236T mutation of Ahnak1, identified in patients with hypertrophic cardiomyopathy,73 increased the ICa as well as shifted slightly leftward its voltage dependence,73 similar to what has been observed after PKA activation. It is proposed that Ahnak1 may be an important target of PKA-mediated phosphorylation in the enhancement of L-type ICa by the β-adrenergic receptor type 2 (β-AR2). Furthermore, three other Ahnak1 variants were identified in hypertrophic and dilated cardiomyopathy patients.74 Contrary to what has been proposed in the former study, it was recently found that Ahnak1 is not essential for β-adrenergic up-regulation of ICa in mice. Instead, Ahnak1 interacts with the Cavβ subunit in order to modulate the β-adrenergic response of ICa.74
4. Potassium channel macromolecular complexes and associated disorders
Cardiac potassium channel proteins are coded by more than 40 different genes.75 In addition, a number of auxiliary subunits and associated proteins are involved in the trafficking, distribution and anchoring of potassium channels at specific microdomains at the plasma membrane, and contribute to their organization in macromolecular complexes.76 Such partners help in the control of potassium channel expression and biophysical properties, thus regulating the plasticity of cardiac electrical activity both under normal conditions and in disease states. In this section we review the interactions of the major potassium channels as part of macromolecular assemblies and the role of such assemblies in cardiac excitation and repolarization.
a. The Strong Inward Rectifying Potassium Channels
Among the three strong inward-rectifying potassium channels (Kir2.1, 2.2 and 2.3) that express in the heart, Kir2.1 is the most abundant in the ventricles. Kir channels are responsible for IK1 and are involved in the depolarization, repolarization and resting phases of the cardiac AP.77 Kir subunits assemble to form tetrameric channels in many cell types, including cardiac myocytes.78, 79 IK1 contributes significant repolarizing current between –30 and –80 mV, and thus is responsible for the terminal phase of the AP.80 In addition, it serves as the primary conductance controlling the resting membrane potential in ventricular myocytes.81 These channels show strong rectification between –50 and 0 mV, which means that they remain closed during the AP plateau; they only open when the membrane potential repolarizes to levels between –30 and –80 mV, which in the normal AP occurs during the late phases of the AP. Rectification is achieved by a voltage-dependent blockade by intracellular magnesium and/or polyamines such as putrescine, spermine and spermidine,82 which interact with at least three amino acid residues located inside the pore of the channel.79
Loss of function mutations of Kir2.1 have been identified in patients affected by Andersen-Tawil Syndrome (ATS), which is also referred to as LQTS type 7 and is characterized by delayed repolarization.83 Since in addition to the heart Kir2.1 is also expressed in other organs such as skeletal muscle, ATS is associated with hypokalemic periodic paralysis and skeletal developmental abnormalities.83 In the heart, reduction of IK1 leads to QT prolongation and predisposes to arrhythmias; yet QT prolongation is less prominent in patients presenting ATS than in other types of LQTS.84 Moreover, while ATS patients do develop ventricular tachyarrhythmias, including torsades de pointes, SCD is rare in these patients.85
Only 3 cases of Kir2.1 gain-of function mutation have been reported. In 2005, Xia et al reported on A Kir2.1 gain-of-function mutation (V93I) in a large Chinese family with atrial fibrillation.86 Subsequently, two different gain-of-function mutations (D172N and E299V) in the KCNJ2 gene were reported in patients with short QT syndrome type 3 (SQTS3).17, 87 Increased IK1 shortens repolarization and the QT interval, and exerts a proarrhythmic effect both in the atria and the ventricles.88
b. Kir2 channels have multiple functional partners
In 2001, Leonoudakis et al identified a direct association of Kir2.1, Kir2.2 and Kir2.3 with SAP97.89 They further demonstrated that a complex composed of members of the MAGUK protein family (SAP97, CASK, Veli, and Mint1) associates with Kir2 channels via the C-terminal PDZ-binding motif.90 Also using in vitro protein interaction assays they showed that SAP97, Veli-1, or Veli-3 binds directly to the Kir2.2 C terminus and recruits CASK and proposed a model whereby Kir2.2 associates with distinct SAP97-CASK-Veli-Mint1 complexes. Subsequently, using immunoaffinity purification and affinity chromatography from skeletal and cardiac muscle and brain, they discovered that α1-, β1-, and β2-syntrophin, dystrophin, and dystrobrevin, all members of the dystrophin-associated protein complex, also interact with Kir2 channels.76 In this regard, cardiomyocytes from the dystrophin-deficient mdx mouse show a small but significant decrease in Kir2.1 protein.32 It is also possible that dystrophin related proteins contribute to determining the subcellular localization of Kir2.x channels in cardiomyocytes, similar to what has been demonstrated for NaV1.5 channels.32 As demonstrated by affinity pull-down experiments Kir2.1-3, and Kir4.1 all bind to scaffolding proteins but with different affinities for the dystrophin-associated protein complex as well as SAP97, CASK, and Veli.90
In 2001 Dart and Leyland showed that Kir2.1 associates with A kinase-anchoring protein (AKAP5), which is a multivalent-anchoring protein that binds PKA, PKC, and calcineurin.91 AKAP5 is targeted to the intracellular N and C terminal domains of Kir2.1 to anchor kinases close to key channel phosphorylation sites and is required for appropriate modulation of channel function.91 More recently, it was suggested that both Kir2.1 and AKAP are part of a macromolecular signaling complex that includes the β1-adrenergic receptor and SAP97.92 Kir2.1 may also associate with caveolin3 (Cav3) in human cardiac cells. Cav3 mutations have been shown to reduce cell surface expression of Kir2.1 with consequent reduction of IK1 density. Such an effect may add to the previously described late INa increase35 and contribute to delayed repolarization and arrhythmia generation in Cav3-mediated LQT9.93
Filamin-A increases the number of functional Kir2.1 channels on membrane in arterial smooth muscle cells.94 It appears to act as a cytoskeletal anchoring protein for the Kir2.1 channel, stabilizing its surface expression. However, while filamin has been shown to localize at the Z lines in cardiomyocytes,95 it is unknown whether pools of Kir2.x channels co-localize with filamin-A. Finally, it has been demonstrated that Kir2.1 interacts with the AP1 adaptin complex through an unusual Golgi exit signal dictated by a tertiary structure, localized within the confluence of the Kir2.1 cytoplasmic NH3 and COOH terminal domains.96 The signal creates allows properly folded Kir2.1 channels to insert into clathrin-coated vesicles at the trans-Golgi for export to the cell surface, which is a critical regulatory step for controlling trafficking and cell surface expression of the Kir2.x channels.96
c. Kir2.1 and the NaV1.5/Kir2.1 channelosome
There is a strong relationship between the inward INa and the inward rectifier potassium current (IK1), the two most important ionic currents controlling ventricular excitability: by controlling the RMP, IK1 modifies Na+ channel availability and therefore, cell excitability.80 In addition, IK1-INa interactions are important in stabilizing and controlling the frequency of the electrical rotors that are responsible for the most dangerous cardiac arrhythmias, including ventricular tachycardia (VT) and VF.88 Recent data demonstrated that the INa-IK1 interplay involves a reciprocal modulation of expression of their respective channel proteins (Kir2.1 and NaV1.5) forming a channelosome within a macromolecular complex (Fig. 6).9 Further, evidence suggests that conditions that result in NaV1.5 protein reduction, such as it occurs in the dystrophin-deficient mdx5cv mice, are accompanied by a concomitant reduction in Kir2.1 protein levels.32 Importantly, the finding that co-expression of NaV1.5 may reduce internalization of Kir2.1 was a central mechanistic observation, with important implications in the control of cardiac excitability and SCD.9
Figure 6.
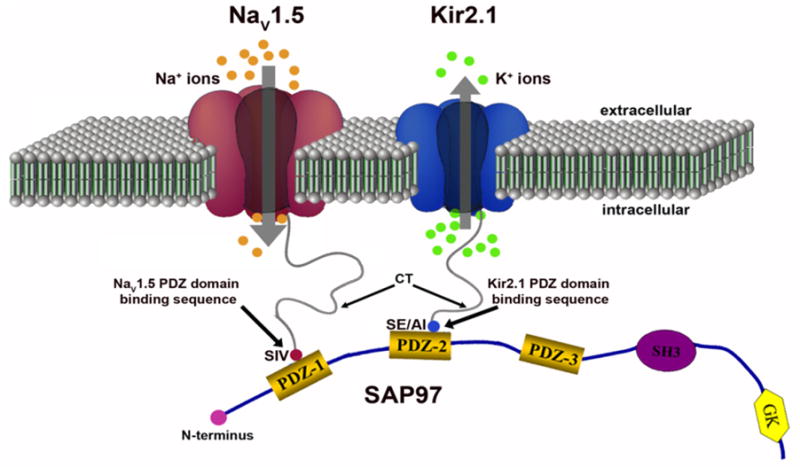
NaV1.5 and Kir2.1 form a macromolecular complex (a channelosome). The subcellular localization and channel activity of both NaV1.5 and Kir2.1 are regulated by protein–protein interactions by their respective carboxyl terminal (CT) PDZ binding motifs with such PDZ domain-containing proteins as SAP97 and syntrophin. The CTs of one NaV1.5 and Kir2.1 molecule each bind to the same SAP97 molecule but at different PDZ domains. These interactions result in changes in the expression of NaV1.5 and/or Kir2.1 and thereby, influence their function in the cell membrane. GK, guanylate kinase-like domain of SAP97; SE/AI, last 3 residues of the Kir2.1 CT, which can be serine and glutamic acid or alanine and isoleucine; SH3, src kinase homology domain of SAP97; SIV, serine, isoleucine, Q:5 valine (last 3 aa of the NaV1.5 CT). Reproduced from Milstein et al, PNAS 2012 (ref 9).
Recently, Gillet et al investigated in vivo the interactions of SAP97 with Kir2.1 and NaV1.5 by generating a genetically modified mouse model in which SAP97 expression was constitutively suppressed in cardiomyocytes.26 As expected, IK1 was reduced in the SAP97 knockout mice (Fig. 7).9, 97 Unexpectedly, INa, and NaV1.5 localization at the intercalated disc, were unaffected by the loss of SAP97 expression. Ostensibly, the data presented by the papers of Gillet et al26 and of Shy et al25 regarding NaV1.5 seem to contradict previous work.8, 9, 97 Yet there are substantial differences between the two mouse models and the previous studies that need to be considered. Most important in both mouse models,25, 26 genetic modification is present early in development, whereas in the other studies,9 SAP97 expression was silenced in adult myocytes that were kept in culture. Therefore it is conceivable that the consequences on INa could be different in an inducible SAP97 knockout mouse model. On the other hand, NaV1.5 is known to interact with other regulatory proteins at the IDs, such as connexin43,98–100 PKP2,45 and ankyrin-G.101 Furthermore, recent studies suggest that there are micro-domains of Nav1.5 at the IDs. In particular a population of Nav1.5 is located at the periphery of gap junctions in a so-called perinexus region that has been proposed to be involved in ephaptic conduction.102 It might be possible that constitutive deletion of SAP97 led to compensatory modifications in the expression and/or organization of one more partner proteins that contributed to maintain NaV1.5 expression.
Figure 7.

Electrophysiological alterations in SAP97 knock-out ventricular cardiac cells (modified with permission from Gillet et al. HRJ 2015 (ref 26). (A) Marked prolongation of the mouse cardiac AP in SAP97-deficient cardiac cells (KO in red and WT in black). Decrease of whole-cell IK1 (B), Ito (C), and Ikur (D) currents in in SAP97-deficient cardiac cells. These decreased repolarization currents are the main causes of the AP prolongation. Phase 0 of the AP, i.e. the dV/dt, was not altered consistent with observation that INa was not modified in the absence of SAP97.
d. The KATP Channel Macromolecular Complex
ATP-sensitive K+ (KATP) channels function as metabolic sensors in many cell types.103 They are an octameric assemblies of a sulfonylurea receptor (SUR) and an ion conducting subunit (Kir6.x).104 This enables them to directly couple their energy metabolism to cellular excitability and function as a crucial regulatory mechanism in the cell response to metabolic demand.103 Genetic manipulation of cardiac KATP subunits has revealed a role of these channels in arrhythmia generation.105 Human KATP mutations underlie different KATP channelopathies and can substantially increase the risk for heart disease.106
The pore forming subunit of the KATP channel is one of two members of the inwardly rectifying family of K+ channels, Kir6.1 and Kir6.2 coded by KCNJ11 and KCNJ8, respectively. The two SUR subunits (SUR1 and SUR2) are respectively coded by ABCC8 and ABCC9.106 Although several SUR splice variants have been described, the most commonly studied are SUR1, SUR2A, and SUR2B.107, 108 Like other Kir channels Kir6.x subunits have a cytoplasmic N and C terminus with two transmembrane domains and a pore forming H5 loop.79, 106 SUR has multiple transmembrane domains with two large intra-cytoplasmic loops, the first and second nucleotide binding domains (NBD1 and NBD2), which contain consensus sequences for the hydrolysis of nucleotides (Walker A and B motifs).109
Co-expression of the two types of subunit is necessary to achieve functional expression of KATP channels and the assembly of a specific Kir6.x with a specific SUR generates currents with a particular single-channel conductance, nucleotide regulation and pharmacology.103 However, accumulating evidence suggest that the KATP channel protein complex is part of a multisubunit macromolecular complex that may also include additional metabolically active protein subunits, including adenylate kinase, creatine kinase, and lactate dehydrogenase.110 In addition, it has been demonstrated that 14-3-3 proteins promote the cell-surface expression of heterologously expressed and native KATP channels by functionally antagonizing the arginine-based endoplasmic reticulum (ER)-localization signal that many ion channels and proteins require to reach the cell surface, and that is present in SUR1.111 Recently it was shown that KATP channels are stalled in the Golgi complex of ventricular, but not atrial, cardiomyocytes.106 It was also demonstrated that PKA-dependent phosphorylation of the C-terminus of Kir6.2 by sustained β-adrenergic stimulation leads SUR1-containing channels to reach the plasma membrane of ventricular cells by silencing the arginine-based retrieval signal. Therefore, it was suggested that sympathetic nervous stimulation might enable adaptation to metabolic challenges by releasing KATP channels from storage in the Golgi.106
In a recent proteomics study, glycolytic enzymes previously described for the KATP channel complex were shown to co-immunoprecipitate with KATP channel subunit from heart, endothelium, and pancreas,108 suggesting that glycolytic ATP production contributes to fine tuning of KATP channel opening in these tissues.
Physical interaction between cardiac KATP channels and the Na+/K+ ATPase has also been suggested, which might provide mechanistic insight into their functional interaction with regards to possibly sharing or competing for the same local pool of submembrane ATP/ADP.112 Finally, Kir6.2 and SUR2A are expressed at a higher density at the IDs in mouse and rat hearts, where they co-localized with PKP2 and plakoglobin. The disruption of the desmosomal complex in PKP2 deficient mice results in downregulation of KATP channels, suggesting a possible role of these channels in cell-to-cell communication.113
5. Diversity of Kv channels
Voltage-gated potassium (Kv) channels are members of the Shal subfamily of voltage-gated K+ channel pore-forming α subunits.114 Kv channels are formed by assemblies of four α-subunits plus accessory subunits.115 They function to control resting membrane potentials, shape AP characteristics and influence the responses to neurotransmitters and neurohormones. There are extensive differences in their kinetics of activation and inactivation among the various Kv channels, and specific channels underlie specific currents in the heart. For example, Kv4.x channels, including KV1.4, KV4.2 and KV4.3, coded by KCNA4, KCND2 and KCND3, respectively, activate and inactivate rapidly and underlie the Ito (transient outward current).114 KV1.5, which is coded by KCNA5 and forms the atria-specific ultrarapid delayed rectifier K+ current (IKur), inactivates much more slowly.116 Kv7.1 is coded by KCNH2 ,117 the human ether-a-go-go-related gene (hERG). In contrast to Kv4.1-3, hERG activates and inactivates rapidly, then conducts most of its current during its recovery from inactivation. Kv2.1, coded by KCNB1, is a slow delayed-rectifier K+ channel that underlies Ik,slow2 in rat cardiomyocytes.118, 119 Targeted elimination of KV2 channels in mouse ventricular myocytes leads to prolongation of the action potential duration and the QT interval.120 KV2.1 may have distinct physiological roles in atrial and ventricular myocytes.119 Finally, IKs, the α subunit (KCNQ1) of the slowly activation delayed-rectifier K+ current activates and deactivates slowly.121
a. Functional interactions of Kv channels in their microenvironment
In the heart, Kv4.2, Kv4.3 and/or Kv1.4 may assemble to generate transient outward currents. Kv4.3/Kv4.2 subunits form the rapidly recovering Ito,f channels, whereas Kv1.4 forms the slowly recovering Ito channel, both of which underlie the early phase of AP repolarization and contribute to the AP plateau.122 Both channel types are differentially expressed in the ventricles, contributing to regional heterogeneities in AP shape and duration.123
Substantial evidence indicates that these channels function as integral components of macromolecular protein complexes,114 and that expressed KV channels can be regulated by post-translational modifications, including phosphorylation.114 Also co-expression with accessory or regulatory proteins in heterologous expression systems modifies cell surface expression, subcellular distribution, channel stability, and biophysical properties of KV4 channels.124 Hence, the specificity of channel-mediated signal transduction is most likely the result of association of these integral membrane proteins with discrete sets of partner proteins or from their assembly into stable macromolecular complexes.125 However, the information available about the functioning of accessory subunits and other regulatory proteins in the generation and regulation of native cardiac Kv channels is limited.126
b. β-Subunits and Voltage-Dependent K+ Channels
The β-subunits of Kv channels (Kvβ) are cytoplasmic proteins that have a mass of ~40 kDa. Nine Kvβ-subunits are encoded by 4 genes. They have been shown to associate with Kv α-subunits. The Kvβ1, Kvβ2, and Kvβ3 proteins, which are coded by different genes, are the only Kvβ proteins expressed in the mammalian heart.114 Additional variability is produced by alternative splicing on the N-terminal region. Kvβ subunits are localized in the cytosol with a conserved carboxyl-terminal and a variable amino-terminal; they form a tetrameric structure and are associated in a 1:1 ratio with the α-subunit. Kvβ1 and Kvβ3 associate with α-subunits early during their biosynthesis in the endoplasmic reticulum and exert a chaperone-like effect enabling their stable expression at the plasma membrane.127 Notably, this chaperone-like property of Kvβ-subunit does not apply to all Kv channels.115
The most important effect of Kvβ1 on the voltage-dependent outward current is to accelerate its rate of inactivation, an effect that is mediated trough a “ball-and-chain” like process whereby the Kvβ1 N-terminal domain blocks the inner cavity of the Kv α-subunit pore.128 In addition, by binding to the C-terminus of the Kv channel Kvβ can accelerate the C-type inactivation.129 In heterologous expression systems co-expression of Kvβ1.3 with Kv1.5 is necessary for the cAMP-dependent PKA-mediated increase in K+ current.130 Moreover, consistent with the presence of multiple phosphorylation sites on the α- and β-subunits, PKC reduces the K+ current of Kv1.5 channels only when co-expressed with Kvβ1.2,131 which may provide an explanation for the effects of the β-adrenergic or PKC stimulation on IKur in human atrial myocytes.132 The duration and the frequency of membrane depolarization can significantly modify the rate of inactivation of Ikur in human atrial myocytes. This effect is modulated by the activation of CaMKII and may also involve the interaction between Kvβ and the Kvα1.5 subunits.133 The contribution of Ikur to the abbreviation of the AP duration during atrial fibrillation134 and the fact that the Kv1.5 channel is more abundantly expressed in atrial than ventricular myocardium are additional examples of the important role played by Kvβ subunits in cardiac pathophysiology.116 Finally, Kvβ subunits have been shown to confer sensitivity to redox modulation and hypoxia to Kv4.2 channels.135
c. Other Ancillary Subunits of KV channels
The best-known partner of Kv4 channels is the cytoplasmic Kv channel-interacting protein KChIP2, which has been shown directly to be an essential component of Ito,f channels in myocardium. The KChIPs were first identified in brain using the Kv4 N-terminus as bait in a yeast two-hybrid screen.136 They were shown to have four EF-hand-like domains and bind calcium ions. The expression of KChIP and Kv4 together reconstitutes several features of native A-type currents by modulating the density, inactivation kinetics and rate of recovery from inactivation of Kv4 channels in heterologous cells.136 All three KChIPs were shown to co-localize and co-immunoprecipitate with brain Kv4 α-subunits, and therefore to be integral components of native Kv4 channel complexes.136 KChIP2 assembles with the N terminus of the pore-forming Kv4 α subunit and acts as a chaperone to regulate both surface expression and electrophysiological properties of the channel.137 In heart, KChIP2 co-immunoprecipitates with α subunits of Kv4.2 and Kv4.3 from adult mouse ventricles, and the targeted deletion of the mouse KChIP2 locus (Kcnip2) abolishes ventricular Ito,f.138 In addition, KChIP2 protein expression is highly reduced in the ventricles of homozygous Kv4.2 knockout mice, suggesting that Kv4 and KChIP2 proteins reciprocally regulate each other’s expression.138, 139 In mouse ventricles the KChIP2 mRNA level is somewhat larger in the epicardium than the endocardium.140 In contrast, large transmural gradients in KChIP2 expression together with large Ito,f density gradients have been demonstrated across the human and the dog ventricular walls.139
Dipeptidyl peptidase–like protein 6 (DPP6) is protein that regulates the activation and inactivation properties of cardiac Kv4 channels.114,141 DPP6 increases heterologously-expressed Kv4 α subunits at the cell surface,142 shifts the voltage dependences of activation and inactivation currents to more negative potentials, and accelerates the rates of current activation, inactivation and recovery.141,143 Notably, when DPP6 is co-expressed with Kv4.3 and KChIP2, it yields Kv currents that closely resemble native cardiac Ito,f.141
Transient outward K+ currents can be modulated by protein kinases.144 The non-receptor protein tyrosine kinase c-Src is a member of a family of nine closely related membrane-bound kinases defined by a common structure with a catalytic kinase domain and amino-terminal regulatory regions termed Src homology 2 (SH2) and 3 (SH3) domains.145 These modular domains mediate intramolecular and intermolecular interactions that are important in signal transduction. The Kv4.3 sequence contains SH2 and SH3 domain binding motifs, making Kv4.3 a strong candidate for direct interaction with and/or phosphorylation by c-Src. Gomes et al have shown through GST pull-down assays and co-immunoprecipitation, that Kv4.3 protein associates with c-Src and that the SH2 and SH3 domains of the kinase mediate this interaction, which may result in enhanced efficiency of Kv4.3 phosphorylation by c-Src leading to rapid modulation of Kv4.3 channel activity.146
SAP97 and Kv1.5 subunits can interact, directly or indirectly, both in the heart and in heterologous systems.137, 147 Adenoviral overexpression of SAP97 in neonatal rat atrial myocytes leads to clustering of endogenous Kv1.5 subunits at myocyte-myocyte contacts and an increase in both IKur and the number of 4-aminopyridine-sensitive potassium channels in cell-attached membrane patches.148 On the other hand, pull-down and coimmunoprecipitation assays in cardiac myocytes showed that the Kv4 channel C terminus, SAP97, and CaMKII interact together, and that the interaction is suppressed by SAP97 silencing and enhanced by SAP97 overexpression.137 In HEK293 cells, SAP97 silencing reproduced the effects of CaMKII inhibition on current kinetics and suppressed Kv4/CaMKII interactions. Altogether, the above data suggest that SAP97 is a major partner for surface expression and CaMKII-dependent regulation of cardiac Kv4 channels.
As reviewed comprehensively elsewhere,114 KCNE genes encode a family of single transmembrane domain proteins called minK-related peptides (MiRPs) that function as accessory beta subunits of Kv channels. When co-expressed in heterologous systems, MiRPs confer changes in Kv channel conductance, gating kinetics and pharmacology.114 Co-expression of Kv4 and Kv4-KChIP2 channels with MiRP1 affects the kinetics and the voltage dependent properties and recapitulates the “overshoot” in peak current amplitude during current recovery,149 that is evident in human epicardial Ito,f.114, 150 Inherited mutations in KCNE genes are associated with diseases of cardiac and skeletal muscle, and the inner ear.151, 152 For example, aspartate to asparagine substitution to yield p.D76N-MinK is linked to cardiac arrhythmia and deafness. Mutation of arginine to histidine (p.R83H) in MiRP2 is associated with periodic paralysis.151 Finally, targeted deletion of Kcne2, which encodes MiRP1, reduced (~25%) ventricular Ito,f densities by 25% with negligible changes in total or surface Kv4.2 expression.153
TheKCNE1 gene encodes a 129 amino acid protein in mouse and human that modifies the currents generated by hERG or KvLQT1. The delayed rectifier K+ currents resulting from expression of KvLQT1 alone are small and activate very rapidly, but IKs is reconstituted when minK is coexpressed with KvLQT1.154 Evidence suggests that KCNE1 may have preferential expression in the conduction system.155 Mutations in KCNE1 have been reported to cause LQTS.156
MiRP2 is a member of the MinK-related peptide family that is coded by KCNE3. It co-immunoprecipitates with Kv4.3 from human atria.157 Interestingly, a missense mutation (p.R99H) in KCNE3 was identified in a family with BrS.157 Co-transfection of MiRP2 (with and without KChIP) decreases Kv4.3 current densities in heterologous expression systems.157, 158 In addition, co-expression of the p.R99H MiRP2 mutant reversed the inhibitory effects of wild type MiRP2 on Kv4.3 currents.157 Altogether, the above data suggest that MiRP2 is required for normal functioning of human Ito,f channels and that gain of function mutations in MiRP2 predispose to BrS through augmentation of Ito,f.114, 157
6.KCNQ1 (KvLQT1)
The KCNQ1 gene, encodes the Kv7.1 channel protein, which can form heteromultimers with two other potassium channel proteins, KCNE1 and KCNE3.159 In the human heart the KCNQ1 encodes the pore-forming α subunit, and KCNE1 (also known as minK) encodes the regulatory β subunit of the KCNQ1- KCNE1 complex responsible for IKs, the slowly activating delayed rectifier K+ repolarizing current.160 Mutations in KCNQ1 are associated with hereditary LQTS1 (also known as Romano-Ward syndrome), Jervell and Lange-Nielsen syndrome, and familial atrial fibrillation.161 In 2002, Marx et al showed that modulation of IKS β-adrenergic receptor stimulation requires targeting of cyclic adenosine 3′,5′-monophosphate (cAMP)–dependent PKA and protein phosphatase 1 (PP1) to hKCNQ1 through the A kinase-anchoring protein (AKAP)-9, also known as yotiao.121 These authors elegantly demonstrated that yotiao binds to the human KCNQ1 by a leucine zipper motif, which is disrupted by an LQTS mutation (hKCNQ1-G589D). Identification of the hKCNQ1 macromolecular complex provides a mechanism for sympathetic nervous system modulation of cardiac APD through IKS.121 These data provided compelling evidence that the cardiac IKs potassium channel is a macromolecular complex consisting of α-(KCNQ1) and β-subunits (KCNE1) and yotiao (AKAP-9), which recruits PKA and protein phosphatase 1 to the channel.121
7. Kv11.1-HERG
The human erg protein (hERG or Kv11.1) is the pore-forming subunit of the rapid component of the cardiac delayed rectifier potassium current (IK) responsible for AP repolarization.162 It is encoded by the hERG gene,163 which comprises three members, erg1, erg2, and erg3, displaying varying expression patterns in different tissues;164 herg1 is the best characterized.163 Structurally, hERG has six transmembrane domains (S1-S6), S4-being the voltage sensor, with cytosolic N and C termini. The N terminus, which contains a PAS domain, strongly affects the biophysical properties of the channel. Functional hERG channels are tetramers with a pore region responsible for K+ current flow through the plasma membrane.162 As reviewed elsewhere, alternative transcription of hERG1 results in two identical proteins, hERG1a and hERG1b, that diverge only in their N-termini.165 hERG1b can form channels alone or co-assemble with hERG1a. A third variant of hERG1, also identical to hERG1a but with a modified C-terminus is termed hERGuso.166 Expression of hERGuso reduces the number of channels at the sarcolemma and the current density. In contrast, co-assembly with hERG1b alters channel kinetics increasing channel availability current magnitude.167 Mutations in hERG lead to long-QT syndrome type 2 (LQT2), a major cause of arrhythmias,168 as well as to short QT syndrome type 2, which results in atrial and ventricular arrhythmias.169
Even though heterologously expressed hERG channels are largely indistinguishable from native cardiac IKr, a role for KCNE1 in this current was suggested by the diminished IKr in an atrial tumor line subjected to minK antisense suppression.170 Subsequently, McDonald et al showed that HERG and minK formed a stable complex, and that the heteromultimerization regulated IKr activity. This provided additional support for the idea that minK, through the formation of heteromeric channel complexes, is central to the control of the heart rate and rhythm.171 hERG has been shown to also co-immunoprecipitate with PKA172, and similar to other cardiac KV channel subunits hERG interact with SNAREs, which are proteins that are critical for synaptic vesicular secretion and possibly membrane protein trafficking.173 Recently, Ma et al168 identified 23 potential interacting proteins that may regulate cardiac IKr through cytoskeletal interactions, G-protein modulation, phosphorylation and downstream second messenger and transcription cascades. Fifteen such proteins were identified as hERG amino terminal (hERG-NT)-interacting proteins, including the caveolin-1, the zinc finger protein FHL2 and protein tyrosine phosphatase non-receptor type 12 (PTPN12). The other 8 proteins were identified as hERG carboxylic terminal (hERG-CT)-interacting proteins, including the NF-κB-interacting protein myotrophin.168 Several unexpected binding partners were identified which greatly enhanced the dynamic modulation of IKr as part of a macromolecular complex.168
8. KCNQ1-HERG interactions
After the pioneering studies of Sanguinetti et al,154 it became clear that IK, the delayed rectifying K current responsible for cardiac repolarization, is mediated by two distinct currents, IKr and IKs, which work together to produce cardiac repolarization and control the APD. Recent results suggest that in addition to their voltage dependent interactions, these two channels also interact at the molecular level.174 For example, studies in both transgenic LQT rabbit cardiomyocytes and stable, heterologous cell lines reported that hERG and KCNQ1 underwent reciprocal, functional downregulation in that co-expression of wild-type or dominant-negative pore mutants of KCNQ1 significantly reduced hERG currents, and vice versa.175 More recently, the same lab conducted acceptor photobleach Förster resonance energy transfer (FRET) experiments and demonstrated that the intermolecular KCNQ1-hERG interactions are direct and mediated by their respective COOH termini.174 In agreement with the above results, another group showed that KCNQ1 preferentially co-immunoprecipitated with mature hERG channels that were localized in the plasma membrane of HEK293 cells.176 On the other hand, the latter group demonstrated that while hERG channels undergo rapid endocytic degradation upon exposure to hypokalemia, KCNQ1 channels are relatively insensitive to extracellular K+ reduction.176 Thus when hERG and KCNQ1 were expressed separately, exposure to 0 mM K+ for 6 h completely eliminated the mature hERG channel expression but had no effect on KCNQ1. However, contrary to the transgenic rabbit data,174 the latter investigators showed that when hERG and KCNQ1 were co-expressed, KCNQ1 significantly delayed the hypokalemia-induced hERG loss.176 Also, hERG degradation led to a significant reduction in KCNQ1 in hypokalemia.176 Therefore while biophysical and pharmacological analyses conducted by both groups indicate that hERG and KCNQ1 closely interact with each other, their respective results seem to go in the opposite direction: the former group concluded that co-expression of KCNQ1 significantly reduced hERG currents and vice versa,174 whereas the latter group concluded that co-expression of KCNQ1 protected hERG against hypokalemia, and hERG reduction reduced KCNQ1.176 Clearly additional studies will be necessary to resolve this controversy.
9. Perspectives and Conclusions
We have briefly reviewed research conducted over the last 20 years showing that cardiac ion channels may function as part of large macromolecular complexes. Such complexes play crucial roles the transcription, translation, oligomerization, trafficking, membrane retention, glycosylation, posttranslational modification, turnover, function and degradation of all cardiac ion channels known to date. In fact, macromolecular complexes are vital to a wide collection of cellular tasks. Some of these require physical contact among partner proteins, others do not. Understanding the structure and signaling dynamics of multiprotein assemblies is vital to understanding their function, and is likely to shed light on how the heart functions in health and disease. However, we are still lacking a detailed knowledge of such processes, and of the role played by the myriad of ion channel molecular assemblies in the compartmentalization of ion channel function and the mechanisms underlying ion channel dysregulation, life-threatening cardiac arrhythmias and SCD. This is a significant problem because both arrhythmias and SCD are among the most important causes of cardiovascular morbidity and mortality in the developed world. Clearly, many more studies are needed to establish new paradigms of cardiac electrophysiology integrating the large diversity of molecular interactions involved in the formation, targeting and regulation of cardiac ion channels and their function, as well as the tissue specific expression of the components of ion channel complexes not only in the working cardiac muscle of the atria and ventricles, but also the specialized pacemaking and conduction systems. Progress likely will come from the use of systems biology approaches, from the nanoscale all the way to the cellular and organ levels. Progress should also derive from the development and application of modern technologies enabling adequate spatio-temporal resolution to visualize and quantify the processes involved in the assembly and dynamic interactions of ion channel macromolecular complexes in living native myocytes from animal models, as well as in human stem-cell derived cardiomyocytes.
Acknowledgments
Funding Sources:
This work was supported in part by Grant HL122352 form the National Heart, Lung and Blood Institute of the National Institutes of Health, by a Transatlantic Networks of Excellence Program grant from the Leducq Foundation (JJ), the Swiss National Science Foundation (310030_14060 to HA) and the European Community's 7th Framework Program FP7/2007-2013 under grant agreement (no. HEALTH-F2-2009-241526, EUTrigTreat to HA).
Nonstandard Abbreviations and Acronyms
- AKAP
A kinase-anchoring protein
- ATS
Andersen-Tawil Syndrome
- β-AR2
β-adrenergic receptor type 2
- BIN1
Bridging Integrator 1
- BrS
Brugada syndrome
- CaM
Calmodulin
- CaMKII
Ca2+/CaM-dependent protein kinase II
- cAMP
Cyclic adenosine 3′,5′-monophosphate (cAMP)
- CASK
Ca2+/calmodulin-dependent serine protein kinase
- Cav3
Caveolin3
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- DPP6
Dipeptidyl peptidase–like protein 6
- e-c coupling
excitation-contraction coupling
- ER
Endoplasmic reticulum
- FGFs
Fibroblast growth factor homologous factors
- FRET
Förster resonance energy transfer
- GPD1L
Glycerol-3-phosphate dehydrogenase like protein
- hERG
Human eter-a-gogo-related
- ID
Intercalated disc
- JPH2
Junctophilin 2
- Kir
Potassium inward rectifier
- KATP channel
ATP-sensitive K+ channel
- LQTS
Long-QT syndrome
- MAGUK
Membrane-associated guanylate kinase
- MirPs
minK-related peptides
- MOG1
Multi-copy suppressor of gsp1
- NBD
Nucleotide binding domain
- PDZ
Post synaptic density protein [PSD95], Drosophila disc large tumor suppressor [Dlg1], and zonula occludens-1 protein [zo-1])
- PKA
Protein Kinase A
- PKC
Protein kinase C
- PKP2
Plakophilin-2
- RyR2
Ryanodine Receptor Type 2
- SAP97
Synapse Associated Protein 97
- SCD
Sudden cardiac death
- SIV-motif
Serine– isoleucine-valine-motif
- SIDS
sudden infant death syndrome
- SH2
Src homology 2
- SH3
Src homology 3
- SQTS
Short-QT syndrome
- SR
Sarcoplasmic Reticulum
- SUNDS
Sudden unexpected nocturnal death syndrome
- SUR
Sulfonylurea receptor
- Veli
Lin-7
- VF
Ventricular fibrillation
- VT
Ventricular tachycardia
Footnotes
This manuscript was sent to Gordon Tomaselli, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
In April 2015, the average time from submission to first decision for all original research papers submitted to Circulation Research was 13.84 days.
Disclosures:
None
10. Literature Cited
- 1.O'Malley HA, Isom LL. Sodium channel beta subunits: Emerging targets in channelopathies. Annu Rev Physiol. 2015;77:481–504. doi: 10.1146/annurev-physiol-021014-071846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patwardhan A, Ashton A, Brandt R, Butcher S, Carzaniga R, Chiu W, Collinson L, Doux P, Duke E, Ellisman MH, Franken E, Grunewald K, Heriche JK, Koster A, Kuhlbrandt W, Lagerstedt I, Larabell C, Lawson CL, Saibil HR, Sanz-Garcia E, Subramaniam S, Verkade P, Swedlow JR, Kleywegt GJ. A 3d cellular context for the macromolecular world. Nature structural & molecular biology. 2014;21:841–845. doi: 10.1038/nsmb.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cebecauer M, Spitaler M, Serge A, Magee AI. Signalling complexes and clusters: Functional advantages and methodological hurdles. J Cell Sci. 2010;123:309–320. doi: 10.1242/jcs.061739. [DOI] [PubMed] [Google Scholar]
- 4.Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15:163–177. doi: 10.1038/nrm3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 6.Abriel H. Cardiac sodium channel na(v)1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. doi: 10.1016/j.yjmcc.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11:414–426. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. Sap97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- 9.Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012;109:E2134–2143. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Priori SG. The fifteen years of discoveries that shaped molecular electrophysiology: Time for appraisal. Circ Res. 2010;107:451–456. doi: 10.1161/CIRCRESAHA.110.226811. [DOI] [PubMed] [Google Scholar]
- 11.Cerrone M, Napolitano C, Priori SG. Genetics of ion-channel disorders. Current opinion in cardiology. 2012;27:242–252. doi: 10.1097/HCO.0b013e328352429d. [DOI] [PubMed] [Google Scholar]
- 12.Zaklyazminskaya EV, Abriel H. Prevalence of significant genetic variants in congenital long qt syndrome is largely underestimated. Frontiers in pharmacology. 2012;3:72. doi: 10.3389/fphar.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behr ER, Dalageorgou C, Christiansen M, Syrris P, Hughes S, Tome Esteban MT, Rowland E, Jeffery S, McKenna WJ. Sudden arrhythmic death syndrome: Familial evaluation identifies inheritable heart disease in the majority of families. European heart journal. 2008;29:1670–1680. doi: 10.1093/eurheartj/ehn219. [DOI] [PubMed] [Google Scholar]
- 14.Benito B, Brugada R, Brugada J, Brugada P. Brugada syndrome. Prog Cardiovasc Dis. 2008;51:1–22. doi: 10.1016/j.pcad.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of brugada syndrome: Depolarization disorder, repolarization disorder, or more? Cardiovasc Res. 2005;67:367–378. doi: 10.1016/j.cardiores.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Liu N, Colombi B, Raytcheva-Buono EV, Bloise R, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Herz. 2007;32:212–217. doi: 10.1007/s00059-007-2975-2. [DOI] [PubMed] [Google Scholar]
- 17.Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M, Noujaim SF, Denegri M, Jalife J, Priori SG. Kcnj2 mutation in short qt syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci U S A. 2013;110:4291–4296. doi: 10.1073/pnas.1218154110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gollob MH, Redpath CJ, Roberts JD. The short qt syndrome: Proposed diagnostic criteria. Journal of the American College of Cardiology. 2011;57:802–812. doi: 10.1016/j.jacc.2010.09.048. [DOI] [PubMed] [Google Scholar]
- 19.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: Different faces of scn5a mutations. Trends in cardiovascular medicine. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Gellens ME, George AL, Jr, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brackenbury WJ, Isom LL. Na channel beta subunits: Overachievers of the ion channel family. Front Pharmacol. 2011;2:53. doi: 10.3389/fphar.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartshorne RP, Catterall WA. The sodium channel from rat brain. Purification and subunit composition. J Biol Chem. 1984;259:1667–1675. [PubMed] [Google Scholar]
- 23.Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884–897. doi: 10.1161/CIRCRESAHA.110.238469. [DOI] [PubMed] [Google Scholar]
- 24.Shy D, Gillet L, Abriel H. Cardiac sodium channel nav1.5 distribution in myocytes via interacting proteins: The multiple pool model. Biochimica et biophysica acta. 2013;1833:886–894. doi: 10.1016/j.bbamcr.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 25.Shy D, Gillet L, Ogrodnik J, Albesa M, Verkerk AO, Wolswinkel R, Rougier JS, Barc J, Essers MC, Syam N, Marsman RF, van Mil AM, Rotman S, Redon R, Bezzina CR, Remme CA, Abriel H. Pdz domain-binding motif regulates cardiomyocyte compartment-specific nav1.5 channel expression and function. Circulation. 2014;130:147–160. doi: 10.1161/CIRCULATIONAHA.113.007852. [DOI] [PubMed] [Google Scholar]
- 26.Gillet L, Rougier JS, Shy D, Sonntag S, Mougenot N, Essers M, Shmerling D, Balse E, Hatem SN, Abriel H. Cardiac-specific ablation of synapse-associated protein sap97 in mice decreases potassium currents but not sodium current. Heart Rhythm. 2015;12:181–192. doi: 10.1016/j.hrthm.2014.09.057. [DOI] [PubMed] [Google Scholar]
- 27.Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Rotin D, Staub O. Role of the ubiquitin system in regulating ion transport. Pflugers Archiv : European journal of physiology. 2011;461:1–21. doi: 10.1007/s00424-010-0893-2. [DOI] [PubMed] [Google Scholar]
- 29.Gabelli SB, Boto A, Kuhns VH, Bianchet MA, Farinelli F, Aripirala S, Yoder J, Jakoncic J, Tomaselli GF, Amzel LM. Regulation of the nav1.5 cytoplasmic domain by calmodulin. Nat Commun. 2014;5:5126. doi: 10.1038/ncomms6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kyle JW, Makielski JC. Diseases caused by mutations in nav1.5 interacting proteins. Card Electrophysiol Clin. 2014;6:797–809. doi: 10.1016/j.ccep.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calhoun JD, Isom LL. The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb Exp Pharmacol. 2014;221:51–89. doi: 10.1007/978-3-642-41588-3_4. [DOI] [PubMed] [Google Scholar]
- 32.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–414. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 33.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long qt syndrome through activation of the nnos-scn5a macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, Abbasi S, Purevjav E, Samani K, Ackerman MJ, Qi M, Moss AJ, Shimizu W, Towbin JA, Cheng J, Vatta M. Alpha-1-syntrophin mutation and the long-qt syndrome: A disease of sodium channel disruption. Circulation. Arrhythmia and electrophysiology. 2008;1:193–201. doi: 10.1161/CIRCEP.108.769224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-qt syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 36.Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Ackerman MJ. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart rhythm : the official journal of the Heart Rhythm Society. 2007;4:161–166. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: Regulation of sodium current amplitude. Circulation Research. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]
- 38.Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002;105:707–713. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 39.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC., Jr Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (gpd1-l) decreases cardiac na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (gpd1-l) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–2259. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. Gpd1l links redox state to cardiac excitability by pkc-dependent phosphorylation of the sodium channel scn5a. American journal of physiology. Heart and circulatory physiology. 2009;297:H1446–1452. doi: 10.1152/ajpheart.00513.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circulation Research. 2010;107:967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu L, Yong SL, Fan C, Ni Y, Yoo S, Zhang T, Zhang X, Obejero-Paz CA, Rho HJ, Ke T, Szafranski P, Jones SW, Chen Q, Wang QK. Identification of a new co-factor, mog1, required for the full function of cardiac sodium channel nav 1.5. The Journal of Biological Chemistry. 2008;283:6968–6978. doi: 10.1074/jbc.M709721200. [DOI] [PubMed] [Google Scholar]
- 44.Kattygnarath D, Maugenre S, Neyroud N, Balse E, Ichai C, Denjoy I, Dilanian G, Martins RP, Fressart V, Berthet M, Schott JJ, Leenhardt A, Probst V, Le Marec H, Hainque B, Coulombe A, Hatem SN, Guicheney P. Mog1: A new susceptibility gene for brugada syndrome. Circulation. Cardiovascular genetics. 2011;4:261–268. doi: 10.1161/CIRCGENETICS.110.959130. [DOI] [PubMed] [Google Scholar]
- 45.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-g, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerrone M, Delmar M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and brugada syndrome. Trends in cardiovascular medicine. 2014;24:184–190. doi: 10.1016/j.tcm.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pablo JL, Pitt GS. Fibroblast growth factor homologous factors: New roles in neuronal health and disease. Neuroscientist. 2014 doi: 10.1177/1073858414562217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C, Hennessey JA, Kirkton RD, Wang C, Graham V, Puranam RS, Rosenberg PB, Bursac N, Pitt GS. Fibroblast growth factor homologous factor 13 regulates na+ channels and conduction velocity in murine hearts. Circ Res. 2011;109:775–782. doi: 10.1161/CIRCRESAHA.111.247957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, Torchio M, Dagradi F, Crotti L, Schwartz PJ, Ackerman MJ, Pitt GS. Fgf12 is a candidate brugada syndrome locus. Heart Rhythm. 2013;10:1886–1894. doi: 10.1016/j.hrthm.2013.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harvey RD, Hell JW. Cav1.2 signaling complexes in the heart. Journal of Molecular and Cellular Cardiology. 2013;58:143–152. doi: 10.1016/j.yjmcc.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang L, Katchman A, Morrow JP, Doshi D, Marx SO. Cardiac l-type calcium channel (cav1.2) associates with gamma subunits. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:928–936. doi: 10.1096/fj.10-172353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buraei Z, Yang J. The ss subunit of voltage-gated ca2+ channels. Physiological reviews. 2010;90:1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rougier JS, Albesa M, Abriel H, Viard P. Neuronal precursor cell-expressed developmentally down-regulated 4–1 (nedd4–1) controls the sorting of newly synthesized ca(v)1.2 calcium channels. The Journal of Biological Chemistry. 2011;286:8829–8838. doi: 10.1074/jbc.M110.166520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pankonien I, Otto A, Dascal N, Morano I, Haase H. Ahnak1 interaction is affected by phosphorylation of ser-296 on cavbeta(2) Biochemical and biophysical research communications. 2012;421:184–189. doi: 10.1016/j.bbrc.2012.03.132. [DOI] [PubMed] [Google Scholar]
- 57.De Jongh KS, Warner C, Catterall WA. Subunits of purified calcium channels. Alpha 2 and delta are encoded by the same gene. The Journal of Biological Chemistry. 1990;265:14738–14741. [PubMed] [Google Scholar]
- 58.Klugbauer N, Marais E, Hofmann F. Calcium channel alpha2delta subunits: Differential expression, function, and drug binding. J Bioenerg Biomembr. 2003;35:639–647. doi: 10.1023/b:jobb.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- 59.Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK, Schwartz A. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. American journal of physiology. Heart and circulatory physiology. 2009;297:H117–124. doi: 10.1152/ajpheart.00122.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rougier JS, Albesa M, Syam N, Halet G, Abriel H, Viard P. Ubiquitin-specific protease usp2–45 acts as a molecular switch to promote alphadelta-1-induced downregulation of ca 1.2 channels. Pflugers Archiv : European journal of physiology. 2014 doi: 10.1007/s00424-014-1636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ, Jr, Vaghy PL, Meissner G, Ferguson DG. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. The Journal of Cell Biology. 1995;129:673–682. doi: 10.1083/jcb.129.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: A novel family of junctional membrane complex proteins. Molecular cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 63.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyers MB, Pickel VM, Sheu SS, Sharma VK, Scotto KW, Fishman GI. Association of sorcin with the cardiac ryanodine receptor. The Journal of Biological Chemistry. 1995;270:26411–26418. doi: 10.1074/jbc.270.44.26411. [DOI] [PubMed] [Google Scholar]
- 65.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. Camkii tethers to l-type ca2+ channels, establishing a local and dedicated integrator of ca2+ signals for facilitation. The Journal of Cell Biology. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, Shaw RM. Bin1 localizes the l-type calcium channel to cardiac t-tubules. PLoS biology. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chase TH, Cox GA, Burzenski L, Foreman O, Shultz LD. Dysferlin deficiency and the development of cardiomyopathy in a mouse model of limb-girdle muscular dystrophy 2b. The American journal of pathology. 2009;175:2299–2308. doi: 10.2353/ajpath.2009.080930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac l-type ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tandan S, Wang Y, Wang TT, Jiang N, Hall DD, Hell JW, Luo X, Rothermel BA, Hill JA. Physical and functional interaction between calcineurin and the cardiac l-type ca2+ channel. Circulation Research. 2009;105:51–60. doi: 10.1161/CIRCRESAHA.109.199828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Best JM, Kamp TJ. Different subcellular populations of l-type ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2012;52:376–387. doi: 10.1016/j.yjmcc.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by st-segment elevation, short qt intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, Sticht H, Rauch A, Puleo C, Hu D, Barajas-Martinez H, Antzelevitch C, Luscher TF, Abriel H, Duru F. Identification of a novel loss-of-function calcium channel gene mutation in short qt syndrome (sqts6) European heart journal. 2011;32:1077–1088. doi: 10.1093/eurheartj/ehr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haase H, Alvarez J, Petzhold D, Doller A, Behlke J, Erdmann J, Hetzer R, Regitz-Zagrosek V, Vassort G, Morano I. Ahnak is critical for cardiac ca(v)1.2 calcium channel function and its beta-adrenergic regulation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:1969–1977. doi: 10.1096/fj.05-3997com. [DOI] [PubMed] [Google Scholar]
- 74.Pankonien I, Alvarez JL, Doller A, Kohncke C, Rotte D, Regitz-Zagrosek V, Morano I, Haase H. Ahnak1 is a tuneable modulator of cardiac ca(v)1.2 calcium channel activity. Journal of muscle research and cell motility. 2011;32:281–290. doi: 10.1007/s10974-011-9269-2. [DOI] [PubMed] [Google Scholar]
- 75.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X International union of pharmacology. Liii. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 76.Leonoudakis D, Conti LR, Anderson S, Radeke CM, McGuire LM, Adams ME, Froehner SC, Yates JR, 3rd, Vandenberg CA. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (kir2.X)-associated proteins. J Biol Chem. 2004;279:22331–22346. doi: 10.1074/jbc.M400285200. [DOI] [PubMed] [Google Scholar]
- 77.Shimoni Y, Clark RB, Giles WR. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol. 1992;448:709–727. doi: 10.1113/jphysiol.1992.sp019066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 79.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 80.Lopatin AN, Nichols CG. Inward rectifiers in the heart: An update on i(k1) J Mol Cell Cardiol. 2001;33:625–638. doi: 10.1006/jmcc.2001.1344. [DOI] [PubMed] [Google Scholar]
- 81.Miake J, Marban E, Nuss HB. Functional role of inward rectifier current in heart probed by kir2.1 overexpression and dominant-negative suppression. J Clin Invest. 2003;111:1529–1536. doi: 10.1172/JCI17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 83.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL, Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in kir2.1 cause the developmental and episodic electrical phenotypes of andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 84.Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of kcnj2 mutations associated with lqt7 (andersen syndrome) J Clin Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tristani-Firouzi M, Etheridge SP. Kir 2.1 channelopathies: The andersen-tawil syndrome. Pflugers Arch. 2010;460:289–294. doi: 10.1007/s00424-010-0820-6. [DOI] [PubMed] [Google Scholar]
- 86.Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J. A kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–1019. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 87.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short qt syndrome (sqt3) is caused by a mutation in the kcnj2 gene. Circ Res. 2005;96:800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 88.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier k+ current (i k1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leonoudakis D, Mailliard W, Wingerd K, Clegg D, Vandenberg C. Inward rectifier potassium channel kir2.2 is associated with synapse-associated protein sap97. J Cell Sci. 2001;114:987–998. doi: 10.1242/jcs.114.5.987. [DOI] [PubMed] [Google Scholar]
- 90.Leonoudakis D, Conti LR, Radeke CM, McGuire LMM, Vandenberg CA. A multiprotein trafficking complex composed of sap97, cask, veli, and mint1 is associated with inward rectifier kir2 potassium channels. J Biol Chem. 2004;279:19051–19063. doi: 10.1074/jbc.M400284200. [DOI] [PubMed] [Google Scholar]
- 91.Dart C, Leyland ML. Targeting of an a kinase-anchoring protein, akap79, to an inwardly rectifying potassium channel, kir2.1. The Journal of Biological Chemistry. 2001;276:20499–20505. doi: 10.1074/jbc.M101425200. [DOI] [PubMed] [Google Scholar]
- 92.Vaidyanathan R, Taffet SM, Vikstrom KL, Anumonwo JM. Regulation of cardiac inward rectifier potassium current (ik1) by synapse associated protein-97. J Biol Chem. 2010 doi: 10.1074/jbc.M110.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaidyanathan R, Vega AL, Song C, Zhou Q, Tan BH, Berger S, Makielski JC, Eckhardt LL. The interaction of caveolin 3 protein with the potassium inward rectifier channel kir2.1: Physiology and pathology related to long qt syndrome 9 (lqt9) J Biol Chem. 2013;288:17472–17480. doi: 10.1074/jbc.M112.435370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sampson LJ, Leyland ML, Dart C. Direct interaction between the actin-binding protein filamin-a and the inwardly rectifying potassium channel, kir2.1. J Biol Chem. 2003;278:41988–41997. doi: 10.1074/jbc.M307479200. [DOI] [PubMed] [Google Scholar]
- 95.Rafizadeh S, Zhang Z, Woltz RL, Kim HJ, Myers RE, Lu L, Tuteja D, Singapuri A, Bigdeli AA, Harchache SB, Knowlton AA, Yarov-Yarovoy V, Yamoah EN, Chiamvimonvat N. Functional interaction with filamin a and intracellular ca2+ enhance the surface membrane expression of a small-conductance ca2+-activated k+ (sk2) channel. Proc Natl Acad Sci U S A. 2014;111:9989–9994. doi: 10.1073/pnas.1323541111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma D, Taneja TK, Hagen BM, Kim BY, Ortega B, Lederer WJ, Welling PA. Golgi export of the kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell. 2011;145:1102–1115. doi: 10.1016/j.cell.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, Wu JC, MacRae CA, Kleber AG, Saffitz JE. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra274. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malhotra JD, Thyagarajan V, Chen C, Isom LL. Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J Biol Chem. 2004;279:40748–40754. doi: 10.1074/jbc.M407243200. [DOI] [PubMed] [Google Scholar]
- 99.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of cx43 results in decreased nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional cx43 knockout mice. Heart Rhythm. 2012;9:600–607. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rhett JM, Ongstad EL, Jourdan J, Gourdie RG. Cx43 associates with na(v)1.5 in the cardiomyocyte perinexus. J Membr Biol. 2012;245:411–422. doi: 10.1007/s00232-012-9465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 e1053k mutation causing brugada syndrome blocks binding to ankyrin-g and expression of nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S. Sodium channels in the cx43 gap junction perinexus may constitute a cardiac ephapse: An experimental and modeling study. Pflugers Arch. 2015 doi: 10.1007/s00424-014-1675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nichols CG. Katp channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 104.Shyng S, Nichols CG. Octameric stoichiometry of the katp channel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nichols CG, Singh GK, Grange DK. Katp channels and cardiovascular disease: Suddenly a syndrome. Circ Res. 2013;112:1059–1072. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arakel EC, Brandenburg S, Uchida K, Zhang H, Lin YW, Kohl T, Schrul B, Sulkin MS, Efimov IR, Nichols CG, Lehnart SE, Schwappach B. Tuning the electrical properties of the heart by differential trafficking of katp ion channel complexes. J Cell Sci. 2014;127:2106–2119. doi: 10.1242/jcs.141440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burke MA, Mutharasan RK, Ardehali H. The sulfonylurea receptor, an atypical atp-binding cassette protein, and its regulation of the katp channel. Circ Res. 2008;102:164–176. doi: 10.1161/CIRCRESAHA.107.165324. [DOI] [PubMed] [Google Scholar]
- 108.Kefaloyianni E, Lyssand JS, Moreno C, Delaroche D, Hong M, Fenyo D, Mobbs CV, Neubert TA, Coetzee WA. Comparative proteomic analysis of the atp-sensitive k+ channel complex in different tissue types. Proteomics. 2013;13:368–378. doi: 10.1002/pmic.201200324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giblin JP, Quinn K, Tinker A. The cytoplasmic c-terminus of the sulfonylurea receptor is important for katp channel function but is not key for complex assembly or trafficking. Eur J Biochem. 2002;269:5303–5313. doi: 10.1046/j.1432-1033.2002.03249.x. [DOI] [PubMed] [Google Scholar]
- 110.Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, Coetzee WA. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the k(atp) channel macromolecular complex and regulate its function. J Biol Chem. 2005;280:38464–38470. doi: 10.1074/jbc.M508744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heusser K, Yuan H, Neagoe I, Tarasov AI, Ashcroft FM, Schwappach B. Scavenging of 14–3–3 proteins reveals their involvement in the cell-surface transport of atp-sensitive k+ channels. J Cell Sci. 2006;119:4353–4363. doi: 10.1242/jcs.03196. [DOI] [PubMed] [Google Scholar]
- 112.Li J, Kline CF, Hund TJ, Anderson ME, Mohler PJ. Ankyrin-b regulates kir6.2 membrane expression and function in heart. J Biol Chem. 2010;285:28723–28730. doi: 10.1074/jbc.M110.147868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hong M, Bao L, Kefaloyianni E, Agullo-Pascual E, Chkourko H, Foster M, Taskin E, Zhandre M, Reid DA, Rothenberg E, Delmar M, Coetzee WA. Heterogeneity of atp-sensitive k+ channels in cardiac myocytes: Enrichment at the intercalated disk. J Biol Chem. 2012;287:41258–41267. doi: 10.1074/jbc.M112.412122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (i(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent k+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- 116.Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: Molecular basis and therapeutic implications. Cardiovasc Res. 2011;89:776–785. doi: 10.1093/cvr/cvq398. [DOI] [PubMed] [Google Scholar]
- 117.Tester DJ, Cronk LB, Carr JL, Schulz V, Salisbury BA, Judson RS, Ackerman MJ. Allelic dropout in long qt syndrome genetic testing: A possible mechanism underlying false-negative results. Heart rhythm : the official journal of the Heart Rhythm Society. 2006;3:815–821. doi: 10.1016/j.hrthm.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 118.Bou-Abboud E, Li H, Nerbonne JM. Molecular diversity of the repolarizing voltage-gated k+ currents in mouse atrial cells. The Journal of physiology. 2000;529(Pt 2):345–358. doi: 10.1111/j.1469-7793.2000.00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.O'Connell KM, Whitesell JD, Tamkun MM. Localization and mobility of the delayed-rectifer k+ channel kv2.1 in adult cardiomyocytes. American journal of physiology. Heart and circulatory physiology. 2008;294:H229–237. doi: 10.1152/ajpheart.01038.2007. [DOI] [PubMed] [Google Scholar]
- 120.Xu H, Barry DM, Li H, Brunet S, Guo W, Nerbonne JM. Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative kv2 alpha subunit. Circulation Research. 1999;85:623–633. doi: 10.1161/01.res.85.7.623. [DOI] [PubMed] [Google Scholar]
- 121.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the kcnq1-kcne1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 122.Nerbonne JM. Molecular basis of functional voltage-gated k+ channel diversity in the mammalian myocardium. J Physiol. 2000;525(Pt 2):285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 124.Jerng HH, Qian Y, Pfaffinger PJ. Modulation of kv4.2 channel expression and gating by dipeptidyl peptidase 10 (dpp10) Biophys J. 2004;87:2380–2396. doi: 10.1529/biophysj.104.042358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schulte U, Muller CS, Fakler B. Ion channels and their molecular environments--glimpses and insights from functional proteomics. Semin Cell Dev Biol. 2011;22:132–144. doi: 10.1016/j.semcdb.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 126.Marionneau C, Townsend RR, Nerbonne JM. Proteomic analysis highlights the molecular complexities of native kv4 channel macromolecular complexes. Semin Cell Dev Biol. 2011;22:145–152. doi: 10.1016/j.semcdb.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shi G, Nakahira K, Hammond S, Rhodes KJ, Schechter LE, Trimmer JS. Beta subunits promote k+ channel surface expression through effects early in biosynthesis. Neuron. 1996;16:843–852. doi: 10.1016/s0896-6273(00)80104-x. [DOI] [PubMed] [Google Scholar]
- 128.Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac k+ channels. Circ Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- 129.Morales MJ, Wee JO, Wang S, Strauss HC, Rasmusson RL. The n-terminal domain of a k+ channel beta subunit increases the rate of c-type inactivation from the cytoplasmic side of the channel. Proc Natl Acad Sci U S A. 1996;93:15119–15123. doi: 10.1073/pnas.93.26.15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kwak YG, Hu N, Wei J, George AL, Jr, Grobaski TD, Tamkun MM, Murray KT. Protein kinase a phosphorylation alters kvbeta1.3 subunit-mediated inactivation of the kv1.5 potassium channel. J Biol Chem. 1999;274:13928–13932. doi: 10.1074/jbc.274.20.13928. [DOI] [PubMed] [Google Scholar]
- 131.Williams CP, Hu N, Shen W, Mashburn AB, Murray KT. Modulation of the human kv1.5 channel by protein kinase c activation: Role of the kvbeta1.2 subunit. J Pharmacol Exp Ther. 2002;302:545–550. doi: 10.1124/jpet.102.033357. [DOI] [PubMed] [Google Scholar]
- 132.Yue L, Feng J, Wang Z, Nattel S. Adrenergic control of the ultrarapid delayed rectifier current in canine atrial myocytes. The Journal of physiology. 1999;516 ( Pt 2):385–398. doi: 10.1111/j.1469-7793.1999.0385v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tessier S, Godreau D, Vranckx R, Lang-Lazdunski L, Mercadier JJ, Hatem SN. Cumulative inactivation of the outward potassium current: A likely mechanism underlying electrical memory in human atrial myocytes. J Mol Cell Cardiol. 2001;33:755–767. doi: 10.1006/jmcc.2001.1345. [DOI] [PubMed] [Google Scholar]
- 134.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–291. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- 135.Perez-Garcia MT, Lopez-Lopez JR, Gonzalez C. Kvbeta1.2 subunit coexpression in hek293 cells confers o2 sensitivity to kv4.2 but not to shaker channels. J Gen Physiol. 1999;113:897–907. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of a-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 137.El-Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, Coulombe A, Jeromin A, Hatem SN. Kv4 potassium channels form a tripartite complex with the anchoring protein sap97 and camkii in cardiac myocytes. Circ Res. 2009;104:758–769. doi: 10.1161/CIRCRESAHA.108.191007. [DOI] [PubMed] [Google Scholar]
- 138.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen-Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the kv channel-interacting protein 2 (kchip2) gene leads to a complete loss of i(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 139.Rosati B, Grau F, Rodriguez S, Li H, Nerbonne JM, McKinnon D. Concordant expression of kchip2 mrna, protein and transient outward current throughout the canine ventricle. J Physiol. 2003;548:815–822. doi: 10.1113/jphysiol.2002.033704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE, McKinnon D. Regulation of kchip2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001;533:119–125. doi: 10.1111/j.1469-7793.2001.0119b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Radicke S, Cotella D, Graf EM, Ravens U, Wettwer E. Expression and function of dipeptidyl-aminopeptidase-like protein 6 as a putative beta-subunit of human cardiac transient outward current encoded by kv4.3. J Physiol. 2005;565:751–756. doi: 10.1113/jphysiol.2005.087312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, Rudy B. The cd26-related dipeptidyl aminopeptidase-like protein dppx is a critical component of neuronal a-type k+ channels. Neuron. 2003;37:449–461. doi: 10.1016/s0896-6273(02)01185-6. [DOI] [PubMed] [Google Scholar]
- 143.Amarillo Y, De Santiago-Castillo JA, Dougherty K, Maffie J, Kwon E, Covarrubias M, Rudy B. Ternary kv4.2 channels recapitulate voltage-dependent inactivation kinetics of a-type k+ channels in cerebellar granule neurons. J Physiol. 2008;586:2093–2106. doi: 10.1113/jphysiol.2007.150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Anderson AE, Adams JP, Qian Y, Cook RG, Pfaffinger PJ, Sweatt JD. Kv4.2 phosphorylation by cyclic amp-dependent protein kinase. J Biol Chem. 2000;275:5337–5346. doi: 10.1074/jbc.275.8.5337. [DOI] [PubMed] [Google Scholar]
- 145.Martin GS. The hunting of the src. Nat Rev Mol Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 146.Gomes P, Saito T, Del Corsso C, Alioua A, Eghbali M, Toro L, Stefani E. Identification of a functional interaction between kv4.3 channels and c-src tyrosine kinase. Biochimica et biophysica acta. 2008;1783:1884–1892. doi: 10.1016/j.bbamcr.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Eldstrom J, Choi WS, Steele DF, Fedida D. Sap97 increases kv1.5 currents through an indirect n-terminal mechanism. FEBS Lett. 2003;547:205–211. doi: 10.1016/s0014-5793(03)00668-9. [DOI] [PubMed] [Google Scholar]
- 148.Abi-Char J, El-Haou S, Balse E, Neyroud N, Vranckx R, Coulombe A, Hatem SN. The anchoring protein sap97 retains kv1.5 channels in the plasma membrane of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H1851–1861. doi: 10.1152/ajpheart.01045.2007. [DOI] [PubMed] [Google Scholar]
- 149.Radicke S, Cotella D, Graf EM, Banse U, Jost N, Varro A, Tseng GN, Ravens U, Wettwer E. Functional modulation of the transient outward current ito by kcne beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc Res. 2006;71:695–703. doi: 10.1016/j.cardiores.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 150.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res. 1994;75:473–482. doi: 10.1161/01.res.75.3.473. [DOI] [PubMed] [Google Scholar]
- 151.Abbott GW, Goldstein SA. Disease-associated mutations in kcne potassium channel subunits (mirps) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J. 2002;16:390–400. doi: 10.1096/fj.01-0520hyp. [DOI] [PubMed] [Google Scholar]
- 152.McCrossan ZA, Abbott GW. The mink-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 153.Roepke TK, Kontogeorgis A, Ovanez C, Xu X, Young JB, Purtell K, Goldstein PA, Christini DJ, Peters NS, Akar FG, Gutstein DE, Lerner DJ, Abbott GW. Targeted deletion of kcne2 impairs ventricular repolarization via disruption of i(k,slow1) and i(to,f) FASEB J. 2008;22:3648–3660. doi: 10.1096/fj.08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of k(v)lqt1 and mink (isk) proteins to form cardiac i(ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 155.Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD, Magnuson MA, Roden DM. Replacement by homologous recombination of the mink gene with lacz reveals restriction of mink expression to the mouse cardiac conduction system. Circulation Research. 1999;84:146–152. doi: 10.1161/01.res.84.2.146. [DOI] [PubMed] [Google Scholar]
- 156.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hmink gene cause long qt syndrome and suppress iks function. Nature Genetics. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 157.Delpon E, Cordeiro JM, Nunez L, Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman-Bang J, Burashnikov E, Christiansen M, Antzelevitch C. Functional effects of kcne3 mutation and its role in the development of brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–218. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lundby A, Olesen SP. Kcne3 is an inhibitory subunit of the kv4.3 potassium channel. Biochem Biophys Res Commun. 2006;346:958–967. doi: 10.1016/j.bbrc.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 159.Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V, Escande D, Demolombe S, Barhanin J. In vitro molecular interactions and distribution of kcne family with kcnq1 in the human heart. Cardiovascular Research. 2005;67:529–538. doi: 10.1016/j.cardiores.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 160.Campuzano O, Brugada R, Iglesias A. Genetics of brugada syndrome. Current opinion in cardiology. 2010;25:210–215. doi: 10.1097/HCO.0b013e32833846ee. [DOI] [PubMed] [Google Scholar]
- 161.Bezzina CR, Wilde AA, Roden DM. The molecular genetics of arrhythmias. Cardiovascular Research. 2005;67:343–346. doi: 10.1016/j.cardiores.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 162.Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of herg channel block. Molecular pharmacology. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- 163.Sanguinetti MC, Tristani-Firouzi M. Herg potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 164.Wimmers S, Wulfsen I, Bauer CK, Schwarz JR. Erg1, erg2 and erg3 k channel subunits are able to form heteromultimers. Pflugers Arch. 2001;441:450–455. doi: 10.1007/s004240000467. [DOI] [PubMed] [Google Scholar]
- 165.Robertson GA, Jones EM, Wang J. Gating and assembly of heteromeric herg1a/1b channels underlying i(kr) in the heart. Novartis Foundation symposium. 2005;266:4–15. discussion 15–18, 44–15. [PubMed] [Google Scholar]
- 166.Kupershmidt S, Snyders DJ, Raes A, Roden DM. A k+ channel splice variant common in human heart lacks a c-terminal domain required for expression of rapidly activating delayed rectifier current. The Journal of Biological Chemistry. 1998;273:27231–27235. doi: 10.1074/jbc.273.42.27231. [DOI] [PubMed] [Google Scholar]
- 167.Jonsson MK, van der Heyden MA, van Veen TA. Deciphering herg channels: Molecular basis of the rapid component of the delayed rectifier potassium current. Journal of Molecular and Cellular Cardiology. 2012;53:369–374. doi: 10.1016/j.yjmcc.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 168.Ma Q, Yu H, Lin J, Sun Y, Shen X, Ren L. Screening for cardiac herg potassium channel interacting proteins using the yeast two-hybrid technique. Cell Biol Int. 2014;38:239–245. doi: 10.1002/cbin.10196. [DOI] [PubMed] [Google Scholar]
- 169.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-qt syndrome linked to mutations in herg. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 170.Yang T, Kupershmidt S, Roden DM. Anti-mink antisense decreases the amplitude of the rapidly activating cardiac delayed rectifier k+ current. Circ Res. 1995;77:1246–1253. doi: 10.1161/01.res.77.6.1246. [DOI] [PubMed] [Google Scholar]
- 171.McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, Fishman GI. A mink-herg complex regulates the cardiac potassium current i(kr) Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- 172.Kagan A, Melman YF, Krumerman A, McDonald TV. 14–3–3 amplifies and prolongs adrenergic stimulation of herg k+ channel activity. The EMBO journal. 2002;21:1889–1898. doi: 10.1093/emboj/21.8.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chao CC, Mihic A, Tsushima RG, Gaisano HY. Snare protein regulation of cardiac potassium channels and atrial natriuretic factor secretion. J Mol Cell Cardiol. 2011;50:401–407. doi: 10.1016/j.yjmcc.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 174.Organ-Darling LE, Vernon AN, Giovanniello JR, Lu Y, Moshal K, Roder K, Li W, Koren G. Interactions between herg and kcnq1 alpha-subunits are mediated by their cooh termini and modulated by camp. Am J Physiol Heart Circ Physiol. 2013;304:H589–599. doi: 10.1152/ajpheart.00385.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ren XQ, Liu GX, Organ-Darling LE, Zheng R, Roder K, Jindal HK, Centracchio J, McDonald TV, Koren G. Pore mutants of herg and kvlqt1 downregulate the reciprocal currents in stable cell lines. Am J Physiol Heart Circ Physiol. 2010;299:H1525–1534. doi: 10.1152/ajpheart.00479.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guo J, Wang T, Yang T, Xu J, Li W, Fridman MD, Fisher JT, Zhang S. Interaction between the cardiac rapidly (ikr) and slowly (iks) activating delayed rectifier potassium channels revealed by low k+-induced herg endocytic degradation. J Biol Chem. 2011;286:34664–34674. doi: 10.1074/jbc.M111.253351. [DOI] [PMC free article] [PubMed] [Google Scholar]