

ABSTRACT
The genomic DNAs of tailed bacteriophages are commonly modified by the attachment of chemical groups. Some forms of DNA modification are known to protect phage DNA from cleavage by restriction enzymes, but others are of unknown function. Recently, the CRISPR-Cas nuclease complexes were shown to mediate bacterial adaptive immunity by RNA-guided target recognition, raising the question of whether phage DNA modifications may also block attack by CRISPR-Cas9. We investigated phage T4 as a model system, where cytosine is replaced with glucosyl-hydroxymethylcytosine (glc-HMC). We first quantified the extent and distribution of covalent modifications in T4 DNA by single-molecule DNA sequencing and enzymatic probing. We then designed CRISPR spacer sequences targeting T4 and found that wild-type T4 containing glc-HMC was insensitive to attack by CRISPR-Cas9 but mutants with unmodified cytosine were sensitive. Phage with HMC showed only intermediate sensitivity. While this work was in progress, another group reported examples of heavily engineered CRISRP-Cas9 complexes that could, in fact, overcome the effects of T4 DNA modification, indicating that modifications can inhibit but do not always fully block attack.
IMPORTANCE
Bacteria were recently found to have a form of adaptive immunity, the CRISPR-Cas systems, which use nucleic acid pairing to recognize and cleave genomic DNA of invaders such as bacteriophage. Historic work with tailed phages has shown that phage DNA is often modified by covalent attachment of large chemical groups. Here we demonstrate that DNA modification in phage T4 inhibits attack by the CRISPR-Cas9 system. This finding provides insight into mechanisms of host-virus competition and also a new set of tools that may be useful in modulating the activity of CRISPR-Cas9 in genome engineering applications.
INTRODUCTION
The functional importance of covalent DNA modification was first demonstrated in 1952 in studies of bacteriophage (1, 2). In bacteriophage T4, genomic DNA contains 5-hydroxymethylcytosine (HMC), which is further modified by the attachment of glucose to yield glucosyl HMC (glc-HMC) (Fig. 1) (3–6). Incorporation of HMC blocks DNA cleavage by many restriction endonucleases, and the glc-HMC modification further blocks attack by the HMC-specific McrABC (Rgl/MspJI) nuclease. Today, more than 10 different types of covalent modification in bacteriophage DNA are known, many of which are of unknown function (7, 8). Eukaryotic DNA can also be modified to methylcytosine, hydroxymethylcytosine, and glucosylated hydroxymethyldeoxyuridine (9, 10).
FIG 1 .
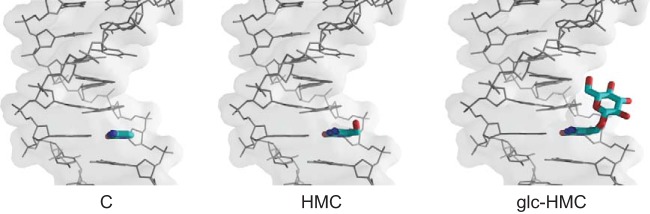
DNA modification in phage T4 showing C-containing DNA (left), HMC-containing DNA (middle), and glc-HMC DNA (right).
Recently, striking studies have revealed a new class of nucleases in bacteria, the CRISPR-Cas systems, which provide bacteria with a form of adaptive immunity against infection with genomic parasites such as phages or plasmids (11–17). Short sequences from genomic parasites are incorporated into CRISPR arrays in the bacterial chromosome; they consist of repeated sequences and unique spacers (typically ~30 nucleotides) that are derived from invaders (18–20). Transcription of the arrays and RNA processing produce spacer RNA sequences (crRNAs) that are bound by a nuclease (Cas9 for the type II CRISPR systems) (21). The crRNA is then used to recognize DNA of invaders by base pairing, allowing subsequent nucleic acid cleavage by Cas9 (22; reviewed in references 23, to ,25). These programmable nuclease systems are now used widely in biotechnology applications (26–31).
The discovery of the CRISPR-Cas9 system raised the question of whether T4 DNA modification might have an additional function—protecting phage DNA from cleavage by CRISPR-Cas9. One previous paper analyzed the effects of a smaller DNA modification—adenine N6 methylation—and showed that phage DNA with this modification was still sensitive (32). Another study demonstrated that genome engineering by Cas9 in eukaryotes is also unaffected by 5-methylcytosine (33). Here we investigated the effects of the larger DNA modifications found in T4. We first characterized T4 DNA modification in detail by single-molecule sequencing and nuclease digestion. We went on to show that the bulkier HMC and glc-HMC modifications can, in fact, inhibit CRISPR-Cas9 attack. While this work was in progress, another group reported examples where T4 with glc-HMC modification could, in fact, be sensitive to attack by CRISPR-Cas9, which we further analyzed and attribute to the use of a heavily engineered and optimized CRISPR-Cas9 system (34).
RESULTS
The extent of modification in T4 and mutant derivatives.
Prior to testing sensitivity to CRISPR-Cas9, wild-type bacteriophage T4 [termed T4(glc-HMC) here] and mutants altered in DNA modification were analyzed to characterize the nature and extent of genomic DNA modification. T4(147) is a mutant with changes in genes encoding the alpha and beta glucosyltransferases that attach glucose to HMC DNA so that genomes contain HMC only [termed T4(HMC) here]. T4(GT7) is a mutant with changes in genes required to substitute HMC for dCTP in nucleotide pools so that genomes contain only unmodified cytosines [termed T4(C) here]. For complete T4 genotypes, see Table S1 in the supplemental material.
Several studies were carried out to verify the presence of the expected DNA modifications in each phage and evaluate the extent of base substitution. First, genomic DNAs were purified from phage stocks and probed by exposure to DNA-modifying enzymes of known specificities. DNA from T4(C), but not T4(HMC) or T4(glc-HMC), was sensitive to digestion by the restriction enzyme AluI (Fig. 2A, top), as expected for DNA containing unmodified cytosines. T4 genomic DNAs were next incubated with MspJI, which cleaves HMC-containing DNA selectively. T4(HMC) DNA was digested, but not T4(glc-HMC) or T4(C) (Fig. 2A, middle). The T4 DNAs were also exposed to a glucosyltransferase and a glucose donor, which resulted in reduced mobility of T4(HMC) DNA, consistent with glucose attachment to HMC, but no changes were observed in faster-migrating T4(C) or slower-migrating T4(glc-HMC) (Fig. 2A, bottom). T4 DNA modification was further probed by infection of Escherichia coli strains expressing the Rgl nuclease, which cleaves HMC-containing DNA selectively. Infection with T4(HMC) was undetectable in Rgl-containing strains, but infection with T4(glc-HMC) or T4(C) was not restricted. These data confirm the expected modification patterns in the T4 DNA stocks studied and indicate that the extents of HMC incorporation and subsequent glucosyl conjugation are great, consistent with an analysis of nucleotides generated after enzymatic degradation of T4(glc-HMC) DNA (35).
FIG 2 .

Characterization of phage T4 DNA modification. (A) Phage T4(glc-HMC), T4(HMC), and T4(C) DNA left untreated (−) or treated with (+) restriction enzymes AluI (top), which cleaves unmodified DNA; MspJI (middle), which cleaves HMC-containing DNA; or T4 glucosyltransferase (bottom), which increases the mobility of HMC-containing DNA by the addition of glucose groups. The arrows indicate the mobility shift due to glucose attachment. (B) Analysis of phage T4 DNA modification by single-molecule sequencing. Results are summarized for each genome by mapping IPD ratios at each base for each of the T4 strains studied. The coloration of each base is shown by the key at the bottom left. The T4 nucleotide sequence runs from top to bottom for each of the four genomes. The distance each colored point is displaced from the center indicates the IPD ratio (scale at bottom; leftward for the reverse strand, rightward for the forward strand). Examples of interpulse distances (indicative of modification) are shown to the right for a short segment of the T4 genome. Bars indicate the magnitude of the IPD ratio (upward for the forward strand and downward for the reverse strand). A 5′ GATC 3′ site of DAM methylation is highlighted in yellow. (C) Violin plot showing IPD ratios of A residues at 5′ GATC 3′ sequences.
Single-molecule sequencing to characterize T4 DNA modification.
To characterize the extent and distribution of DNA modifications in more detail, we subjected each T4 DNA sample to analysis by single-molecule real-time (SMRT) sequencing by Pacific Biosciences technology (36, 37). In this method, single DNA molecules are sequenced by synthesis on immobilized DNA polymerase enzymes. Sequence information is acquired by detection of fluorescently labeled nucleotides during each incorporation step. The presence of DNA modifications in the template can slow the kinetics of incorporation, allowing DNA modification to be quantified as an increase in interpulse duration (IPD). IPD values are calculated for each position in the template sequence and are compared to an in silico model of IPD values for an unmodified sequence (IPD ratio) (38). In favorable cases, different forms of DNA modification show distinguishable kinetic profiles (38–41).
Figure 2B (see also Fig. S1A and B in the supplemental material) summarizes the SMRT sequencing results for T4(glc-HMC), T4(HMC), T4(C), and a T4 genome lacking all forms of modification made by copying wild-type T4 DNA with a DNA polymerase in vitro (whole-genome amplification [WGA]). Kinetic profiles of T4(glc-HMC) showed many increased IPD ratios associated with expected positions of glc-HMC (Fig. 2B, top). Particularly increased IPDs were seen for potential glc-HMC sites 3′ of a G residue or 5′ of a pyrimidine (see Fig. S2 in the supplemental material). The mechanism of these sequence context effects is unknown—they could reflect either different extents of glucose attachment dependent on the local sequence or differential effects of the sequence on polymerase kinetics. Kinetic perturbations were also seen at additional base positions, commonly those near C residues in the sequence, suggesting that modifications may contact polymerase from nearby positions in the DNA chain. For T4(HMC), kinetic lags were also associated with sites of potential C modification (Fig. 2B, middle), but the magnitude of the effects was typically less than for T4(glc-HMC). For T4(HMC) modification, IPD ratios were particularly high for pairs of expected HMC residues (see Fig. S2). T4(C) and the WGA DNA showed no notable alterations in kinetics at C residues (Fig. 2B, bottom two panels).
T4 also encodes an (N6-adenine)-methyltransferase that methylates A residues at 5′ GATC 3′ sequences. The role of this modification is unknown, but it may help protect T4 DNA from the cellular methyl-directed mismatch repair system, which carries out double-strand cleavage when mismatches are detected near unmodified 5′ GATC 3′ sequences (42–44). Methylation was evident as increased IPD values at A in 5′ GATC 3′ in all three T4 genomic DNAs but not in the WGA control. However, the extents of modification differed (Fig. 2C). The extent of 5′ GATC 3′ adenine methylation was higher in both T4(C) and T4(HMC) than in T4(glc-HMC) DNA, paralleling a previous report (45). This is consistent with a steric interference model, which posits that glucosylation of HMC obstructs access of the adenine methyltransferase to T4 DNA and thereby reduces the extent of 5′ GATC 3′ adenine methylation.
Sequencing data for T4(C) showed a high proportion of reads that mapped to the E. coli genome (56.7%; see Fig. S3 in the supplemental material). Far fewer E. coli reads were detected in the T4(glc-HMC) and T4(HMC) samples (1.5 and 0.8%, respectively). The T4(C) strain has been widely used in generalized transduction for genetic mapping in E. coli (46–48). T4(C) contains a mutation that inactivates the gene encoding the DenB nuclease, which normally degrades host cell DNA, thus allowing E. coli DNA to compete for packaging in T4 particles, as well as mutations inactivating the T4-encoded dCTPase and dCMP hydroxymethylase enzymes. In T4(C), different segments of the E. coli DNA were not packaged with uniform frequency (see Fig. S3), suggesting the possible involvement of sequence-specific recognition during T4 packaging (49).
Inhibition of CRISPR-Cas9 by T4 DNA modification.
Given that the densities of DNA modifications in T4(glc-HMC) and T4(HMC) are high, we sought to investigate whether the CRISPR-Cas system was blocked by T4 DNA modification. As a representative CRISPR-Cas system, we chose the type II system of Streptococcus pyogenes because it has been widely used in biotechnology applications and functions well in E. coli (27). CRISPR spacers (targeting sequences) were designed to target four regions of the T4 genome (termed protospacers), each proximal to the required downstream 5′ NGG 3′ protospacer-adjacent motif (PAM) in the T4 target.
Protospacer sequences and T4 DNA modification densities in these regions are shown in Fig. 3. The spacers were designed to target regions of the T4 genome with various cytosine arrangements and densities. None of the spacers contain the adenine methylation target sequence, GATC. Spacer T4 CRISPR1 was designed to target a region of the T4 genome that maximizes the number of cytosines on the target strand and in the seed sequence, which is the inferred 3′ region of the CRISPR RNA that is reported to be most important for recognition (50). Spacer T4 CRISPR2 maximizes the number of cytosines or modified derivatives on both the target and complementary strands in the T4 protospacer. Spacer T4 CRISPR3 minimizes the number of cytosines in the target strand but maximizes the number of cytosines on the complementary strand of the T4 protospacer. Spacer T4 CRISPR4 minimized the numbers of cytosine residues on both the target and complementary strands of the T4 protospacer. IPD ratio analysis of T4(glc-HMC) and T4(HMC) DNA showed slowed kinetics at C residues in the complement of the 5′ NGG 3′ PAM sequence and at internal cytosines, indicative of DNA modification.
FIG 3 .

IPD modification profiles of T4(glc-HMC), T4(HMC), and T4(C) phage protospacers. IPD ratios for the forward strand (blue) and reverse strand (red) of T4(glc-HMC), T4(HMC), and T4(C) are depicted for the regions of the T4 genome targeted by spacers 1 to 4 along with the surrounding nucleotides. The nucleotide sequences of the phage protospacer (orange), the PAM (green), and the surrounding nucleotides (black) are along the x axis. The top strand of the protospacer is identical in sequence to the crRNA/spacer, and the bottom strand is the target strand, which is complementary to the spacer and will base pair with the crRNA.
The efficiency of phage infection was then tested on the CRISPR-Cas9-containing strains. To confirm that the CRISPR system was active with our engineered spacers, we transformed the CRISPR-Cas9-containing bacteria with pUC19-derived plasmids encoding either the corresponding T4 protospacers and PAM sequences or a nonspecific control sequence (Fig. 4A). Using spectinomycin antibiotic selection for the target plasmid, we quantified the efficiency of transformation by comparing the number of bacteria containing the incoming plasmid with a target matching the T4 protospacer with a control plasmid lacking the target (Fig. 4B). All of the CRISPR-Cas9-containing bacteria showed reduced acquisition of the target-containing plasmid, indicating that the CRISPR systems are functional and reduce transformation by at least 2 logs (Fig. 4B).
FIG 4 .

Glc-HMC and HMC modifications inhibit attack by the CRISPR-Cas9 system on phage T4. (A) Diagram of the strategy used to validate CRISPR spacers in a transformation assay. Bacteria containing the type II CRISPR system were transformed with a pUC19 plasmid containing either a T4 protospacer and PAM sequence or a nonspecific DNA sequence. Antibiotic selection for the pUC19 plasmid and quantification of the efficiency of transformation reveal the efficacy of CRISPR system cleavage of unmodified DNA containing a protospacer and PAM. (B) Results of plasmid challenge tests. The efficiency of transformation is the ratio of colony counts of cells transformed with equal amounts of pUC19 that contain a protospacer targeting the plasmid (numerator) to the colony counts of cells transformed with pUC19 (denominator). (C) Diagram of plaque assays to assess inhibition of T4 infection with CRISPR-Cas9. (D to F) Results of plaque assays in which the E. coli strains indicated were infected with up to 1 × 104 PFU of T4(C) (panel D), T4(glc-HMC) (panel E), or T4(HMC) (panel F). E. coli strains expressed Cas9 and crRNAs targeting T4 or controls. Starting from the left in each panel, None indicates no crRNA or Cas9, non-sp indicates nonspecific crRNA, 1 contained the maximum number of cytosines in the target strand and seed sequence, 2 contained the maximum number of cytosines in the target and complementary strands, 3 contained no cytosines in the target strand and seven cytosines in the complementary strand, and 4 contained the fewest cytosines in the target and complementary strands. Mean values were compared with the Kruskal-Wallis test. *, P < 0.01; ***, P < 0.0001; ns, not significant.
The abilities of T4 and mutant derivatives to infect CRISPR-Cas9-containing bacteria were then scored in plaque assays (Fig. 4C). Figure 4D to F show illustrative experiments in which T4 phage were plated on CRISPR-Cas9-containing strains or controls and the efficiency of plating was quantified. Infection with cytosine-only strain T4(C) resulted in reduced or undetectable plaque formation in the presence of the T4-targeting spacers (Fig. 4D, rightmost four spacers). Plaque formation was not affected by the presence of Cas9 and a nonspecific spacer or in E. coli with no CRISPR-Cas9 system (Fig. 4D, left two samples, marked None and non-sp). T4 CRISPR1 showed the weakest activity, possibly because of high G/C content or the presence of homopolymeric sequences in the crRNA, which were previously reported to inhibit function (51). Titration studies of strains containing Cas9 and CRISPR2, 3, and 4 showed the efficiency of plating to be reduced by >10,000-fold. CRISPR1 was weaker, showing a reduction of only about 3-fold, paralleling many studies showing variation in the efficiency of CRISPR targeting.
T4(glc-HMC), in contrast, formed plaques on strains expressing T4 CRISPR1 to 4 and Cas9 efficiently (Fig. 4E). Infection of three of the four CRISPR-containing strains was as efficient as for the strains with control nonspecific spacers. The fourth (T4 CRISPR4) contained the spacer with the fewest modified C resides on both DNA strands and therefore the lowest modification density. Infection with T4(glc-HMC) was reduced 2- to 10-fold in repeated assays, and plaque size was reduced by about two-thirds, indicating some sensitivity. Note that two modified cytosines are present in the T4 CRISPR4 target in the DNA complementary to the 5′ NGG 3′ PAM, and glucosylation of these likely exerted some inhibition. A previous study showed that both DNA strands of the PAM are important for target recognition in other CRISPR systems (52). T4(glc-HMC) infection was not inhibited in strains expressing T4 CRISPR3, which contains no cytosines on the target DNA strand but seven on the complementary strand, indicating that modifications on either strand can interfere with CRISPR attack. Thus, glucosylation of HMC mostly protects T4 from attack by the CRISPR-Cas9 system, but a region with few glc-HMC residues showed modest but detectable sensitivity.
For T4(HMC) (Fig. 4F), the T4-targeting CRISPR1 to 3 constructs did not inhibit infection, indicating that substitution of cytosine with HMC was also sufficient to block CRISPR-Cas9 attack. However, for T4 CRISPR4, which has the fewest C residues on both the target and complementary strands, T4(HMC) was highly sensitive—efficiency of plating was reduced by at least 10,000-fold (Fig. 4F). This indicates that the HMC modification alone on the cytosines on the complementary strand of the 5′ NGG 3′ PAM is not enough to inhibit CRISPR-Cas9 attack. Evidently, the HMC modification is a less effective blocker than the glc-HMC modification, though both suffice at a high enough density.
Comparison to results of Yaung et al.
While this work was in progress, Yaung et al. reported three spacers in an engineered type II CRISPR system that were functional against wild-type T4(glc-HMC) phage and a T4 mutant containing HMC DNA (34). We obtained their spacer plasmids and confirmed that they were able to restrict the growth of T4(HMC) and T4(glc-HMC) phage in plaque assays as reported (see Fig. S4A and B in the supplemental material). These spacers differed from ours in that the crRNAs were engineered so that they were fused to tracrRNAs also known as single-guide RNAs (26). The tracrRNA is a small RNA bound by Cas9 that is required for crRNA processing and as a cofactor for Cas9 nuclease activity (53). Fusion of the two RNAs is convenient in some genome engineering applications (26).
We cloned the spacers of Yaung et al. into the type II CRISPR system used in our studies, where crRNAs are not fused to tracrRNA, as in the natural S. pyogenes CRISPR-Cas9 system. We found that two of the three spacers were ineffective against the modified DNA of phage T4(glc-HMC) (see Fig. S4D), but all three spacers were effective against unmodified DNA (see Fig. S5). Two of the spacers restricted the growth of T4(HMC), while the third showed partial activity (see Fig. S4C). We confirmed that the Cas9 nucleases used here and by Yaung et al. functioned similarly in side-by-side tests, indicating that the CRISPR RNAs and not the Cas9 nuclease were responsible for the functional differences (see Fig. S5E and F). Evidently, the spacers of Yaung et al., with the synthetic single-guide RNA fusion, show higher activity against modified T4 DNA (see Fig. S4). These data indicate that DNA modifications can inhibit a biologically occurring type II CRISPR system but that particularly potent crRNAs can overcome this inhibition.
With the model system available to study CRISPR-Cas9 attack on T4, we were able to address further questions of T4 biology as described below.
Testing the role of T4 IP proteins.
Three T4 proteins are injected into E. coli along with T4 DNA early during infection (IPI to IPIII) and bind the T4 genome (54). IPI protects T4 from the GmrS/GmrD restriction enzyme, but the functions of IPII and IPIII are unknown (55)—we thus asked whether any of the IP proteins contribute to evasion of the CRISPR-Cas9 system. This study was motivated in part by a previous report in which Pseudomonas phages were shown to encode protein inhibitors of a CRISPR-Cas system (56). For T4, such proteins would hypothetically be required for DNA modifications to exert their protective effect. A T4(glc-HMC) mutant strain in which all three IP genes are altered was tested by infection of strains containing the T4-targeting CRISPRs. No difference in infectivity was observed, indicating that the IP proteins are not cofactors required to allow DNA modification to inhibit attack by CRISPR-Cas9 (see Fig. S6 in the supplemental material). However, we note that there are multiple types of CRISPR-Cas systems that are quite different from each other, and T4 can infect Escherichia, Shigella, and Yersinia (57), so it would be of interest to test possible inhibition of additional CRISPR-Cas systems from these organisms.
Characterization of a revertant of T4(C) with reduced sensitivity to CRISPR-Cas9.
We observed a T4(C) revertant that reduced sensitivity to CRISPR-Cas9, and so we characterized it further. Normally, T4(C) plaques are small and turbid. During growth, we observed the appearance of a new variant generating large clear plaques resembling T4(glc-HMC) plaques. Further tests showed reduced sensitivity to CRISPR-Cas9 (see Fig. S7C in the supplemental material). We sequenced the revertant phage, named T4(C)R, and identified three mutations (see Fig. S7A and B). One point mutation eliminated the stop codon in gp42, which encodes dCMP hydroxymethylase, an enzyme necessary to synthesize HMC. A second mutation introduced a single nucleotide deletion into the deoxycytidylate deaminase gene, yielding a stop codon that truncated the encoded protein. Deoxycytidylate deaminase converts dCMP (a precursor of HMC) to dUMP. Both of these mutations favor the synthesis of HMC, which can be incorporated into the T4(C)R genome and then further modified to glc-HMC with α- and β-glucosyltransferases. The third mutation was a nonsynonymous point mutation in the uncharacterized, hypothetical protein NrdC.5 and is of unknown significance.
T4(C)R was resistant to spacers 1, 2, and 3 in the CRISPR-Cas9 system but sensitive to spacer 4 (see Fig. S7C), thus showing slightly greater sensitivity than wild-type T4(glc-HMC). These and other results are consistent with the idea that T4(C)R contains HMC or glc-HMC, though potentially not at every position in the genome because of larger cellular dCTP pools competing for incorporation. These findings again support the idea that DNA modifications can block CRISPR-Cas9 activity.
DISCUSSION
These data show that modification of T4 DNA to HMC or glc-HMC reduces sensitivity to attack by CRISPR-Cas9. A previous study showed that adenine methylation at a 5′ GATC 3′ sequence did not block CRISPR-Cas-mediated inhibition (32), and data presented here show that low-density modification with HMC also was not protective. Evidently, protection against CRISPR-Cas9 attack can be achieved by either the addition of bulkier glucosyl-HMC modifications or the addition of a high density of less bulky HMC modifications.
While this work was in progress, and contrary to our developing data, Yaung et al. reported spacers that could, in fact, target glc-HMC-modified T4 DNA efficiently (34). Our own tests with the reagents of Yaung et al. confirmed their conclusions. The Cas9 enzymes used were identical in both studies, specifying the RNA component as the origin of the different potency. Yaung et al. used crRNAs fused to tracrRNAs, which could have potentially improved activity by favoring RNA loading onto Cas9 or increased specific activity of the loaded sgRNA/Cas9 complex. One of the crRNAs of Yaung et al. was notably potent even without the tracrRNA fusion, suggesting that for this spacer, fusion with the crRNA did not explain its potency. Another candidate explanation is that the positions of base modifications in the recognition site may be important and that the rules for this are not fully clarified. For all of the spacers studied here, we have not investigated whether cleavage mediating T4 inhibition is, in fact, due to on- or off-target cleavage, so increased off-target specificity is another possible explanation for the increased inhibition seen (58).
Classic studies of the tailed DNA phages have identified more than 10 different forms of covalent DNA modification, and modification is commonly found in the DNA of these viruses (7, 8). Recent metagenomic studies also emphasize the ubiquity of CRISPR systems targeting phage in natural environments such as the human microbiome (59–61). There are even examples of phage from the human gut that themselves encode CRISPR spacers targeting other phage from the same individual, indicating that phages may compete with each other by using the CRISPR-Cas system (60, 61). Given these observations and data shown here that modification of T4 DNA to HMC or glc-HMC can reduce sensitivity to attack by CRISPR-Cas9, it seems probable that many of the bulkier forms of DNA modification seen in tailed DNA phage have evolved, at least in part, to reduce sensitivity to cleavage by CRISPR-Cas systems.
MATERIALS AND METHODS
Propagation of phage strains.
Manipulation of phage T4 was carried out as described in reference 62. Phage T4(glc-HMC), T4(HMC), and T4(C) were provided by Lindsay Black. For their genotypes, see Table S1 in the supplemental material. T4(C) contains amber mutations in several DNA-modifying genes (see Table S1). The amber mutations are known to revert easily, so T4(C) was propagated in the amber suppressor strain E. coli CR63 to prevent genotype reversion. Experiments with T4(C) were carried out in nonsuppressor E. coli strain DH10B to ensure that cytosines in T4(C) were unmodified. Experiments and propagation of T4(glc-HMC) and T4(HMC) were carried out with DH10B. Experiments with T4(IP0) were carried out with E. coli B834.
CRISPR system and spacer design.
Design of CRISPR spacers was carried out with custom code in R (see Text S1 in the supplemental material). CRISPR-targeting plasmids were constructed with the system described by L. Marraffini and coworkers (27), which consists of two plasmids, pCas9 and pCRISPR. pCas9 contains the Cas9 nuclease and tracrRNA (Addgene no. 42876). Spacers in this study were cloned into the CRISPR array on pCRISPR (Addgene no. 42875) with the Marraffini lab protocol available at Addgene. Comparison of our work with that of Yaung et al. was carried out with plasmids DS-SPCas (Addgene no. 48645) and PM-SP!TB (Addgene no. 48650) and plasmids provided by Yaung et al. For the oligonucleotides used for cloning, see Table S2 in the supplemental material.
Plasmid transformation assays.
The T4 protospacer and PAM sequences used in this study were individually cloned into pUC19 plasmids. One hundred nanograms of protospacer/PAM-containing pUC19 was transformed into chemically competent E. coli DH10B containing a CRISPR-Cas9 system targeting the corresponding protospacer. As a transformation control, 100 ng of pUC19 without a protospacer was transformed into DH10B containing a CRISPR-Cas9 expression system. Transformation mixtures were incubated at 37°C for 1 h in 200 µl of S.O.C. medium without antibiotic selection and then plated on LB plates containing carbenicillin at 100 µg/ml to select for pUC19. Efficiency of transformation was determined by dividing the number of CFU observed in the protospacer-containing pUC19 transformation mixture by the number of CFU observed in the control pUC19 transformation mixture.
Plaque assays.
Plaque assays were used to determine the ability of phage to infect DH10B bacteria containing the CRISPR-Cas9 system. Up to 104 PFU of phage in a volume of 10 µl were added to 200 µl of log-phase E. coli DH10B and incubated at room temperature for 10 min. Three milliliters of 0.4% LB top agarose was added to the bacterium-phage mixture, mixed, and poured onto LB plates containing the appropriate antibiotics (100 µg/ml kanamycin for pCRISPR, 50 µg/ml chloramphenicol for pCas9, 100 µg/ml ampicillin for DS-SPcas, and 50 µg/ml chloramphenicol for PM-SP!TB). Plates were incubated at 37°C overnight. Three biological replicates, each with three technical replicates, were prepared per experiment. The efficiency of plating was determined by dividing the number of plaques on an experimental plate by the number of plaques on a control plate containing E. coli with no CRISPR system. A Kruskal-Wallis nonparametric comparison of means was carried out with GraphPad Prism software for each experiment.
Phage DNA isolation and sequencing.
Phage lysates were grown at a multiplicity of infection of 0.01 on DH10B. Phage T4 DNAs were isolated with the Norgen phage DNA isolation kit (Norgen Biotek Corp., Thorold, Canada). Chloroform-treated phage lysates were concentrated by 4% precipitation in PEG 4000–500 mM NaCl, resuspended in Tris-EDTA buffer, and purified as recommended. The concentration of isolated T4 phage DNAs was measured with the Quant-iT PicoGreen dsDNA Assay kit (Life Technologies, Carlsbad, CA). For single-molecule sequencing, purified phage DNA was fragmented to an average size of 1.5 kb via adaptive focused acoustics (Covaris, Woburn, MA). SMRTbell template sequencing libraries were prepared as previously described (63). Sequencing was carried out on a PacBio RS II (Pacific Biosciences, Menlo Park, CA) by using P4/C2 sequencing chemistry and standard protocols for large insert libraries. Consensus sequences were generated by Quiver, and kinetic data were generated with SMRT Analysis Software v2.0 (Pacific Biosciences). For further details of the methods used, see Text S1 in the supplemental material. Libraries for the sequencing of T4(C) and T4(C)R were made with Illumina’s Nextera XT DNA Sample Preparation kit with 1 ng of input DNA, generating paired-end fragments. Metagenomic sequencing was performed on an Illumina MiSeq instrument. Paired-end reads from the MiSeq instrument were quality trimmed. Reads were aligned with the NCBI T4 genome sequence by Geneious to form consensus sequences for T4(C) and T4(C)R.
Nuclease assays.
One microgram of T4(C), T4(HMC), or T4(glc-HMC) was digested with AluI (R0137s; NEB), MspJI (R0661S; NEB), or T4 phage β-glucosyltransferase (M0357S; NEB) in accordance with NEB-specified protocols.
Nucleotide sequence accession numbers.
The T4 genome sequences have been deposited in GenBank at NCBI under accession numbers KJ477684.1 for T4(glc-HMC)/T4 wild type, KJ477685.1 for T4(HMC)/T4(147), and KJ477686.1 for T4(C)/T4(GT7).
SUPPLEMENTAL MATERIAL
Code used to design CRISPR spacers. The program (in R) takes as input a DNA sequence file (fasta) and outputs protospacer targets. Download
(A) Read lengths in the single-molecule sequence data sets. (B) Mapped subread lengths in the single-molecule sequence data sets. Download
Heat map summarizing the effects of local sequences on IPD ratios at C residues. The base preceding the C residue in the sequence is marked 5′-base, and that following the C residue is marked 3′-base. The scale at the bottom summarizes the IPD ratios. Download
Sequence coverage maps for T4 strains comparing T4 (top) to E. coli B834 (bottom). Download
Comparison of our work with that of Yaung et al. (A and B) Results of plaque assays in which the E. coli strains indicated containing previously studied CRISPR spacers (C1 to C3) in the Yaung-Church CRISPR system (in C) were infected with up to 100 PFU of T4(HMC) (panel A) or T4(glc-HMC) (panel B). CRISPR spacer labeling: None, no crRNA or Cas9; non-sp, nonspecific crRNA. E.O.P., efficiency of plating. (C and D) Results of plaque assays in which the E. coli strains indicated containing previously studied CRISPR spacers (C1 to C3) in the Marraffini CRISPR system (in M) were infected with up to 100 PFU of T4(HMC) (panel C) or T4(glc-HMC) (panel D). CRISPR spacer labeling: None, no crRNA or Cas9; non-sp, nonspecific crRNA. (E and F) Plaque assay results of the Church laboratory Cas9 expression vector with the Marraffiini CRISPR array on the T4 CRISPR spacers studied here. The Church Cas9 expression vector and the Marraffini CRISPR array containing the spacers studied in this investigation (spacers 1 to 4) and the previously studied spacers (C1 to C3) were tested for efficacy against T4(HMC) (panel E) and T4(glc-HMC) (panel F). Mean efficiency of transformation was compared to that of a nonspecific control with a t test. **, P < 0.001; ***, P < 0.0001; ns, not significant. Download
Results of plasmid transformation assay comparing the efficacies of all of the CRISPR spacers studied in this investigation against unmodified DNA. Efficiency of transformation was normalized to 1 by the transformation of a plasmid not targeted by the CRISPR system (control). Spacers 1 to 4 are from this study. C1 to C3 are the spacers from Yaung et al. cloned into the CRISPR-Cas9 system developed by the Church lab (in C) or the Marraffini lab (in M). Mean efficiency of transformation was compared to the nonspecific control with a t test. ***, P < 0.0001. E.O.P., efficiency of plating. Download
Mutation of IP1-3 genes encoding the three T4 proteins that are injected along with the phage DNA does not reduce resistance to attack by CRISPR-Cas9. Shown are replicate infections with wild-type T4(glc-HMC) (A) and the triple mutant T4(IP0) (B), which show no differences in infectivity for the strains tested. Plaque assay with approximately 100 PFU of T4(glc-HMC) or T4(IP0) infecting E. coli not expressing CRISPR-Cas9 (None) or E. coli expressing CRISPR-Cas9 with spacers targeting a protospacer in the T4 genome with the maximum number of cytosines in the target sequence and seed sequence (1), the maximum number of cytosines in the target and complementary strands (2), the fewest cytosines in the target strand (3), the fewest cytosines in the target and complementary strands (4), or a nonspecific spacer that does not target the T4 genome (non-sp). The mean efficiency of plating (E.O.P.) for infection of cells with each spacer was compared to that of no-CRISPR-Cas9 control with a t test. No statistically significant differences were found. Download
Phenotype of a revertant of T4(C) named T4(C)R with reduced CRISPR sensitivity. (A) Genetic map of T4(C) and the revertant obtained by Illumina deep sequencing. The bottom black line represents the T4(glc-HMC) genome length, and black arrows indicate genes and the direction of transcription. Variations in the T4(C) genome compared to T4(glc-HMC) are blue for single nucleotide deletions, red for large deletions, and pink for nonsynonymous substitutions; red ×’s represent early stop codons. The top black line represents the T4(C)R genome. Variations in T4(C)R compared to T4(C) are green for nonsynonymous mutations and blue for deletions. The green circle indicates reversion of a stop codon, and the green × indicates a stop codon. (B) Glycosylated hydroxymethylcytosine synthesis pathway in T4(glc-HMC) phage and mutations in T4(C) and T4(C)R. Proteins mutated in T4(C) are shown by red ×’s. T4(C)R acquired a mutation designated by a green × and reverted a previous amber mutation denoted by a green circle. (C) Reduced sensitivity to CRISPR attack in the revertant. The mean efficiency of plating (E.O.P.) for infection of cells with each spacer was compared with that of the no-CRISPR-Cas9 control with a t test. ***, P < 0.0001; ns, not significant. Download
Genotypes of the bacteria and bacteriophages used in this study.
Oligonucleotides used in this study.
ACKNOWLEDGMENTS
We thank M. Boitano for assistance with DNA sequencing and J. Korlach for comments on the manuscript. Thanks to Alice Laughlin for assistance, to Kushol Gupta for help with Fig. 1A, and to L. Marraffini for providing plasmids pCas9 and pCRISPR and useful discussions.
This work was supported by Project UH2DK083981, NIH (AI39368 to G.D.W.), the Penn Digestive Disease Center (P30 DK050306), and The Joint Penn-CHOP Center for Digestive, Liver and Pancreatic Medicine (S10RR024525, UL1RR024134, and K24-DK078228).
Tyson Clark is an employee of Pacific Biosciences. The rest of us have no competing interests.
Footnotes
Citation Bryson AL, Hwang Y, Sherrill-Mix S, Wu GD, Lewis JD, Black L, Clark TA, Bushman FD. 2015. Covalent modification of bacteriophage T4 DNA inhibits CRISPR-Cas9. mBio 6(3):e00648-15. doi:10.1128/mBio.00648-15.
REFERENCES
- 1.Luria SE, Human ML. 1952. A nonhereditary, host-induced variation of bacterial viruses. J Bacteriol 64:557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hattman S. 2009. The first recognized epigenetic signal: DNA glucosylation of T-even bacteriopages [sic]. Epigenetics 4:150–151. doi: 10.4161/epi.4.3.8444. [DOI] [PubMed] [Google Scholar]
- 3.Karam JD, Drake JW. 1994. Molecular biology of bacteriophage T4. American Society for Microbiology, Washington, DC. [Google Scholar]
- 4.Josse J, Kornberg A. 1962. Glucosylation of deoxyribonucleic acid. III. Alpha- and beta-glucosyl transferases from T4-infected Escherichia coli. J Biol Chem 237:1968–1976. [PubMed] [Google Scholar]
- 5.Pratt EA, Kuo S, Lehman IR. 1963. Glucosylation of the deoxyribonucleic acid in hybrids of coliphages T2 and T4. Biochim Biophys Acta 68:108–111. doi: 10.1016/0926-6550(63)90413-4. [DOI] [PubMed] [Google Scholar]
- 6.Kornberg A, Baker T. 1991. DNA replication. W. H. Freeman and Company, New York, NY. [Google Scholar]
- 7.Warren RA. 1980. Modified bases in bacteriophage DNAs. Annu Rev Microbiol 34:137–158. doi: 10.1146/annurev.mi.34.100180.001033. [DOI] [PubMed] [Google Scholar]
- 8.Loenen WA, Raleigh EA. 2014. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res 42:56–69 doi: 10.1093/nar/gkt747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gommers-Ampt JH, Van Leeuwen F, de Beer AL, Vliegenthart JF, Dizdaroglu M, Kowalak JA, Crain PF, Borst P. 1993. Beta-d-glucosyl-hydroxymethyluracil: a novel modified base present in the DNA of the parasitic protozoan T. brucei. Cell 75:1129–1136. doi: 10.1016/0092-8674(93)90322-H. [DOI] [PubMed] [Google Scholar]
- 10.Kriaucionis S, Heintz N. 2009. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrangou R, Coûté-Monvoisin AC, Stahl B, Chavichvily I, Damange F, Romero DA, Boyaval P, Fremaux C, Horvath P. 2013. Genomic impact of CRISPR immunization against bacteriophages. Biochem Soc Trans 41:1383–1391. doi: 10.1042/BST20130160. [DOI] [PubMed] [Google Scholar]
- 12.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 14.Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. 1987. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tyson GW, Banfield JF. 2008. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol 10:200–207. doi: 10.1111/j.1462-2920.2007.01444.x. [DOI] [PubMed] [Google Scholar]
- 16.Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 17.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 19.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 20.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. 2011. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA. 2014. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- 24.Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. 2012. The CRISPRs, they are a-changin’: how prokaryotes generate adaptive immunity. Annu Rev Genet 46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 25.Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 26.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrangou R, May AP. 2015. Unraveling the potential of CRISPR-Cas9 for gene therapy. Expert Opin Biol Ther 15:311–314. doi: 10.1517/14712598.2015.994501. [DOI] [PubMed] [Google Scholar]
- 30.Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, Anderson DG. 2014. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dupuis MÈ, Villion M, Magadán AH, Moineau S. 2013. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat Commun 4:2087. doi: 10.1038/ncomms3087. [DOI] [PubMed] [Google Scholar]
- 33.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaung SJ, Esvelt KM, Church GM. 2014. CRISPR/Cas9-mediated phage resistance is not impeded by the DNA modifications of phage T4. PLoS One 9:e98811. doi: 10.1371/journal.pone.0098811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehman IR, Pratt EA. 1960. On the structure of the glucosylated hydroxymethylcytosine nucleotides of coliphages T2, T4, and T6. J Biol Chem 235:3254–3259. [PubMed] [Google Scholar]
- 36.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 37.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. 2012. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat Methods 9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE. 2012. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol 30:1232–1239. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, Kislyuk A, Clark TA, Luong K, Keren-Paz A, Chess A, Kumar V, Chen-Plotkin A, Sondheimer N, Korlach J, Kasarskis A. 2013. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res 23:129–141. doi: 10.1101/gr.136739.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark TA, Spittle KE, Turner SW, Korlach J. 2011. Direct detection and sequencing of damaged DNA bases. Genome Integr 2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doutriaux MP, Wagner R, Radman M. 1986. Mismatch-stimulated killing. Proc Natl Acad Sci U S A 83:2576–2578. doi: 10.1073/pnas.83.8.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deschavanne P, Radman M. 1991. Counterselection of GATC sequences in enterobacteriophages by the components of the methyl-directed mismatch repair system. J Mol Evol 33:125–132. doi: 10.1007/BF02193626. [DOI] [PubMed] [Google Scholar]
- 44.Au KG, Welsh K, Modrich P. 1992. Initiation of methyl-directed mismatch repair. J Biol Chem 267:12142–12148. [PubMed] [Google Scholar]
- 45.Hattman S. 1970. DNA methylation of T-even bacteriophages and of their nonglucosylated mutants: its role in P1-directed restriction. Virology 42:359–367. doi: 10.1016/0042-6822(70)90279-5. [DOI] [PubMed] [Google Scholar]
- 46.Wilson GG, Young KY, Edlin GJ, Konigsberg W. 1979. High-frequency generalised transduction by bacteriophage T4. Nature 280:80–82. doi: 10.1038/280080a0. [DOI] [PubMed] [Google Scholar]
- 47.Young KK, Edlin GJ, Wilson GG. 1982. Genetic analysis of bacteriophage T4 transducing bacteriophages. J Virol 41:345–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young KK, Edlin G. 1983. Physical and genetical analysis of bacteriophage T4 generalized transduction. Mol Gen Genet 192:241–246. doi: 10.1007/BF00327673. [DOI] [PubMed] [Google Scholar]
- 49.Lin H, Black LW. 1998. DNA requirements in vivo for phage T4 packaging. Virology 242:118–127. doi: 10.1006/viro.1997.9019. [DOI] [PubMed] [Google Scholar]
- 50.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A 108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rollins MF, Schuman JT, Paulus K, Bukhari HS, Wiedenheft B. 2015. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res 43:2216–2222. doi: 10.1093/nar/gkv094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benchetrit LC, Bachrach U. 1980. Studies on phage internal proteins. VI. Interaction of bacteriophage T4 internal proteins with T4 DNA in vivo and in vitro. Rev Bras Pesqui Med Biol 13:41–45. [PubMed] [Google Scholar]
- 55.Rifat D, Wright NT, Varney KM, Weber DJ, Black LW. 2008. Restriction endonuclease inhibitor IPI* of bacteriophage T4: a novel structure for a dedicated target. J Mol Biol 375:720–734. doi: 10.1016/j.jmb.2007.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. 2013. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tétart F, Repoila F, Monod C, Krisch HM. 1996. Bacteriophage T4 host range is expanded by duplications of a small domain of the tail fiber adhesin. J Mol Biol 258:726–731. doi: 10.1006/jmbi.1996.0281. [DOI] [PubMed] [Google Scholar]
- 58.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. 2013. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stern A, Mick E, Tirosh I, Sagy O, Sorek R. 2012. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res 22:1985–1994. doi: 10.1101/gr.138297.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. 2011. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 21:1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. 2013. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A 110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karam JD, Drake JW, Kreuzer KN. 1994. Molecular biology of bacteriophage T4. ASM Press, Washington, DC. [Google Scholar]
- 63.Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. 2012. The methylomes of six bacteria. Nucleic Acids Res 40:11450–11462. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Code used to design CRISPR spacers. The program (in R) takes as input a DNA sequence file (fasta) and outputs protospacer targets. Download
(A) Read lengths in the single-molecule sequence data sets. (B) Mapped subread lengths in the single-molecule sequence data sets. Download
Heat map summarizing the effects of local sequences on IPD ratios at C residues. The base preceding the C residue in the sequence is marked 5′-base, and that following the C residue is marked 3′-base. The scale at the bottom summarizes the IPD ratios. Download
Sequence coverage maps for T4 strains comparing T4 (top) to E. coli B834 (bottom). Download
Comparison of our work with that of Yaung et al. (A and B) Results of plaque assays in which the E. coli strains indicated containing previously studied CRISPR spacers (C1 to C3) in the Yaung-Church CRISPR system (in C) were infected with up to 100 PFU of T4(HMC) (panel A) or T4(glc-HMC) (panel B). CRISPR spacer labeling: None, no crRNA or Cas9; non-sp, nonspecific crRNA. E.O.P., efficiency of plating. (C and D) Results of plaque assays in which the E. coli strains indicated containing previously studied CRISPR spacers (C1 to C3) in the Marraffini CRISPR system (in M) were infected with up to 100 PFU of T4(HMC) (panel C) or T4(glc-HMC) (panel D). CRISPR spacer labeling: None, no crRNA or Cas9; non-sp, nonspecific crRNA. (E and F) Plaque assay results of the Church laboratory Cas9 expression vector with the Marraffiini CRISPR array on the T4 CRISPR spacers studied here. The Church Cas9 expression vector and the Marraffini CRISPR array containing the spacers studied in this investigation (spacers 1 to 4) and the previously studied spacers (C1 to C3) were tested for efficacy against T4(HMC) (panel E) and T4(glc-HMC) (panel F). Mean efficiency of transformation was compared to that of a nonspecific control with a t test. **, P < 0.001; ***, P < 0.0001; ns, not significant. Download
Results of plasmid transformation assay comparing the efficacies of all of the CRISPR spacers studied in this investigation against unmodified DNA. Efficiency of transformation was normalized to 1 by the transformation of a plasmid not targeted by the CRISPR system (control). Spacers 1 to 4 are from this study. C1 to C3 are the spacers from Yaung et al. cloned into the CRISPR-Cas9 system developed by the Church lab (in C) or the Marraffini lab (in M). Mean efficiency of transformation was compared to the nonspecific control with a t test. ***, P < 0.0001. E.O.P., efficiency of plating. Download
Mutation of IP1-3 genes encoding the three T4 proteins that are injected along with the phage DNA does not reduce resistance to attack by CRISPR-Cas9. Shown are replicate infections with wild-type T4(glc-HMC) (A) and the triple mutant T4(IP0) (B), which show no differences in infectivity for the strains tested. Plaque assay with approximately 100 PFU of T4(glc-HMC) or T4(IP0) infecting E. coli not expressing CRISPR-Cas9 (None) or E. coli expressing CRISPR-Cas9 with spacers targeting a protospacer in the T4 genome with the maximum number of cytosines in the target sequence and seed sequence (1), the maximum number of cytosines in the target and complementary strands (2), the fewest cytosines in the target strand (3), the fewest cytosines in the target and complementary strands (4), or a nonspecific spacer that does not target the T4 genome (non-sp). The mean efficiency of plating (E.O.P.) for infection of cells with each spacer was compared to that of no-CRISPR-Cas9 control with a t test. No statistically significant differences were found. Download
Phenotype of a revertant of T4(C) named T4(C)R with reduced CRISPR sensitivity. (A) Genetic map of T4(C) and the revertant obtained by Illumina deep sequencing. The bottom black line represents the T4(glc-HMC) genome length, and black arrows indicate genes and the direction of transcription. Variations in the T4(C) genome compared to T4(glc-HMC) are blue for single nucleotide deletions, red for large deletions, and pink for nonsynonymous substitutions; red ×’s represent early stop codons. The top black line represents the T4(C)R genome. Variations in T4(C)R compared to T4(C) are green for nonsynonymous mutations and blue for deletions. The green circle indicates reversion of a stop codon, and the green × indicates a stop codon. (B) Glycosylated hydroxymethylcytosine synthesis pathway in T4(glc-HMC) phage and mutations in T4(C) and T4(C)R. Proteins mutated in T4(C) are shown by red ×’s. T4(C)R acquired a mutation designated by a green × and reverted a previous amber mutation denoted by a green circle. (C) Reduced sensitivity to CRISPR attack in the revertant. The mean efficiency of plating (E.O.P.) for infection of cells with each spacer was compared with that of the no-CRISPR-Cas9 control with a t test. ***, P < 0.0001; ns, not significant. Download
Genotypes of the bacteria and bacteriophages used in this study.
Oligonucleotides used in this study.