

Abstract
In this review the stereochemistry of palladium-catalyzed addition of nucleophiles to alkenes is discussed, and examples of these reactions in organic synthesis are given. Most of the reactions discussed involve oxygen and nitrogen nucleophiles; the Wacker oxidation of ethylene has been reviewed in detail. An anti-hydroxypalladation in the Wacker oxidation has strong support from both experimental and computational studies. From the reviewed material it is clear that anti-addition of oxygen and nitrogen nucleophiles is strongly favored in intermolecular addition to olefin–palladium complexes even if the nucleophile is coordinated to the metal. On the other hand, syn-addition is common in the case of intramolecular oxy- and amidopalladation as a result of the initial coordination of the internal nucleophile to the metal.
Keywords: alkenes, catalysis, nucleophilic addition, palladium, stereochemistry
1. Introduction
Electrophilic addition to olefins is one of the fundamental reactions in organic chemistry. Thus, bromine or hypobromous acid are readily added across an electron-rich olefinic double bond (1) in a process that is initiated by an electrophilic attack to generate the corresponding bromonium ion 2, which is then opened by Br− (in the case of Br2) or H2O (when HOBr is employed) from the opposite side, to afford the anti-addition product (Scheme 1).[1,2] Electrophilic metal cations Mn+, such as Hg2+ and Tl3+, follow a similar pattern in both inter- and intramolecular reactions.[1,3] By contrast, owing to the nature of the electron-rich C=C bonds, nucleophilic additions are rare and typically limited to intramolecular additions of an alkoxide moiety, generated from a suitable polycyclic alkenol by deprotonation with NaH or tBuOK.[4] Transition metals are known to form η2-complexes 3 that can be attacked by nucleophiles to effect a formal nucleophilic addition upon the removal of the metal.[5] However, the transition-metal-promoted process is more complicated, since the nucleophile can, a priori, attack the η2-species either from the opposite side (3) in analogy to bromonium ions 2 and their congeners, or could first coordinate to the metal and then be delivered in a syn-fashion to the original alkene (4).[5]
Scheme 1.
Addition of bromine and transition metals to alkenes.
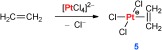
The first η2-olefin metal complex 5 was prepared by Zeise (Scheme 2) via coordination of ethylene (generated by an in situ dehydrogenation of boiling ethanol) to K2PtCl4.[6] However, this complex was long considered as a rarity and it took more than 100 years before the nature of the π bond between an alkene and a metal was understood and its potential had been realized.
Scheme 2.
Preparation of Zeisse’s salt.
The present review is concerned with the stereochemistry of nucleophilic additions to η2-alkene metal complexes of palladium (analogous to 5) and application of these reactions in synthetic organic chemistry. The experimental and theoretical findings, accumulated over the years, are submitted to a thorough mechanistic analysis in light of the most recent results, which are not included in previous reviews. The new results have allowed us to draw conclusions that were not explicitly expressed in previous reviews.[7]
2. Wacker Oxidation
Apart from catalytic hydrogenation, the first large-scale industrial application of palladium was developed by Smidt and his colleagues at the Wacker company in the 1950s.[8] This process, which converts ethylene into acetaldehyde, is known as the Wacker oxidation (Scheme 3). Here, the tetrachloropalladate first generates the corresponding η2-ethylene-Pd complex (in analogy to the formation of Zeise’s salt 5), which then reacts with water to produce acetaldehyde, Pd0, and two equivalents each of hydrogen chloride and chloride ions [Eq. (1)]. In order to make the process catalytic, the resulting Pd0 needs to be reoxidized to its active form, that is, PdII, which is effected by the reaction with CuCl2 (2 equiv), using the two equivalents of Cl− generated in the previous step [Eq. (2)]. The latter reaction produces CuCl, which is then reoxidized to CuCl2 by molecular oxygen, consuming the two equivalents of HCl generated in the first step [Eq. (3)]. The reaction is thus catalytic in both PdII and CuII, with molecular oxygen serving as the terminal, stoichiometric oxidant. Note that direct reoxidation of Pd0 to PdII by molecular oxygen in water under Wacker conditions is too slow and would not prevent aggregation and formation of metallic palladium.
Scheme 3.
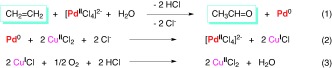
Wacker oxidation.
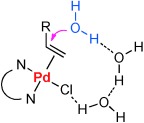
After the original publication on the Wacker process, this reaction became the subject of numerous mechanistic studies.[7a It was proposed that the intermediate η2-complex 7 reacts with water to produce the 2-hydroxyethylpalladium complex 8, which would undergo a β-H elimination to generate vinyl alcohol 10 (via 9 a), tautomerization of which would produce acetaldehyde 11 (Scheme 4). However, deuterium labeling studies clearly demonstrated that the final stages of the cascade do not adopt this route: the η2-complex 9 a, primarily arising from 8 by the expected β-H elimination, does not dissociate to produce 10, but rather undergoes an insertion reaction to generate complex 12 (via rotamer 9 b), which now uses the O=H (rather than C=H) for the final β-H elimination (12 a or 12 b) to produce acetaldehyde (11).[9] An alternative pathway, where a lone-pair on oxygen ejects Pd0 as a leaving group (12 b), was also considered,[10] which was later supported by theoretical calculations.[11]
Scheme 4.
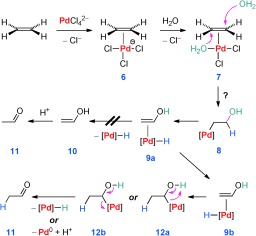
Wacker oxidation.
The latter mechanism has been inferred from isotopic labeling as follows (Scheme 5): If the reaction proceeded through enol 10, the molecule would abstract a proton from the environment [note that there is free HCl available, according to Equation (1). However, the experiment with perdeuteriated ethylene [Eq. (4)] showed that all the label was retained, which is consistent with the mechanism depicted in Scheme 4, namely with the isomerization 9 a → 9 b → 12. The isotope effect [compare Eqs. (1) and (4) in Scheme 5] was found to be marginal.[9]
Scheme 5.
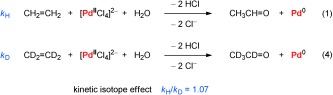
Deuterium labeling in Wacker oxidation.
In a complementary experiment, the specifically dideuteriated ethylenes 13 and 14 (Scheme 6) were found to exhibit an internal competitive isotope effect (H vs D shift), demonstrating that the rate-limiting step must occur before the β-H elimination and formation of acetaldehyde.[12,13] It was further argued that the rate-limiting step is the hydroxypalladation, and the results are better explained by a syn-migration since an anti-hydroxypalladation would seem to require a rate constant larger than diffusion control.[9,12]
Scheme 6.
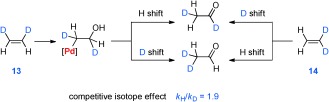
Isotope effect in Wacker oxidation.
The proposed syn-migration of an oxygen nucleophile has been experimentally observed in a stoichiometric experiment with a Pt–olefin complex (Scheme 7). In this experiment the extremely electron-deficient tetrafluoroethylene (15) was treated with the Pt–OMe complex 16. The formation of intermediate 17 and product 18, as well as the kinetics, were monitored by NMR spectroscopy in [D8]THF with added CD3OD.[14] Here, an external attack by CD3OD (which should proceed with anti stereochemistry due to the steric hindrance from the Pt side) has not been observed, nor was the exchange of OCH3 with OCD3, so that the product 18 can only arise by the syn-migration of OCH3 from Pt. This may seem to support the syn-migration pathway in the Wacker process. However, it can be argued that the starting olefin 15 in this experiment is rather special, so that generalization of these findings, in particular to the Wacker oxidation of the electron-rich ethylene, cannot be made directly.
Scheme 7.
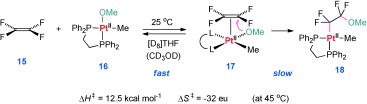
Experimentally observed syn-migration of MeO from platinum.
In order to elucidate the stereochemistry of hydroxypalladation, the trans-dideuteriated ethylene 19 was employed as a model compound (Scheme 8).[10,15] Under catalytic hydroxypalladation conditions, using a mixture of PdCl2, LiCl, and CuCl2, the latter alkene was expected to generate the η2-complex 20, where the water molecule would have to be cis-coordinated toward the ethylene ligand (otherwise the syn-transfer would not be possible at all). The anti-attack by an external water molecule on 20 should generate complex 21, which in the presence of chloride ions should produce chlorohydrin 22 as a result of the SN2 displacement of palladium. Treatment of 22 with a base would then produce epoxide 23. The latter epoxide was found to be cis-configured (as shown), which is consistent with the pathway involving three inversions of configuration, that is, in each step starting with complex 20. Since the replacement of palladium by Cl− was known from the previous work[16] to proceed with inversion of configuration at carbon, and the stereochemistry of the chlorohydrin transformation into the corresponding epoxide is a textbook example of inversion, the key hydration 20 → 21 must also occur with inversion, that is, via an external attack as shown. This study thus provides evidence for the anti-addition mechanism for the key hydroxypalladation step.
Scheme 8.
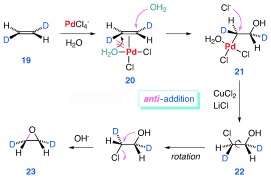
Stereochemistry of hydroxypalladation in the presence of chloride ions.
An extended theoretical study[17] then reconciled the fact that depending on the nature of the nucleophile the nucleophilic attack may occur either in a syn or anti fashion (Scheme 9). Essentially, two types of nucleophiles can be discerned: NuA, which can, a priori, coordinate the metal but prefer the external anti-addition to the olefinic ligand (pathway a), and NuB, which prefer an intramolecular, that is, syn-transfer (pathway b).
Scheme 9.
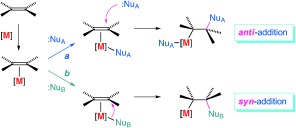
Stereochemistry of the reactions of metal complexes with nucleophiles.
Analysis of the orbital interactions in four model systems, namely with OH−, F−, H−, and CH3− as representative nucleophiles coordinated to Pd, revealed the following (Figure 1):[17b] the HOMO orbitals of the first two complexes lie rather low, so that their interaction with the LUMO of the (coordinated) olefin will be weak and result in little stabilization. By contrast, the two latter complexes have high energy HOMO orbitals for the Pd=Nu bond (Nu = H− and CH3−), so that the interaction between the HOMO orbital of these complexes and the LUMO of the alkene should be stronger and lead to a considerable lowering of the energy of the system. Hence, it can be anticipated that the reactivity of H− and CH3− will be controlled mainly by orbital interactions, so that the transfer from the metal should be preferred. On the other hand, the orbital effects in the case of OH− and F− are likely to be weak, so that the reaction should be dominated by ionic effects and thus proceed via anti-addition.[17b]1
Figure 1.
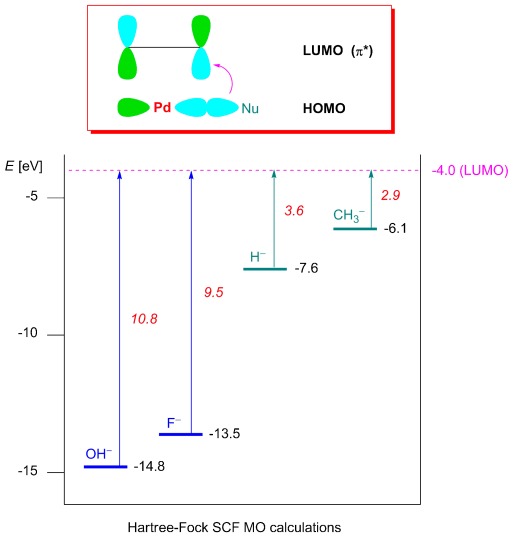
Orbital interactions in the palladium η2-complexes.
Furthermore, the reactions controlled by ionic interaction (favoring the anti-pathway) can be expected to obey the Markovnikov rule like any other electrophilic addition and afford products, where the incoming nucleophile is planted on the carbon that can better stabilize a partial positive charge (24 in Scheme 10). On the other hand, in the orbital-controlled reactions (favoring the syn-pathway) the nucleophile should add to the less-substituted carbon (25), as the LUMO orbital at that carbon should be larger.[17]
Scheme 10.
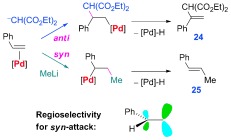
Regioselectivity as a result of the reaction stereochemistry.
A possible supramolecular interaction, involving several molecules of water and hydrogen bonding to the chloride bound to Pd, has also been considered for the Wacker oxidation (Figure 2).[18]
Figure 2.
Calculated supramolecular interaction of water with the η2-complex.
It has been argued that the chloride concentration in the experiments shown in Scheme 8 is higher (3 m) than in the Wacker process and in the kinetic experiments[9] (≤1 m) and that the stereochemistry may not be the same at the different concentrations of Cl−.[7,19,20] However, the chemistry of 1,4-difunctionalization of dienes, catalyzed by Pd(OAc)2, shows that while under chloride-free conditions, the acetate group is delivered preferentially to the intermediate allyl group from Pd (i.e., in the syn-fashion), addition of only catalytic amounts of LiCl (4 equiv per Pd) shuts down this reaction pathway. The chloride is replacing coordinated acetate on Pd, and in this way acetate can only be delivered in an anti-fashion by an intermolecular reaction.[21] Further increase of the chloride concentration does not alter the latter stereochemistry and only increases the proportion of the product resulting from the Cl− attack on the η3-complex (rather than AcO− attack). This is in direct analogy to the formation of chlorohydrin 22 in the Wacker process, lending additional support for the argument that the stereochemistry should be the same at high and moderate concentrations of chloride ions.
Kinetic studies have demonstrated that the Wacker oxidation is actually inhibited by an increasing concentration of chloride ions [Eqs. (5)–(7) in Scheme 11].[7, 17, 19] In fact, at ≤1 m concentration of Cl− and CuCl2 (industrial conditions for Wacker oxidation), acetaldehyde is the predominantly formed product (with small amounts of the corresponding chlorohydrin). By contrast, at 3 m concentration of Cl− (conditions used in the mechanistic study shown in Scheme 8[15]), chlorohydrin becomes the predominant product (Scheme 12). It is highly unlikely that a change of the chloride ion concentration from 1 m to 3 m would change the stereochemistry of the hydroxypalladation of ethylene.
Scheme 11.
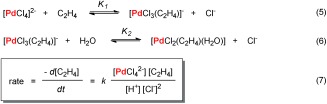
Chloride effect: rate inhibition.
Scheme 12.
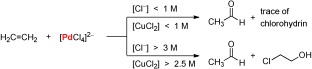
Chloride effect: change of mechanism and product distribution.
Finally, strong support for the anti-addition of AcO− and PdII under the chloride-free conditions has been obtained from the acetoxypalladation of deuteriated 3,3-dimethyl-1-butene 26 (Scheme 13).[22] Here, the isomeric products 30 and 31 were shown to arise from predominant anti-attack of AcO− on the η2-Pd complex 27 (via the Markovnikov pathway a and anti-Markovnikov route b), followed by the stereospecific β-hydrogen elimination from the respective intermediates 28 and 29 that occurs solely in a syn-fashion. Some loss of the stereochemical integrity observed for this process was attributed to a partial isomerization of the Markovnikov product (confirmed by a control experiment), so that the (E)- and (Z)-vinyl acetates 30 a and 30 b were obtained as a 3.5:1 mixture. The anti-Markovnikov product was obtained as a 9:1 mixture of (E)-isomers 31 a and 31 b. Here, the control experiment with non-deuteriated 3,3-dimethyl-1-butene showed that the Markovnikov product was a pure (E)-isomer, apparently arising from a conformation of the non-deuteriated congener of 29, where the bulky tBu and AcO groups avoid the gauche interaction. The 9:1 ratio of 31 a and 31 b thus suggests that the initial acetoxypalladation proceeds mainly (but not solely) via the anti-mechanism. Complementary results were obtained with the (E)-isomer of 26.[22]
Scheme 13.
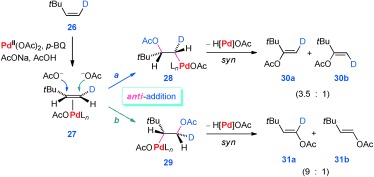
Stereochemistry of acetoxypalladation under chloride-free conditions.
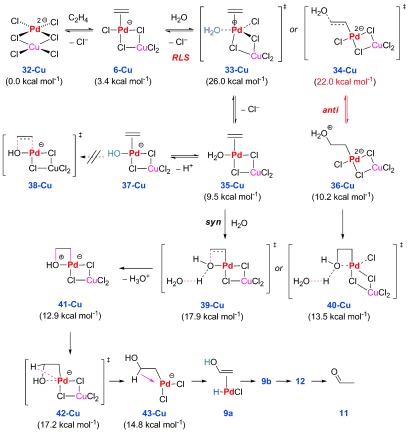
In a computational study,[11] the energy of various intermediates and transition states that can be considered for the hydration pathway was assessed (Scheme 14). For the scenario with low concentration of Cl−, the starting tetrachloropalladate 32 is first converted into the η2-complex 6, which can be attacked by water either at Pd or at the alkene ligand. The energy of the corresponding transition states 33 and 34 has been calculated to be almost identical but the Pd-hydrated product 35, arising from the former TS≠, is by 10.7 kcal mol−1 lower in energy than 36, arising from the latter TS≠. Deprotonation of 35 to generate 37, which in principle could transfer the hydroxy group to the alkene ligand, would proceed through the transition state 38 but this species is rather high in energy (33.4 kcal mol−1), well above the experimentally overall observed value (22.4 kcal mol−1).
Scheme 14.
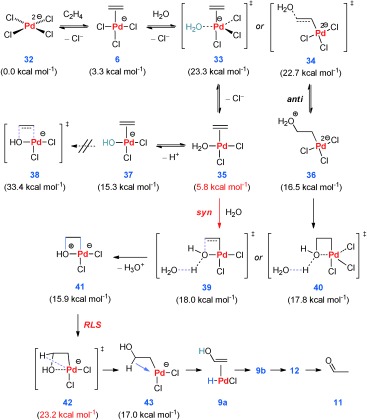
Energies of various intermediates in the proposed syn-mechanism of the Wacker oxidation in the absence of CuCl2.
However, an alternative pathway, namely the water-assisted deprotonation of 35 with concomitant syn-migration, eventually leading to 41, would proceed through the transition state 39 that is half-way down on the energy scale compared to 38, indicating that this pathway would be favored if the syn addition mechanism operated. Conversely, the anti-delivery of water (34) generates intermediate 36. Either of the transition states (39 or 40) would give rise to 41 by water-assisted deprotonation. Dissociation of the Pd=O bond in 41 would generate complex 43 ready for the expected β-H elimination. The calculations further found the energy of the corresponding transition state 42 (including the agostic Pd-H interaction) to be 23.2 kcal mol−1, which makes this species highest in energy in the whole cascade (via 39/40),[11] so that this step can be regarded as rate-limiting.
However, there is a problem with this calculation since the transition state for the anti-attack is calculated on the negatively charged species 6. The kinetics of the Wacker process shows that two chlorides are displaced, so an anti-attack would occur on the neutral species PdCl2(OH2)(CH2=CH2). It can be expected that the latter neutral species reacts much faster than the negatively charged species 6 in external anti-attack by water. This has also been confirmed later in other calculations (vide infra).
Subsequent calculations[23] suggested that the anti-addition mechanism is strongly favored over the syn pathway; this was disputed in a subsequent article[24] and the quality of the calculation was questioned.
Calculations including CuCl2 gave a slightly different picture,[11] as shown in Scheme 15 (the structures here bear the same numbers as in Scheme 14 but with added “Cu” so that a direct comparison can be made). Here, the initial PdCl2/CuCl2 complex 32-Cu first coordinates ethylene to produce 6-Cu (upon a loss of Cl−), which then can be attacked by water either at Pd or at the carbon via transition states 33-Cu and 34-Cu, respectively, of which the latter is lower in energy by 4.0 kcal mol−1, indicating that the anti-pathway is preferred. Furthermore, the energy of the TS≠ 33-Cu is 26.0 kcal mol−1, which is slightly higher than the experimentally observed value (22.4 kcal mol−1). The two transition states would generate the hydrated species 35-Cu and 36-Cu that are almost equal in energy but the subsequent gradual conversion into 41-Cu should preferably proceed through TS≠ 40-Cu that is by 4.4 kcal mol−1 lower in energy than its congener 39-Cu. The end-game from 41-Cu via 42-Cu and 43-Cu has been calculated to require lower energy in each step than the experimental value. Hence, the highest energy in this scenario (not exceeding the experimental value) is that of the transition state 34-Cu, so that the conversion of 6-Cu into 36-Cu can be regarded as the rate-limiting step (RLS) and the reaction should proceed predominantly via an anti-addition.[11] Again, one can argue that external attack on an (ethylen)palladium complex, where water has coordinated to Pd to remove negative charge (vide supra), should react faster than the negatively charged complex 6-Cu (34-Cu).
Scheme 15.
Energies of various intermediates in the proposed syn-mechanism of the Wacker oxidation in the presence of CuCl2.
Recent computational studies, which included aqueous medium, namely a cubic box containing up to 26 molecules of water as a model that is more closely related to the “real” experimental conditions, arrived at the conclusion that anti-hydroxypalladation is the favored stereochemical pathway (Scheme 16):[25] First, the initial coordination of tetrachloropalladate to ethylene (44) is known to produce the η2-complex 6, which was taken as the starting point. The latter complex should then undergo a ligand exchange if syn-migration of the nucleophile from Pd is to be allowed. In view of the geometrical restriction, previous study[11] only considered the cis-complex 35. However, as the new study shows, the calculated activation barrier for its formation from 6 is 35 kcal mol−1, which is more than twice as high as that for the pathway leading to its trans-isomer 45 (14 kcal mol−1). Since the experimental value for the whole Wacker process has been found to be only 22.4 kcal mol−1, the formation of 35 appears unlikely. Note that the conversion of 6 into 45 will be assisted by the trans-effect (absent in the formation of 35), which should considerably lower the activation energy, consistent with the calculations. Furthermore, the activation energy required for the syn-migration to generate complex 47 from 35 has been calculated to be 60 kcal mol−1, far above the experimental value. By contrast, the activation barrier for the anti-attack by water on the trans-complex 45 to produce 46 was calculated to be only 19 kcal mol−1. Hence, it can be clearly seen that none of the activation barriers in the anti-pathway (6 → 45 → 46) exceeds the experimentally established value[11] for the whole process (22.4 kcal mol−1), also indicating that the rate-limiting step should occur after those initial steps, which is consistent with the previous findings.[11] The mechanism in Scheme 16 is also consistent with the rate expression of the Wacker reaction [Eq. (7)], where the rate is inversely dependent on [H+] and the square of [Cl−]. These advanced calculations thus allow to conclude that the anti-hydroxypalladation should be the preferred pathway.[25]
Scheme 16.
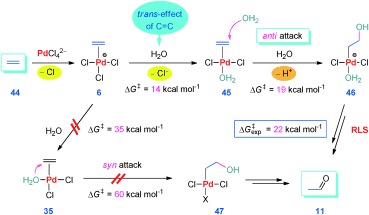
Molecular dynamics calculations including a cubic box of 26 molecules of H2O, which simulates the “real” reaction medium and low Cl− concentration.
Another theoretical study, in combination with experimental results (Scheme 17), provides additional support for the anti-hydroxypalladation pathway.[26] In the latter study, the equilibria in the system of ethylene, tetracholoropalladate, and water were investigated. Starting with the tetrachloropalladate (32), there are two initial reactions to be considered: a ligand exchange with ethylene, generating η2-complex 6, and partial hydrolysis, producing complex 48. Calculations suggest that the equilibrium should favor 48, as the energy of its formation is lower than that of 6 (by 5.1 kcal mol−1). The experimentally established difference was found to be smaller (0.5 kcal mol−1), indicating that both species should be considered for further reactions. Ligand exchange with water in the case of 6 would produce either the trans-isomer 45 or its cis-counterpart 35. As discussed in the previous paragraph,[25] conversion of 6 into 45 should be associated with a barrier of 14.4 kcal mol−1; the reversed process would require 17.0 kcal mol−1, so that this equilibrium should be shifted toward 45. Formation of the cis-complex 35 (from 6) would require 22.6 kcal mol−1, according to these calculations, whereas the reversed reaction can proceed with a barrier of only 14.4 kcal mol−1, so that the equilibrium should favor the starting complex 6. In other words, the displacement of Cl− in 6 by water should preferentially produce the trans-complex 45. The other possible mechanism would be the ligand exchange starting with the aqua-complex 48. Its reaction with ethylene, generating the trans-complex 45 was found to be associated with the activation energy of 23.8 and 24.5 kcal mol−1 for the forward and reversed process, respectively. The latter difference is sufficiently small to allow the existence of both species in the equilibrium mixture. A completely different scenario was found for the conversion of 48 into the cis-complex 35. Here, the forward process would require 19.4 kcal mol−1, whereas the reversed reaction would be associated with merely a 6.1 kcal mol−1 barrier, showing that the equilibrium should be heavily shifted toward 48. These finding are thus consistent with the previously suggested dominance of the trans-complex 45 over its cis-isomer 35. Since the barrier for the anti-attack on 45 was previously calculated[25a] to be 19 kcal mol−1, whereas the syn-migration from 35 would require[25a] 60 kcal mol−1, the syn-pathway can be ruled out.[26]
Scheme 17.
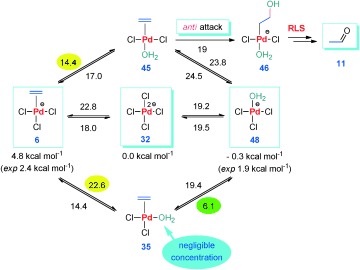
Equilibrium study of ethylene, tetrachloropalladate, and water (ΔG≠ in kcal mol−1).
When extended beyond the original conversion of ethylene to acetaldehyde, the Wacker oxidation has served, with various modifications, as a standard method for oxidation of terminal olefins to produce methyl ketones[27,28] (e.g., 49 → 50 in Scheme 18);[29] this process is referred to as the Wacker–Tsuji oxidation.[27] However, alteration of the regioselectivity in favor of the corresponding aldehydes 52 has recently been achieved simply by using tBuOH rather than water. The reaction presumably proceeds via the vinyl ether 51, resulting from the anti-Markovnikov attack of the bulky nucleophile at the sterically less hindered terminal carbon.[30]
Scheme 18.
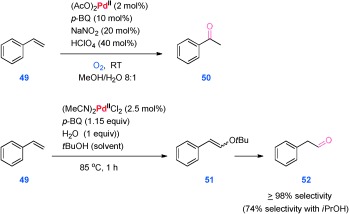
Recent modifications of the Wacker oxidation of terminal olefins and alteration of its regioselectivity.
3. Amination of Olefins
Related to Wacker oxidation, where the stereochemistry of the initial hydroxypalladation was discussed in detail in the previous chapter, is the amination of olefins. However, in contrast to water and alcohols, amines are not normally considered for analogous Pd-catalyzed reaction, as they are easily oxidized. Nevertheless, a stoichiometric Pd-mediated amination of (E)-2-butene (53) with dimethyl amine has been successfully investigated at low temperature (Scheme 19).[31] The initially formed aminopalladation product 54 was reduced in situ with LiAlD4 (with retention of configuration) to afford the deuteriated amine 55.[31b] The relative configuration of the latter derivative was established in two steps, involving N-oxidation (55 → 56) followed by Cope elimination, which is known to proceed with a syn-elimination mechanism. The product analysis (57–59) was consistent with an initial anti-addition of PdII and Me2NH across the C=C bond,[31b] in analogy to the mechanism discussed for the Wacker oxidation. Complementary experiments with (Z)-2-butene led to the same conclusions.[31,32] Note that before the amine attacks the coordinated olefin, two amine molecules coordinate to palladium. A syn-migration of the amine from Pd to the coordinated olefin should be precluded, as this process apparently would be much higher in energy (Figure 1) than the intermolecular anti-attack by Me2NH, generating 54. This is in contrast to the Pd-OAc η3-species, where the syn-delivery via a cyclic transition state is allowed.[33] However, analogous syn delivery in the case of the corresponding η2-complexes is disfavored (Scheme 13).[22]
Scheme 19.
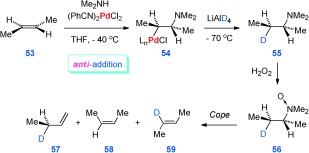
Stereochemistry of the PdII-catalyzed amination of 2-butene.
4. Intramolecular Oxypalladation
The stereocontrolled intramolecular nucleophilic attack on a metal–olefin complex (as in 60), is an important reaction in synthetic organic chemistry,[34] and is analogous to halolactonization, haloetherification, and related reactions employing S, Se, Hg, Tl, Au, and other electrophiles [Scheme 20, Eq. (8)].[1,35] When metals, such as Pd[1,3,5] or Hg,[1,3] are employed as the electrophilic triggers of the reaction, the initially generated organometallic product 61 can be utilized in a subsequent reaction that would allow the construction of a new C=C bond from the C=M bond. The overall result would then be the formation of a C-X and C=C bond, where X is introduced as a nucleophile, and the new C-substituent formally as an electrophile (62).[1] Aside from this anti-mechanism [Eq. (8)], the syn-addition 63 → 64 [Eq. (9)] may also operate (see also Scheme 1), due to the initial coordination (60 → 63), known for instance from the vanadium-catalyzed epoxidation.[1c] Examples of both mechanisms and the effects favoring one or the other will be discussed and analyzed in this and subsequent sections.
Scheme 20.
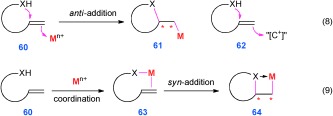
Neighboring group effects in the metal-catalyzed functionalization of olefins.
4.1. Intramolecular Oxypalladation Followed by Carbonylation
The first intramolecular oxypalladation was carried out in conjunction with carbonylation (Scheme 21).[36] Here, the reaction of alkenol 65 with CO and MeOH, catalyzed by PdII, commenced with the η2-coordination, followed by an anti-attack by the neighboring hydroxyl group. Of the two facial stereoisomers, one is consumed faster (66) and generates the tetrahydropyran derivative 67 with high stereocontrol exercised by the original chiral center (note the “equatorial” methyl in 66). Coordination of carbon monoxide (68), followed by migratory insertion then generated complex 69, which on reaction with methanol produced the desired methyl ester 70 and Pd0, whose reoxidation with CuII completed the catalytic cycle.
Scheme 21.
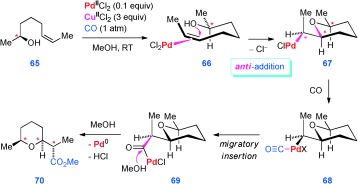
Intramolecular hydroxypalladation controlled by a residing chiral center followed by carbonylation.
Several other examples followed shortly, all featuring the initial anti-addition of the electrophilic PdII and the neighboring OH group across the C=C bond,[37–39] such as the cyclization of 71 and 73 (Scheme 22),[39] and 76 (Scheme 23), followed by carbonylation, as detailed in the previous paragraph.[3a]
Scheme 22.
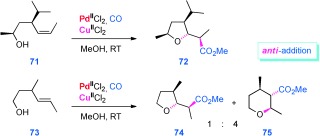
Carbonylative cyclization: the effect of alkene geometry on the regioselectivity.
Scheme 23.
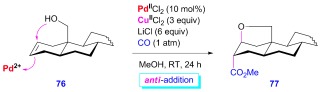
Carbonylative cyclization occuring with pure anti-sterechemistry.
All those reactions (Schemes 21—23) occurred with alkenols, whose structures allow 5-exo- or 6-exo-cyclizations. If the only option is a 4-exo process, as in the case of the homoallylic alcohol 78, the reaction has been found to take a different course (Scheme 24).[40] Here, the C=C bond coordination to PdII (as described in Scheme 21) becomes unproductive, as cyclization to produce the corresponding oxetane 79 would be too high in energy. Instead, the reaction proceeds through a different channel, namely that involving the initial coordination of CO to Pd, followed by a reaction with the OH group and additional coordination to the C=C bond (80). These events are followed by reductive elimination to generate lactone 81 with Pd chelated to the exocyclic carbon in an η1-fashion (which formally corresponds to a syn-addition across the C=C bond). The cascade is then completed by the second carbonylation to produce 82 (Scheme 24). The trans-configured alcohol 83 reacted in the same way to produce the diastereoisomeric lactone 84. The reaction conditions for these transformations are noteworthy: aside from the standard reoxidation of the resulting Pd0 by CuII, propene epoxide was employed to consume the Brønsted acid generated by the reaction, whereas trimethyl orthoacetate was added to secure anhydrous conditions.[40]
Scheme 24.
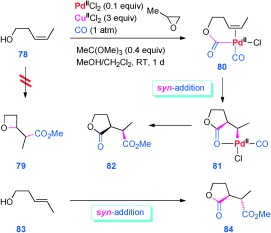
Carbonylative cyclization occuring with pure syn-sterechemistry.
Other examples of this reaction course, where the cyclization involving intramolecular oxypalladation is precluded by structural restrictions in the homoallylic arrangement, have been reported but without addressing the stereochemistry issues.[41]
4.2. Intramolecular Oxypalladation Followed by β-Hydride Elimination
The stereochemistry of the PdII-catalyzed ring closure of alkenols has been investigated with the aid of the stereospecifically deuteriated substrate 85, using p-benzoquinone (p-BQ) as the terminal oxidant (Scheme 25).[42] Since intermediates in a catalytic reaction are difficult to isolate, the steric course was inferred from the products of the subsequent β-H elimination, which is known to occur as a syn-process. With the weakly coordinating BF4− anion at PdII, the free phenolic hydroxyl group tends to coordinate to the metal [as in Eq. (9), Scheme 20], which leads to a π-olefin complex where Pd is bound to the top face of cyclohexene. A syn-oxypalladation then produces 86, which on subsequent β-hydride elimination removed the cis-positioned deuterium. By contrast, coordination to the neighboring hydroxyl is precluded in the case the strongly coordinating Cl− at PdII (especially when additional LiCl is present) and the reaction thus proceeded with anti-stereochemistry to give 87, as revealed by the β-H elimination of the cis-disposed proton.[33,42,43]
Scheme 25.
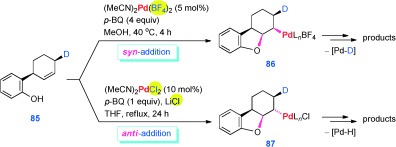
Switching between syn- and anti-mechanism in the intramolecular hydroxypalladation as a function of the anion coordinated to Pd.
The latter study used phenolic hydroxyl as a neighboring group, whose coordination capability can differ from that of an ordinary alcohol due to the difference in the pKa. Therefore, two diastereoisomerically deuteriated substrates 88 and 89 were investigated by Stoltz and co-workers (Scheme 26).[44] The catalyst employed possessed the weakly coordinating trifluoroacetate anions and the reaction was found, in both cases, to follow the syn-pathway, as revealed by the β-hydride elimination of 90 and 91 to give 92 and 93, respectively. These results are in full agreement with the first reaction shown in Scheme 25. Notably, the catalyst in Scheme 26 allowed the use of molecular oxygen as the stoichiometric oxidant, avoiding the requirement to employ a mediator, such as CuII or quinone/metal macrocycle.[44] The use of the bipyridine ligand presumably keeps the reduced palladium in solution and gives it time to be reoxidized. This study clearly demonstrates that for intramolecular alkoxypalladation, coordination of the neighboring hydroxy group is a powerful process that favors syn-addition. When the alkoxy group is first coordinated to palladium in the π-olefin complex of 88 and 89, there will be no competing pathway via anti-attack.
Scheme 26.
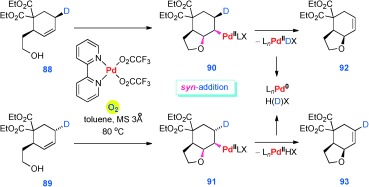
syn-Mechanism in the intramolecular hydroxypalladation.
Solvents (e.g., MeOH vs MeCN) have been found to have an effect on the regiochemistry of the β-hydride elimination but that study did not address the stereochemical issues.[45]
4.3. Intramolecular Oxypalladation Followed by Heck Addition
Phenolic olefin 94 has been reported to undergo a Pd-catalyzed cyclization (with p-BQ as the terminal oxidant) in the presence of Michael acceptors, such as methyl vinyl ketone, to produce 96 with high enantioselectivity, owing to the chiral ligand 97 (Scheme 27). Although the steric course of the initial step (94 → 95) has not been investigated, it can be assumed to proceed via a syn-mechanism, owing to the weakly coordinating anion (CF3CO2−) and the presence of 97, whose ligating properties are likely to mirror those of the bipyridine, featured in Scheme 26.[46]
Scheme 27.
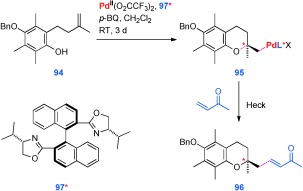
PdII-Catalyzed ring closure followed by Heck addition.
4.4. Intramolecular Oxypalladation Followed by Arylation
The PdII species 100, generated in the catalytic cycle from the Pd0 complex 98 on an oxidative addition to aryl halide 99, has been shown to react with the alkoxide generated from the bishomoallylic alcohol 101, giving rise to the cyclization product 104 (Scheme 28).[47] The corresponding alkenyl aryl ether that would be normally expected for these conditions (typical for the Hartwig–Buchwald coupling) has not been observed. The reaction has been suggested to proceed via the η2-complex 102 (with pentacoordinated Pd), predisposed to the syn-cyclopalladation, generating the tetrahydrofuran intermediate 103. The latter species then undergoes reductive elimination to afford 104 as the final arylated product, thereby regenerating Pd0 98 for the next catalytic cycle. Aldehyde 106 has been isolated as a byproduct,[47] apparently arising from 105 via a β-hydride elimination.
Scheme 28.
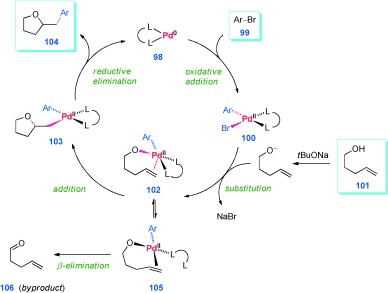
Pd0-Catalyzed ring closure followed by arylation.
The stereochemistry of the latter reaction was investigated with the aid of the cis-alkenol 107 (Scheme 29).[48] As expected (based on the discussion in the previous paragraph), the reaction proceeded as a syn-addition to produce 108. However, by using DPPE as a strongly chelating ligand, which apparently prevents the alkoxide coordination to palladium, the mechanism was pushed toward anti-addition, giving rise to the diastereoisomeric product 109. Amine 110 reacted in the same way.[48]
Scheme 29.
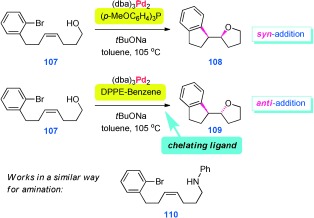
Stereochemistry variation in the Pd0-catalyzed ring closure followed by arylation.
The syn-addition mechanism has been masterly utilized in the cyclization, where the stereochemistry was controlled by the residing chiral center in the secondary alcohol 111 (Scheme 30).[48] Here, the initial oxidative addition to a Pd0 catalyst produces 112, where the C=C bond is coordinated to Pd. Two diastereofacial ways of this coordination can be considered, so that a mixture might be expected. However, the subsequent replacement of bromide in the Pd coordination sphere with the neighboring alkoxy group (to generate 113) is apparently faster for one stereoisomer (as, e.g., in Scheme 21), which in view of the reversibility of the η2-coordination results in dynamic stereodifferentiation.[49] Hence, isomer 114 is thus formed preferentially and the subsequent reductive elimination affords the cyclization product 115 of high diastereopurity.[48]
Scheme 30.
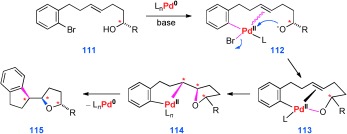
Stereocontrol of the cyclization by a residing chiral center.
5. Intramolecular Amidopalladation
The first amidopalladation, analogous to alkoxypalladation, was explored in a stoichiometric experiment with enamide 116 and found to produce the azepine derivative 117 with the new C=N and C=Pd bonds related in a cis-fashion, which correspond to syn-addition (Scheme 31).[50]
Scheme 31.
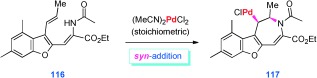
Ring-closing amidopalladation occuring with syn-stereochemistry.
5.1. Intramolecular Amidopalladation Followed by β-Hydride Elimination
Assuming a similar mechanism for the PdII-catalyzed cyclization of the alkenyl sulfonamide 118 (Scheme 32), two transition states 120 and 121, differing in the mode of Pd coordination can be proposed. The former species (with Pd coordinated to the nitrogen) was found to be much lower in energy than the latter by quantum chemistry calculations.[51,52]
Scheme 32.
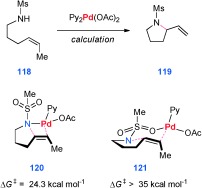
Computational analysis of ring-closing amidopalladation proceeding with syn-sterechemistry.
Further investigation revealed that the stereochemistry of amidopalladation is subjected to delicate effects of the anion in the original PdX2 catalyst and ligand (Scheme 33).[52,53] With the monodeuteriated substrate 122, the cyclization was catalyzed by two PdII salts in the presence or absence of ligand 125 and the products were analyzed for the content of deuterium in the resulting cyclic olefins 123 and 124. The latter analysis demonstrated that the chelation of Pd (and thus the syn-pathway) is suppressed when the combination of (CF3CO2)2Pd and ligand 125 is used, whereas the remaining reactions were dominated by the syn-pathway.[53b]
Scheme 33.
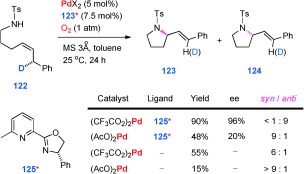
Anion effect on the stereochemistry of the sulfonamidopalladation.
To add to the complexity of the mosaic, the cyclic, stereospecifically deuteriated sulfonamide 126 was found to be cyclized via the anti-pathway (with 127 and 128 as intermediates), giving rise to 129, which lacks the label (Scheme 34).[54] In this instance the reaction was catalyzed by Pd(OAc)2 and would be expected to occur via the syn-addition pathway according to Scheme 33. However, in the previous study the final oxidant was molecular oxygen, whereas in Scheme 34 it was p-BQ, which is known to coordinate to Pd in an η2-intermediates[55] and this should be regarded as a contributing factor. Also, the fact that the reaction was run under slightly acidic conditions may inhibit coordination of the tosylamide. Note that a stoichiometric experiment with the cyclopentene analogue of 126, N-coordinated to (tBu2bipy)PdCl, has been found to cyclize via the syn-pathway in DMSO in the presence of molecular oxygen (to simulate the catalytic conditions).[53a] Clearly, more studies are required to define the relationship between the reaction conditions and the mechanism.
Scheme 34.
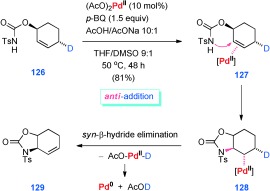
Stereochemistry of sulfonamidopalladation with cyclic substrates.
The syn-mechanism has been assumed (but not proven) for the cyclization of 130[56] and 131[57] (Scheme 35), where the chiral control is exercised by a chiral sulfinimide group and chiral ligand 132, respectively.
Scheme 35.
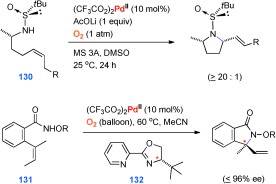
Asymmetric amidopalladation.
Two other amidation reactions have been reported recently (Scheme 36) but their stereochemistry has not yet been elucidated.[58] In accord with the previous observations (vide supra), the hydroxylamine derivative 133 afforded the cis-configured isoxazolidine 134 (≥30:1 dr), whereas its hydrazine analogue 135 produced the trans-configured pyrrazolidine derivative 136 (≥30:1 dr).[58a] The latter products can be regarded as surrogates of 1,3-amino alcohols and diamines, respectively.
Scheme 36.
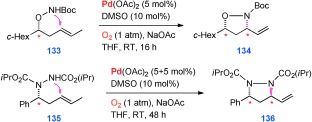
Diastereoselective 1,3-amidation.
Finally, the O-allyl hemiaminal 137, prepared from the corresponding allylic alcohol and AcOCH2NHCbz, has been shown to undergo an analogous cyclization, giving rise to the trans-configured oxazolidine 138 (Scheme 37) but again without specification as to the actual stereochemistry of the cyclopalladation step.[59] This strategy has been developed as an approach to 1,2-syn-amino alcohols and employed in the synthesis of acosamine.[59]
Scheme 37.

Diastereoselective 1,2-amidation.
5.2. Intramolecular Amidopalladation Followed by Arylation
The cascade of catalytic amidopalladation of olefins and arylation has been developed in parallel with oxypalladation (cf. Section 4.4). Thus, the Pd0 species, generated from Pd(OAc)2 and the diphosphine ligand 139 (Scheme 38),[60] has been shown to catalyze the cyclization of the Boc-functionalized aminoalkene 142 to produce the piperidine derivative 145 with high diastereoselectvity. The latter outcome has been rationalized in a similar way as in the case of oxypalladation, namely by the initial oxidative addition to generate the PdII complex 141, whose reaction with 142 (upon deprotonation with Cs2CO3) generates the Pd=N complex 143, where Pd is also coordinated to the C=C bond. The key addition across the C=C bond thus occurs with a syn-mechanism to generate the η1-complex 144, which affords the final product 145 by reductive elimination and Pd0 that enters the next catalytic cycle.[60,61]
Scheme 38.
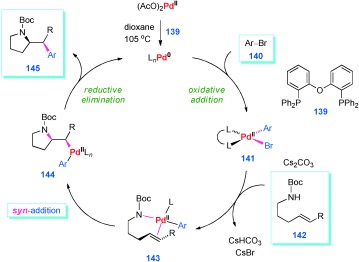
Intramolecular amidopalladation combined with arylation.
The cyclization of the isomeric derivatives of hydroxylamine 146 and 148 (Scheme 39) was expected to proceed via the syn-mechanism[62] in analogy to the related cyclizations highlighted in Scheme 38 (see also the evidence presented in Scheme 40). The syn-mechanism was than proved by isotopic labeling at the terminus of the double bond. Note, however, that while 146 gave the cis-disubstituted isoxazolidine 147 (typically with ≥20:1 dr), its positional isomer 148 that reacts by forming the C=O (rather than C=N) bond, furnished mainly the trans-product 149 (7:1 dr). This change of stereochemistry was attributed to the steric interference by the Boc group 1,2-related to the phenyl in 148; a transition state leading to 149 is believed to be lower in energy than that producing the cis-diastereoisomer (both using the syn-addition mechanism).[62]
Scheme 39.
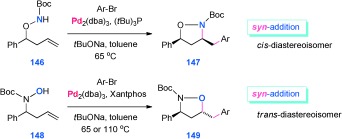
Intramolecular amidopalladation/arylation of hydroxylamine derivatives.
Scheme 40.
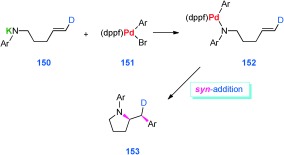
Mechanism of the stoichiometric aminopalladatio/arylation elucidated by isotopic labeling (Ar=p-CF3C6H4).
Stoichiometric experiments (Scheme 40),[63] employing the potassium salt 150, stereospecifically deuteriated at the terminus of the double bond, and the ArPdBr complex 151, lend further credence to the syn-mechanism (via 152).[63] The same conclusion has been arrived at for an intermolecular, stoichiometric amination of (Z)-CHD=CHD with Ph2N-[Pd][64] and for an intramolecular amidation of a deuterated cyclopentene substrate followed by β-H elimination.[53a]
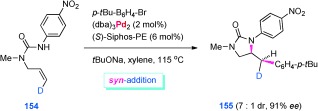
Another piece of complementary evidence was obtained from the catalytic cyclization of the deuteriated urea derivative 154. The relative configuration of the major product 155 was found to be consistent with the syn-mechanism of the addition (Scheme 41).[65]
Scheme 41.
Asymmetric aminopalladatio/arylation.
On the other hand, cyclization of the deuteriated acetamide 156 a, carried out in the absence of a strong base, afforded the arylated pyrrolidine derivative 158 (Scheme 42) as a result of an anti-amidopalladation (156 a → 157 a). The PdII species 157 a thus generated is oxidized with N-fluorosulfonimide to afford the corresponding PdIV complex. The latter intermediate then effects an electrophilic attack on toluene with retention of configuration to produce the C-arylated derivative 158 (thus accomplishing a C=H activation). The stereochemistry of the anti-aminopalladation has been confirmed by a stoichiometric experiment, in which 156 b was converted into the stable bipy complex 159, characterized by NMR spectroscopy.[66] The dramatic difference in the two mechanisms described in Schemes 41 and 42 apparently originates in the actual reaction conditions: in the former case, the amide-type nitrogen in 154 is deprotonated by a strong base, which increases its propensity to coordinate the palladium catalyst. In the latter case the base is absent and the palladium apparently prefers to first coordinate to the C=C bond; the resulting species then undergoes a traditional attack from the opposite face as in any classical electrophilic addition.
Scheme 42.
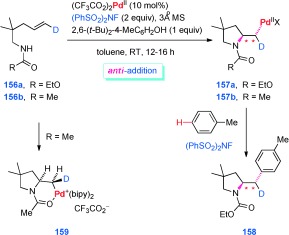
Intermolecular amidopalladation followed arylation with C=H activation.
Numerous other examples highlight the application of this palladium-catalyzed intramolecular amidoarylation of alkenes in the synthesis of various nitrogen heterocycles.[67,68] In the most recent examples both amidation and arylation were carried out as intramolecular processes to produce polycyclic structures.[69] This area has also been reviewed.[70]
5.3. Intramolecular Amidopalladation Followed by a Second Amidation
The amine-type nitrogen, being trivalent, offers another dimension in synthetic strategy that is not available to the divalent oxygen: while intramolecular alkoxypalladation produces a cyclic ether that cannot be further elaborated on the oxygen, the analogous amidopalladation features an additional N-substituent that can be involved in the subsequent events. Thus, the urea derivative 160 has been shown to undergo double cyclization to give the trans-configured bicyclic product 163 (Scheme 43).[71] The stereochemistry of this cascade was rationalized as follows: the initial amidopalladation (presumably proceeding with the established syn-mechanism) generates the Pd-chelate 161, in which Pd is replaced by bromide from CuBr2 with SN2 inversion and the resulting bromo derivative 162 undergoes cyclization with a second inversion to produce 163.[71] However, an alternative pathway, involving anti-amidopalladation, followed by oxidative cleavage of the C=Pd bond by SN 2 displacement with the nitrogen, cannot be excluded.
Scheme 43.
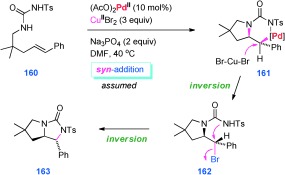
Catalytic intramolecular bis-amidation.
The related urea derivative 164 (with an ester group in the place of a phenyl of the previous example), also underwent a successful cyclization (Scheme 44) but producing the cis-derivative 168 (in contrast to the trans-isomer resulting from the previous example). The discrepancy was reconciled by assuming just one inversion: here, complex 165 (analogous to 161), instead of undergoing the Br− initiated inversion, is believed to coordinate CuBr2 (167), and Pd itself then serves as a leaving group in the ring closure,[72] so that 168 is formed with a single inversion. The difference between the reactivities of 161 and 165 has been attributed to the enolate-type equilibrium 165 ⇄ 166 that is not available to 161.[71] Nevertheless, an alternative anti-amidopalladation, followed by an oxidative cleavage of the C-Pd by CuBr2 with inversion, can also be considered.
Scheme 44.
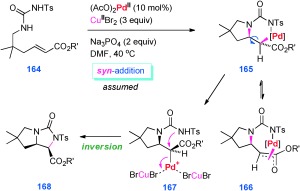
Catalytic intramolecular bis-amidation.
A different type of bis-amidation has been reported for the stilbene-derived bis-sulfonamide 169 (Scheme 45).[73] Here, the first cyclization presumably proceeds with anti-stereochemistry[74] and the arising PdII σ-complex is believed to be oxidized by PhI(OAc)2 to generate PdIV, which enables its replacement with the second sulfonamide group to produce 170.[73]
Scheme 45.

Catalytic intramolecular double-amidation.
The deuteriated substrate 156 b was used again to elucidate the diamidation process that afforded the pyrrolidine derivative 172 (Scheme 46). The latter outcome corresponds to the initial anti amidopalladation generating the PdII species 157 b (as in Scheme 42). The latter complex then undergoes oxidation with (PhSO2)2NF, giving rise to the PdIV species 171, which is then converted into the final product 172 on reaction with the imide anion via an SN2 inversion.[66]
Scheme 46.
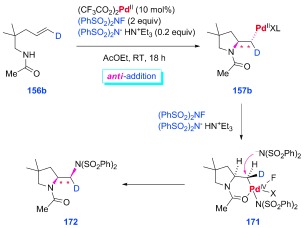
Intermolecular amidopalladation followed another amidation.
5.4. Intramolecular Amidopalladation Followed by Carbonylation
Intramolecular amidopalladation of olefinic amides, sulfonamides, and ureas 173–177 in the presence of carbon monoxide and methanol (Scheme 47)[75] has been reported to afford products corresponding to the anti-mechanism, irrespective of the configuration of the C=C bond in the starting molecule. The reaction conditions are mild: atmospheric pressure of CO, room temperature, and CuCl2 as the terminal oxidant. Both 5-exo and 6-exo cyclizations were attained. In the case of the urea derivatives 175 and 176, trapping of the acyl–Pd intermediate with the second nitrogen was observed in the absence of methanol to give 180 and 181. In the latter instance, the chloro derivative 182 was also obtained, apparently as a product of the SN2 substitution of Pd in the intermediate by chloride ion (as in the case of 21 and 161).[75]
Scheme 47.
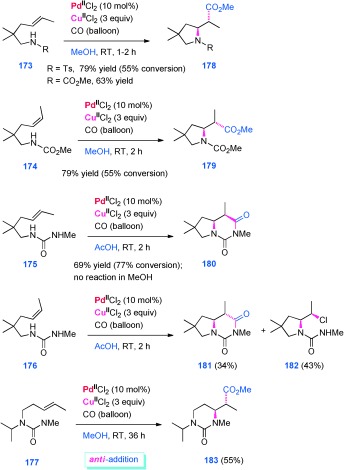
Amidopalladation followed by carbonylation.
With a free hydroxy group in the molecule as in 184, the initial amidation has been found to continue by lactonization to the neighboring hydroxyl (Scheme 48).[76] The yield of the resulting lactone 186 was maximized by replacing MeOH (as a solvent and competitor) with AcOH. While the stereochemistry of the initial amidopalladation has not been investigated in this instance, it was assumed to correspond to the anti-delivery. The diastereoselectivity is controlled by the hydroxy group, presumably by Pd coordination (185).
Scheme 48.
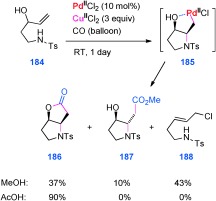
Amidopalladation followed by lactonization.
Finally, the Boc-protected hydroxylamine derivative 189 has been shown to undergo the cyclization/carbonylation with an overall syn-addition across the double bond, giving rise to the isoxazoline derivative 191 (Scheme 49).[77] The stereochemical outcome[78] apparently originates from the initial coordination of the Boc group to PdII (190), followed by syn-amidopalladation and subsequent carbon monoxide cleavage of the Pd=carbon bond with retention of configuration. The striking contrast between the stereochemistry of the carbonylative cyclization of the amides shown in Scheme 47 and the latter case is puzzling, as the reaction conditions are very similar. One explanation is that CuCl2 was used in Scheme 47, whereas Cu(OAc)2 was employed in the cyclization of 189 (Scheme 49). The use of CuCl2 leads to a high chloride concentration, which makes it more difficult for the amide to coordinate to palladium, and hence the anti-pathway should be favored. Furthermore, the latter reaction was carried out in the presence MeC(OMe)3, which could modify the pH of the mixture and thus improve the propensity of the ONHCO2(tBu) group to coordinate to palladium.
Scheme 49.
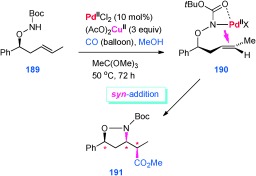
Amidopalladation of hydroxylamine derivatives followed by carbonylation.
6. Intermolecular Amidopalladation
The intramolecular amidopalladation has been shown to mostly proceed as a syn-addition, which apparently stems from the considerable entropic advantage of the neighboring group over an external nucleophile. This factor is absent in intermolecular additions, which may change the stereochemistry, as demonstrated convincingly for the Wacker oxidation (see Chapter 2).
6.1. Amidoacetoxylation
While terminal olefins have been shown to readily undergo intermolecular Pd-catalyzed amidoacetoxylation,[79] their internal counterparts resisted a number of attempts. Finally, cis-olefins, such as 192 (Scheme 50), have been successfully converted into the amidoacetoxylation products on reaction with phthalimide in the presence of PhI(OAc)2 as the oxidizing agent of palladium. The reaction was originally formulated as proceeding via an initial syn-amidopalladation. However, recent re-investigation,[80] prompted by the results of the related diamidation (see the next subchapter), demonstrated that the original structural assignment of the product[81] was incorrect. This finding led to a revision of the mechanism, according to which the initial amidopalladation of 192, catalyzed by PdII, proceeds as a pure anti process (rather than syn) to generate the palladium(II) intermediate 193 that is subsequently oxidized by the hypervalent iodine reagent to produce the PdIV species 194. The latter reaction prevents the usual β-elimination and is followed by the SN2 replacement of palladium with acetate (i.e., with inversion of configuration[82]) to afford the final product 195 that is anti-configured (rather than syn), according to X-ray analysis.[80,83] Noteworthy is also the high regioselectivity of this cascade, owing to the preferential attaching of the palladium moiety to the benzylic position (193).[84]
Scheme 50.
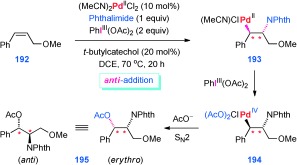
Intermolecular amidoacetoxylation of internal olefins.
By contrast, the behavior of terminal olefins has been shown to be more complicated: Thus, with the aid of the stereospecifically deuteriated olefin 196 (Scheme 51), the reaction was found not to be stereoselective, giving rise to a ∼4:3 mixture of diastereoisomeric amidopalladation products (analogous to 195). The latter outcome has been attributed to the poor stereoselectivity of the replacement of Pd with acetate in the final step. On the other hand, under aerobic conditions (i.e., in the absence of the hypervalent iodine reagent), the formation of the (E)-enamide 198 was ascribed to the syn-addition, generating 197, followed by the ordinary syn-stereoselective elimination of [PdH]. However, it is pertinent to note that the product 198 was isolated in merely 5 % yield (together with 70 % of the recovered starting material).[80]
Scheme 51.
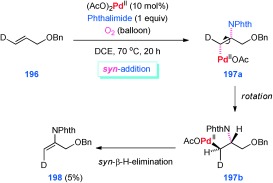
Intermolecular aerobic amidoacetoxylation of terminnal olefins.
6.2. Diamidation
In analogy to acetoxyamidation, the recently developed diamidation (Scheme 52) has also been found to be specific to cis-olefins, such as 199. The formation of the anti-configured of final product 202 has been rationalized by the initial anti-addition of PdII and phthalimide to generate 200, which is then oxidized with the hypervalent iodine reagent to produce the PdIV intermediate 201. The final displacement of Pd with ditosyl imide is believed to proceed with inversion (as in the previous case[80,82]) to produce the vicinal diamido derivative 202.[85]
Scheme 52.
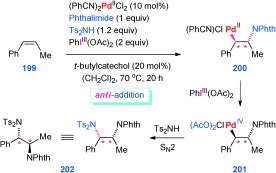
Intermolecular diamidation of internal olefins.
7. Overview of the Stereochemical Outcome in Nucleophilic Addition
The syn/anti-dichotomy in the nucleophilic additions was discussed mainly for oxygen and nitrogen nucleophiles. The stereochemical trends are summarized in Table 1, which covers cyclization processes (entries 1–28) and intermolecular reactions (entries 29–33).
Table 1.
Stereochemistry of the PdII-catalyzed addition of nucleophiles to C=C bond: intramolecular (entries 1–28) and intermolecular (entries 29–33).
| Entry | Reaction[a] | Scheme | syn/anti | PdX2 | Ligand | Oxidant | Solvent | Additive | T [°C] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A[b] | 21 | anti | PdCl2 | – | CuCl2 | MeOH | CO | RT | [36] |
| 2 | A[b] | 22 | anti | PdCl2 | – | CuCl2 | MeOH | CO | RT | [39] |
| 3 | A[b] | 23 | anti | PdCl2 | – | CuCl2 | MeOH | CO, LiCl | RT | [3a] |
| 4 | A[b,c] | 24 | syn | PdCl2 | – | CuCl2 | MeOH, CH2Cl2 | CO, MeC(OMe)3 | RT | [40] |
| 5 | B[b] | 25 | anti | PdCl2 | MeCN | p-BQ | THF | LiCl | reflux | [42] |
| 6 | B[b] | 25 | syn | Pd(BF4)2 | MeCN | p-BQ | MeOH | – | 40 | [42] |
| 7 | B[b] | 26 | syn | Pd(O2CCF3)2 | bipy[h] | O2 | toluene | 3 Å MS | 80 | [44] |
| 8 | C[b] | 28 | syn | Pd2(dba)3 | dpe-phos (139)[h] | – | THF | tBuONa, ArBr | 65 | [47] |
| 9 | C[b] | 29 | syn | Pd2(dba)3 | (4-MeOC6H4)3P | – | toluene | tBuONa, ArBr[l] | 105 | [48] |
| 10 | C[b] | 29 | anti | Pd2(dba)3 | dppe-C6H6[h] | – | toluene | tBuONa, ArBr[l] | 105 | [48] |
| 11 | C[b] | 30 | syn | Pd2(dba)3 | (4-MeOC6H4)3P | – | toluene | tBuONa, ArBr[l, m] | 105 | [48] |
| 12 | D[b,d] | 31 | syn | PdCl2 | MeCN | – | MeCN | – | RT | [50] |
| 13 | E[b] | 32 | syn | Pd(OAc)2 | pyridine | – | - | calculations | RT | [51,52] |
| 14 | E[b] | 33 | syn | Pd(OAc)2 | 125[h] | O2 | toluene | 3 Å MS | 25 | [52,53] |
| 15 | E[b] | 33 | anti | Pd(O2CCF3)2 | 125[h] | O2 | toluene | 3 Å MS | 25 | [52,53] |
| 16 | E[b] | 33 | syn | Pd(O2CCF3)2 | – | O2 | toluene | 3 Å MS | 25 | [52,53] |
| 17 | E[b] | 34 | anti | Pd(OAc)2 | – | p-BQ | THF, DMSO | AcOH, AcONa | 50 | [54] |
| 18 | F[b] | 38 | syn | Pd(OAc)2 | 139[h] | – | dioxane | Cs2CO3, ArBr | 105 | [60,61] |
| 19 | F[b] | 39 | syn | Pd2(dba)3 | (tBu)3P | – | toluene | tBuONa, ArBr | 65 | [62] |
| 20 | F[b] | 39 | syn | Pd2(dba)3 | Xanpthos[h] | – | toluene | tBuONa, ArBr | 65 or 110 | [62] |
| 21 | F[b,d,e] | 40 | syn | Pd2(dba)3 | dppf[h] | – | THF | (Me3Si)2NK, ArBr[l] | 23–60 | [63,64] |
| 22 | F[b] | 41 | syn | Pd2(dba)3 | Siphos-PE | – | xylene | tBuONa, ArBr | 115 | [65] |
| 23 | F[b] | 42 | anti | Pd(O2CCF3)2 | – | (PhSO2)2NF[i] | toluene | ArOH, toluene | RT | [66] |
| 24 | G[b] | 43 | syn[g] | Pd(OAc)2 | – | CuBr2 | DMF | Na3PO4 | 40 | [71] |
| 25 | G[b] | 44 | syn[g] | Pd(OAc)2 | – | CuBr2 | DMF | Na3PO4 | 40 | [72] |
| 26 | G[b] | 46 | anti | Pd(O2CCF3)2 | – | (PhSO2)2NF[j] | toluene | (PhSO2)2N− | RT | [66] |
| 27 | H[b] | 47 | anti | PdCl2 | – | CuCl2 | MeOH or AcOH | CO | RT | [75] |
| 28 | H[b] | 49 | syn | PdCl2 | – | Cu(OAc)2 | MeOH | CO, MeC(OMe)3 | 50 | [77] |
| 29 | I[f] | 13 | anti | Pd(OAc)2 | – | p-BQ | AcOH | AcONa | RT | [22] |
| 30 | J[d,f] | 19 | anti | PdCl2 | MeCN | – | THF | Me2NH | −40 | [31] |
| 31 | K[f] | 50 | anti | PdCl2 | MeCN | PhI(OAc)2[k] | (CH2Cl)2 | phthalimide, ArOH | 70 | [80] |
| 32 | E[f] | 51 | syn[n] | Pd(OAc)2 | – | O2 | (CH2Cl)2 | phthalimide | 70 | [80] |
| 33 | G[f] | 52 | anti | PdCl2 | PhCN | PhI(OAc)2[j] | (CH2Cl)2 | phthalimide, Ts2NH | 70 | [85] |
A=alkoxypalladation-carbonylation; B=alkoxypalladation-HPdX elimination; C=alkoxypalladation-arylation; D=amidopalladation; E=amidopalladation-HPdX elimination; F=amidopalladation–arylation; G=diamidopalladation; H=amidopalladation-carbonylation; I=acetoxypalladation; J=aminopalladation; K=amidoacetoxylation.
Intramolecular reaction.
Note that the reaction cannot proceed via 4(O)n-exo-trig cyclization.
Stoichiometric reaction.
The starting amide was first deprotonated.
Intermolecular reaction.
A different interpretation suggests anti-stereochemistry; see the comments in the text.
Chelating ligand.
Required in the second step, where the PdII intermediate 157 is oxidized to generate a PdIV species that then reacts with toluene via a C=H activation.
Required in the second step for the oxidation 157 (PdII) → 171 (PdIV); the latter species then undergoes an SN2-type introduction of the second nitrogen group.
Required in the second step for the oxidation 193 (PdII) → 194 (PdIV); the latter species then undergoes an SN2-type introduction of AcO.
ArBr is connected to the C=C bond by a linker.
Note the dynamic stereodifferentiaion by the residing chiral center.
Only 5 % yield.
The intramolecular alkoxypalladation-carbonylation cascade with carbon monoxide and alcohol as the stoichiometric reactants (reaction A) preferentially proceeds via the anti-pathway (entries 1–3), except for the examples, where this mechanism is disfavored by geometrical restrictions in the substrate (entry 4). On the other hand, anti/syn dichotomy has been observed for the analogous amidopalladation–carbonylation (reaction H; entries 27 and 28). Here, the dramatic difference in the stereochemistry can be tentatively attributed to the difference in the pH (neutral vs acidic), which is likely to influence the coordination capabilities of the participating amidic group.
Stereochemistry of the intramolecular alkoxypalladation-β-elimination cascade (reaction B; entries 5–7) can be controlled by the anion: thus, with PdCl2, that is, with strongly coordinating chlorides, especially in the presence of additional LiCl, the potential coordination of the participating alcohol group to Pd is disfavored and the reaction proceeds as an anti-addition (entry 5). On the other hand, with a weekly coordinating anion (BF4− or CF3CO2−), PdII can become coordinated to the participating OH group, which results in the preferential syn-addition (entries 6 and 7), regardless of the oxidizing reagents or solvent.
The analogous intramolecular amidopalladation-β-elimination cascade (reaction E; entries13–17) also exhibits the syn/anti dichotomy, depending on the actual conditions: thus, the reactions catalyzed by Pd(OAc)2 or Pd(O2CCF3)2 follow the syn-pathway (entries 13, 14, and 16); however, the presence of ligand 125 has been found to drive the reaction catalyzed by Pd(O2CCF3)2 toward the anti-mechanism (entry 15). By contrast, this ligand effect was not observed in the case of Pd(OAc)2 (entry14), which is rather intriguing. The change of the oxidizing agent (p-BQ vs O2) and the solvent (THF-DMSO vs toluene) and additives, namely AcOH/AcONa, has been found to also drive the reaction toward anti-mechanism (compare entries 14 and 16 with 17). Calculations predict the syn-mechanism (entry 13), which was also observed for the stoichiometric cyclization using PdCl2 (reaction D; entry 12), where the palladated product was isolated.
The intramolecular alkoxypalladation-arylation cascade (reaction C; entries 8–11) requires a strong base (as in the Hartwig-Buchwald arylation), which converts the participating alcohol group into a strongly coordinating alkoxide. As a result, the initial alkoxypalladation favors the syn-mechanism (entries 9 and 11). Addition of a chelating ligand may change the stereochemical course to anti (entry 10) but apparently not always (entry 8), especially when THF is used as a solvent (entry 8) instead of toluene (entry 10).
In analogy, the intramolecular amidopalladation-arylation cascade (reaction F; entries 18–22), also occurring in the presence of a base, invariably proceeds as a syn-addition, regardless of the solvent or the nature of the ligand employed. On the other hand, the reaction that involves oxidation to PdIV in the second step (required for the C=H activation of the “nucleophile”) has been shown to proceed with anti-stereochemistry, even when catalyzed by Pd(O2CCF3)2 (entry 23). The outcome in the latter case can be attributed to the absence of the base, which renders the participating N-nucleophile less prone to coordination of the PdII catalyst.
Intramolecular diamidopalladation (reaction G; entries 24 and 25) has been found to prefer the syn-mechanism, apparently due to the same effects as those discussed for entries 18–22. Again, this reaction, involving the PdII → PdIV oxidation and proceeding in the absence of a base, favors the anti-pathway (entry 26).
Intermolecular hydroxypalladation clearly prefers the anti-mechanism, as shown by the recent studies of the Wacker oxidation (Schemes 16–17). In analogy, intermolecular catalytic acetoxypalladation (reaction I; entry 29) also follows the anti-pathway. The same stereochemistry has been demonstrated for the stoichiometric intermolecular aminopalladation with Me2NH (reaction J; entry 30).
Intermolecular amidoacetoxylation (reaction K; entry 31) and diamidopalladation (reaction G; entry 33), catalyzed by PdCl2 (which disfavors coordination of the nucleophile to Pd), give the anti-addition products. By contrast, the intermolecular amidopalladation-β-elimination cascade (reaction E; entry 32), catalyzed by Pd(OAc)2, favors the syn-mechanism, which seems to be the only experimentally proven example of syn-migration in intermolecular nucleopalladation with O- and N-nucleophiles (although only in 5 % yield) to date. Note that this reaction proceeds with a different oxidizing agent (O2) than those cited in entries 31 and 33, and that it does not involve the PdIV species. This, however is unlikely to have a major effect on the mechanism of the first step; it appears that the key point here is the use of Pd(OAc)2 rather than PdCl2 as the catalyst.
8. Conclusion
This review has discussed the stereochemistry of the palladium-catalyzed addition of nucleophiles to alkenes and application of these processes in organic synthetic transformations. The syn/anti-dichotomy in the nucleophilic additions was discussed, mainly with oxygen and nitrogen nucleophiles. A general picture is emerging that in intermolecular reactions the anti-addition of the oxygen and nitrogen nucleophiles to (alkene)PdII complexes is strongly favored. However, in intramolecular reactions a special situation arises when the nitrogen or oxygen nucleophile coordinates to PdII. In this case there is no nucleophile available for external attack since there is a 1:1 ratio between the nucleophilic site and substrate. Therefore, the syn-attack is tremendously favored in the intramolecular cases where the nucleophile is coordinated to the metal. However, stereochemistry of the intramolecular reactions is dependent on the coordination capability of the internal nucleophile, which can be modified by the reaction conditions, so that the whole process can be driven either to the syn- or anti-pathway. In the intermolecular process, there will always be a considerable amount of free nucleophile in solution and therefore coordination of the nucleophile does not shut down the external anti-pathway as is done in the intramolecular case. As a result, external anti-attack is the predominant pathway in the intermolecular nucleophilic addition to (alkene)PdII complexes.
Acknowledgments
Financial support from the Swedish Research Council, the European Research Council (ERC AdG 247014), and the Wenner-Gren Foundation is gratefully acknowledged.
Biographies
Pavel Kočovský was raised and educated in Prague, Czechoslovakia (now Czech Republic). He received an MSc in 1974 from the Institute of Chemical Technology (ICT), Technical University, Prague, where he did his diploma work with Prof. O. Červinka in the area of asymmetric reactions. He obtained a PhD in 1977 from the Czechoslovak Academy of Sciences, Institute of Organic Chemistry and Biochemistry (IOCB), Prague, where he worked on steroid chemistry under the guidance of Dr. V. Černý and Prof. F. Šorm. He was then appointed to an academic position at the same Institute, which he held for thirteen years (1977–1990). He did his postdoctoral work with Prof. J. E. McMurry at Cornell University, Ithaca, NY, USA (1983–84), and later spent a sabbatical year with Prof. J.-E. Bäckvall at the University of Uppsala, Sweden (1989–1990). In January 1991 he moved to the University of Leicester, UK, where he spent almost nine years, obtained a DSc (1993), and raised in the ranks to full professor. In 1999 he moved to the University of Glasgow as the Sir William Ramsay Professor of Chemistry and in 2010 was elected a Fellow of the Royal Society of Edinburgh. He left Glasgow in the spring of 2014 and now holds a dual appointment at Stockholm University and Charles University in Prague, so that he is currently “delocalized” between the two cities. His research interests span organic and organometallic chemistry, stereoselective reactions, asymmetric catalysis, reaction mechanisms, and synthesis of functional molecules.

Jan-Erling Bäckvall was born in Malung, Sweden, in 1947. He received his Ph.D. from the Royal Institute of Technology, Stockholm, in 1975 with Prof. B. Åkermark. After postdoctoral work (1975–76) with Prof. K. B. Sharpless at Massachusetts Institute of Technology he joined the faculty at the Royal Institute of Technology. He was appointed Professor of Organic Chemistry at Uppsala University in 1986. In 1997 he moved to Stockholm University where he is currently Professor of Organic Chemistry. He is a member of the Royal Swedish Academy of Sciences, Finnish Academy of Science and Letters, and Academia Europaea. He is a member of the Nobel Committee for Chemistry. He is a member of a number of Editorial Boards of journals and he is the Chairman of the Editorial Board of Chemistry—A European Journal. He has edited the book “Modern Oxidation Methods” Wiley-VCH, 2004, 2nd ed., 2010. He has been visiting Professor in Lyon (France), Rennes (France), Utrecht University (the Netherlands), Strasbourg (France), Alicante (Spain), Paris (France), Mülheim (Germany), and Caltech, Pasadena (USA). He recently (2014) became honorary doctor at Åbo Akademi University, Finland. His current research interests include transition metal-catalyzed organic transformations, biomimetic oxidations, and enzyme catalysis.

References
- [1].p. 2.
- [1a].Schmid GH, Garratt DG, Bonds ElectrophilicAdditionstoC=X. In: The Chemistry of Double Bonded Functional Groups. Morrison JD, editor; Patai S, editor. New York: Wiley; (Ed.: (Ed.:, Part, p. [Google Scholar]
- [1b].Kočovský P, Bonds ElectrophilicAdditionstoC=X. In: Chemistry of Functional Groups, Supp. A3: The Chemistry of Double-Bonded Functional Groups. Patai S, editor. Chichester: Wiley; 1977. (Ed.:; [Google Scholar]
- [1c].Finn MG, Sharpless KB. Asymmetric Synthesis. New York: Academic; 1997. 247, pVol1985. [Google Scholar]
- [2].p. 1084. The only claim that iodoetherification of homoallylic alcohols and the related iodolactonization of the corresponding acids may proceed as a syn-process proved to be incorrect:
- [2a].Lipshutz BH, Barton JC. J. Am. Chem. Soc. 114 [Google Scholar]
- [2b].Mihelich ED, Hite GA. J. Am. Chem. Soc. 1992;114 [Google Scholar]
- [3].p. 5580.
- [3a].Kočovský P, Pour M. J. Org. Chem. 55 [Google Scholar]
- [3b].Kočovský P, Langer V, Gogoll A. J. Chem. Soc. Chem. Commun. 1990 [Google Scholar]
- [3c].Kočovský P. Organometallics. 1993;12 [Google Scholar]
- [4].p. 228.
- [4a].Pfund RA, Ganter C. Helv. Chim. Acta. 62 [Google Scholar]
- [4b].Tombo GMR, Ganter C. Helv. Chim. Acta. 1979;68 [Google Scholar]
- [4c].Tombo GMR, Ganter C. Helv. Chim. Acta. 1985;85 and references therein. [Google Scholar]
- [5].p. 2119.
- [5a].Hegedus LS. Transition Metals in the Synthesis of Complex Organic Molecules. Mill Valley: University Science Books; [Google Scholar]
- [5b].Negishi E. Organopalladium Chemistry for Organic Synthesis. New York: Wiley; 1994. Vol. 2p. 2002. [Google Scholar]
- [6].Zeise WC. Ann. Phys. Chem. 1827;6:632. [Google Scholar]
- [7].p. 9038. For previous reviews on this matter, see.
- [7a].Keith JA, Henry PM. Angew. Chem. Int. Ed. 48 [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- [7b].McDonald RI, Liu G, Stahl SS. Chem. Rev. 2009;111 doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].p. 176.
- [8a].Smidt J, Hafner W, Jira R, Sedlmeier J, Sieber R, Rüttinger R, Kojer H. Angew. Chem. 71 [Google Scholar]
- [8b].Smidt J, Hafner W, Jira R, Sieber R, Sedlmeier J, Sabel A. Angew. Chem. Int. Engl. Ed. 1959;1 [Google Scholar]; Angew. Chem. 1962;74 ; for a historical overview of the discovery of Wacker oxidation, see: [Google Scholar]
- [8c].Jira R. Angew. Chem. Int. Ed. 1962;48 [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- [9].Henry PM. J. Am. Chem. Soc. 1964;86:3246. [Google Scholar]
- [10].Bäckvall J-E, Åkermark B, Ljunggren SO. J. Am. Chem. Soc. 1979;101:2411. [Google Scholar]
- [11].Keith JA, Nielsen RJ, Oxgaard J, Goddard WA. J. Am. Chem. Soc. 2007;129:12342. doi: 10.1021/ja072400t. [DOI] [PubMed] [Google Scholar]
- [12].Henry PM. J. Org. Chem. 1973;38:2415. [Google Scholar]
- [13].p. 351.
- [13a].Kosaki M, Isemura M, Kitaura K, Shinoda S, Saito I. J. Mol. Catal. 2 [Google Scholar]
- [13b].Saito I, Shinoda S. J. Mol. Catal. 1977;9 [Google Scholar]
- [14].Bryndza HE. Organometallics. 1985;4:406. [Google Scholar]
- [15].Bäckvall J-E, Åkermark B, Ljunggren SO. J. Chem. Soc. Chem. Commun. 1977:264. [Google Scholar]
- [16].Bäckvall J-E. Tetrahedron Lett. 1977;18:467. [Google Scholar]
- [17].p. 4369.
- [17a].Bäckvall J-E, Björkman EE, Petersson L, Siegbahn P. J. Am. Chem. Soc. 106 [Google Scholar]
- [17b].Bäckvall J-E, Björkman EE, Petersson L, Siegbahn P. J. Am. Chem. Soc. 1984;107 [Google Scholar]
- [17c].Siegbahn P. J. Am. Chem. Soc. 1985;117 [Google Scholar]
- [18].p. 14672.
- [18a].Siegbahn P. J. Phys. Chem. 100 [Google Scholar]
- [18b].Anderson BJ, Keith JA, Sigman MS. J. Am. Chem. Soc. 1996;132 doi: 10.1021/ja1057218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].p. 6735. Note that the species with the 1,4-disposition of [Pd] and a hydroxy group, proposed as an intermediate in another reaction (including the same catalytic mixture of PdCl2224−2122CuCl, and LiCl, or Li PdCl in a stoichiometric experiment, both in DME at RT), undergoes a spontaneous ring closure (obviously with inversion of configuration). The latter complex does not require an initial replacement of [Pd] with Cl that would generate the corresponding 4-chloro-1-hydroxyderivative (which would be stable under the reaction conditions and would not undergo a spontaneous ring closure). Hence, similar behavior might be anticipated for the 1,2-hydroxypalladated species, and it is interesting that the chlorohydrin was actually isolated (Scheme 8). For a full discussion, see.
- [19a].Kočovský P, Pour M, Gogoll A, Hanuš V, Smrčina M. J. Am. Chem. Soc. 112 [Google Scholar]
- [19b].Kočovský P, Šrogl J, Pour M, Gogoll A. J. Am. Chem. Soc. 1990;116 [Google Scholar]
- [20]. See also discussion in Section 4.2., Scheme 24.
- [21].p. 4959.
- [21a].Bäckvall J-E, Nordberg RE. J. Am. Chem. Soc. 103 [Google Scholar]
- [21b].Bäckvall J-E, Byström SE, Nordberg RE. J. Org. Chem. 1981;49 [Google Scholar]
- [21c].Bäckvall J-E, Nordberg RE, Wilhelm D. J. Am. Chem. Soc. 1984;107 [Google Scholar]
- [21d].Bäckvall J-E, Andersson PG. J. Am. Chem. Soc. 1985;112 [Google Scholar]
- [22].Andell OS, Bäckvall J-E. J. Organomet. Chem. 1983;244:401. [Google Scholar]
- [23].Beyramabadi SA, Eshtiagh-Hosseini H, Housaindokht MR, Morsali A. Organometallics. 2008;27:72. [Google Scholar]
- [24].Keith JA, Nielsen RJ, Oxgaard J, Goddard WA, Henry PM. Organometallics. 2009;28:1618. [Google Scholar]
- [25].p. 8738.
- [25a].Comas-Vives A, Stirling A, Lledós A, Ujaque G. Chem. Eur. J. 16 doi: 10.1002/chem.200903522. [DOI] [PubMed] [Google Scholar]
- [25b].Nair NN. J. Phys. Chem. B. 2010;115 doi: 10.1021/jp110168p. [DOI] [PubMed] [Google Scholar]
- [25c].Imandi V, Kunnikuruvan S, Nair NN. Chem. Eur. J. 2011;19 doi: 10.1002/chem.201204342. [DOI] [PubMed] [Google Scholar]
- [26].Kovács G, Stirling A, Lledós A, Ujaque G. Chem. Eur. J. 2012;18:5612. doi: 10.1002/chem.201102138. [DOI] [PubMed] [Google Scholar]
- [27].Tsuji J, Nagashima H, Nemoto H. Org. Synth. 1990;7:137. [Google Scholar]
- [28].Keinan Y, Seth KK, Lamed R. J. Am. Chem. Soc. 1986;108:3474. For an example, where the Wacker-type oxidation catalyzed by PdCl22anti-II/CuCl (producing a ketone) was assumed to proceed as an addition, with an initial binding of Pd to a neighboring ether oxygen in the substrate molecule, see. [Google Scholar]
- [29].p. 2885. Originally, this transformation required PdCl2−p4II0or the presence of Cl as in the original Wacker protocol. The chloride-free oxidation (with -BQ as the terminal oxidant) was first shown to require the addition of a strong Brønsted acid (namely HClO ), which prevents the precipitation of Pd-black by pushing the equilibrium HPd X ⇄ Pd + HX to the left:
- [29a].Bäckvall J-E, Hopkins RB. Tetrahedron Lett. 29 [Google Scholar]
- [29b].Bäckvall J-E, Hopkins RB, Grennberg H, Mader M, Awasthi AK. J. Am. Chem. Soc. 1988;112 2The same observation was also reported recently, where NaNO is believed to have a role: [Google Scholar]
- [29c].Zhang G, Xie X, Wang Y, Wen X, Zhao Y, Ding C. Org. Biomol. Chem. 1990;11 doi: 10.1039/c3ob40277k. [DOI] [PubMed] [Google Scholar]
- [30].p. 3237.
- [30a].Teo P, Wickens ZK, Dong G, Grubbs RH. Org. Lett. 14 ; for an earlier work on altering the regioselectivity, see: [Google Scholar]
- [30b].Feringa BL. J. Chem. Soc. Chem. Commun. 2012 [Google Scholar]
- [30c].Kiers NH, Feringa BL, van Leeuwen PWNM. Tetrahedron Lett. 1986;33 [Google Scholar]
- [30d].Kiers NH, Feringa BL, Kooijman H, Spek AL, van Leeuwen PWNM. J. Chem. Soc. Chem. Commun. 1992 [Google Scholar]
- [30e].Wenzel TT. J. Chem. Soc. Chem. Commun. 1992 [Google Scholar]
- [31].p. 1363.
- [31a].Åkermark B, Bäckvall J-E, Hansén KS, Sjöberg K, Zetterberg K. Tetrahedron Lett. 15 [Google Scholar]
- [31b].Åkermark B, Bäckvall J-E. Tetrahedron Lett. 1974;16 [Google Scholar]
- [31c].Åkermark B, Zetterberg K. J. Am. Chem. Soc. 1975;106 [Google Scholar]
- [32].p. 295. An analogous mechanism was also demonstrated for the related Pt-promoted amination.
- [32a].Panuzi A, de Renzi A, Paiaro G. J. Am. Chem. Soc. 92 [Google Scholar]
- [32b].Stille JK, Fox DB. J. Am. Chem. Soc. 1970;92 [Google Scholar]
- [33].p. 335.
- [33a].Bäckvall J-E. Acc. Chem. Res. 16 [Google Scholar]
- [33b].Bäckvall J-E. Pure Appl. Chem. 1983;64 [Google Scholar]
- [34].Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. New York: Wiley; [Google Scholar]
- [34a].Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. Weinheim: VCH; 1989. [Google Scholar]
- [34b].Nicolaou KC, Snyder SA. Classics in Total Synthesis. Wiley-VCH, Weinheim: II; 1996. [Google Scholar]
- [34c].Kürti L, Czakó B. Strategic Applications of Named Reactions in Organic Synthesis. Amsterdam: Elsevier; 2003. [Google Scholar]
- [34d].Hudlický T, Reed JW. The Way of Synthesis. Weinheim: Wiley-VCH; 2005. [Google Scholar]
- [34e].Kočovský P, Tureček F, Hájíček J. Synthesis of Natural Products: Problems of Stereoselectivity. Boca Raton, Florida: CRC Press; 2007. Vols. 1 and 21986. [Google Scholar]
- [35].p. 3. For representative reviews on halolactonization and related reactions, see.
- [35a].Bartlett PA. Tetrahedron. 36 [Google Scholar]
- [35b].Cardillo G, Orena M. Tetrahedron. 1980;46 [Google Scholar]
- [35c].Robin S, Rousseau G. Eur. J. Org. Chem. 1990 [Google Scholar]
- [36].p. 1496.
- [36a].Semmelhack MF, Bodurow C. J. Am. Chem. Soc. 106 [Google Scholar]
- [36b].J. Am. Chem. Soc. 1984;106 For correction of some of the structures, see: [Google Scholar]
- [37].Tamaru Y, Kobayashi T, Kawamura S, Ochiachi H, Hojo M, Yoshida Z. Tetrahedron Lett. 1985;26:3207. [Google Scholar]
- [38].Semmelhack MF, Zhang N. J. Org. Chem. 1989;54:4483. [Google Scholar]
- [39].McCormick M, Mohanan R, Soria J, Goldsmith D, Liotta D. J. Org. Chem. 1989;54:4485. [Google Scholar]
- [40].p. 325.
- [40a].Tamaru Y, Hojo M, Yoshida Z. Tetrahedron Lett. 28 [Google Scholar]
- [40b].Tamaru Y, Hojo M, Yoshida Z. J. Org. Chem. 1987;56 [Google Scholar]
- [41].Ferguson J, Zeng F, Alper H. Org. Lett. 2012;14:5602. doi: 10.1021/ol302725x. [DOI] [PubMed] [Google Scholar]
- [42].Hayashi T, Yamasaki K, Mimura M, Uozumi Y. J. Am. Chem. Soc. 2004;126:3036. doi: 10.1021/ja031946m. [DOI] [PubMed] [Google Scholar]
- [43].Andersson PG, Bäckvall J-E. In: Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. New York: Wiley; 2002. p. 1859. Vol. 2-catalyzed 1,4-functionalization of conjugated dienes, see ref. [21] and: (Ed.:, C-O and C-N Bond Formation Involving Conjugated Dienes and Allylpalladium IntermediatesFor a similar effect of chloride on the stereochemistry of the PdII. [Google Scholar]
- [44].Trend RM, Ramtohul YK, Stoltz BM. J. Am. Chem. Soc. 2005;127:17778. doi: 10.1021/ja055534k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Semmelhack MF, Kim CR, Dobler W, Meier M. Tetrahedron Lett. 1989;30:4925. [Google Scholar]
- [46].p. 262.
- [46a].Tietze L, Sommer KM, Zinngrebe J, Stecker F. Angew. Chem. 117 doi: 10.1002/anie.200461629. [DOI] [PubMed] [Google Scholar]
- [46b].Tietze L, Stecker F, Zinngrebe J, Sommer KM. Chem. Eur. J. 2005;12 ; for related but simpler exmaples, see: [Google Scholar]
- [46c].Semmelhack MF, Epa WR. Tetrahedron Lett. 2006;34 [Google Scholar]
- [47].p. 1620.
- [47a].Wolfe JP, Rossi MA. J. Am. Chem. Soc. 126 doi: 10.1021/ja0394838. [DOI] [PubMed] [Google Scholar]
- [47b].Hay MB, Hardin AR, Wolfe JP. J. Org. Chem. 2004;70 doi: 10.1021/jo050022+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47c].Hay MB, Wolfe JP. J. Am. Chem. Soc. 2005;127 doi: 10.1021/ja054754v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47d].Hay MB, Wolfe JP. Tetrahedron Lett. 2005;47 doi: 10.1016/j.tetlet.2006.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47e].Ward AF, Xu Y, Wolfe JP. Chem. Commun. 2006;48 doi: 10.1039/c1cc15880e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakhla JS, Kampf JW, Wolfe JP. J. Am. Chem. Soc. 2006;128:2893. doi: 10.1021/ja057489m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].p. 1969. For a similar demonstration of the control of facial selectivity of electrophilic additions across a C=C bond by neighboring groups, see, e.g., ref. [1b,3a] and the following.
- [49a].Kočovský P, Stieborová I. J. Chem. Soc. Perkin Trans. 1 [Google Scholar]
- [49b].Kočovský P, Starý I, Zajíček J, Tureček F, Vašíčková S. J. Chem. Soc. Perkin Trans. 1. 1987 and references therein. [Google Scholar]
- [50].Isomura K, Okada N, Saruwatari M, Yamasaki H, Taniguchi H. Chem. Lett. 1985:385. [Google Scholar]
- [51].Ye X, Liu G, Popp BV, Stahl SS. J. Org. Chem. 2011;76:1031. doi: 10.1021/jo102338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. J. Org. Chem. 2013;78:2083. doi: 10.1021/jo302266t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].p. 18594.
- [53a].White PB, Stahl SS. J. Am. Chem. Soc. 133 doi: 10.1021/ja208560h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53b].Weinstein AB, Stahl SS. Angew. Chem. Int. Ed. 2011;51 doi: 10.1002/anie.201206702. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 ; for related cyclizations, where stereochemistry has not been addressed, see: [Google Scholar]
- [53c].Lu Z, Stahl SS. Org. Lett. 2012;14 doi: 10.1021/ol300030w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Joosten A, Persson AKÅ, Millet R, Johnson MT, Bäckvall J-E. Chem. Eur. J. 2012;18:15151. doi: 10.1002/chem.201202359. [DOI] [PubMed] [Google Scholar]
- [55].p. 2243.
- [55a].Bäckvall J-E, Gogoll A. Tetrahedron Lett. 29 [Google Scholar]
- [55b].Grennberg H, Gogoll A, Bäckvall JE. Organometallics. 1988;12 [Google Scholar]
- [56].p. 1242.
- [56a].Redford JE, McDonald RI, Rigsby KL, Wiensch JD, Stahl SS. Org. Lett. 14 doi: 10.1021/ol3000519. ; for the use of DMSO, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56b].Diao T, White P, Guzei I, Stahl SS. Inorg. Chem. 2012;51 doi: 10.1021/ic301799p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yang G, Shen C, Zhang W. Angew. Chem. Int. Ed. 51:9141. doi: 10.1002/anie.201203693. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [58].p. 4901.
- [58a].Malkov AV, Lee DS, Barłóg M, Elsegood MRJ, Kočovský P. Chem. Eur. J. 20 doi: 10.1002/chem.201400123. ; See also: [DOI] [PubMed] [Google Scholar]
- [58b].Malkov AV, Derrien N, Barłóg M, Kočovský P. Chem. Eur. J. 2014;20 doi: 10.1002/chem.201304798. [DOI] [PubMed] [Google Scholar]
- [59].Weinstein AB, Schuman DP, Tan ZX, Stahl SS. Angew. Chem. Int. Ed. 52:11867. doi: 10.1002/anie.201305926. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- [60].p. 3605.
- [60a].Ney JE, Wolfe JP. Angew. Chem. Int. Ed. 43 doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004;116 [Google Scholar]
- [60b].Bertrand MB, Neukom JD, Wolfe JP. J. Org. Chem. 2004;71 doi: 10.1021/jo801631v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].For a related cascade that features vinylation (with trans3. Babij NR, Wolfe JP. Angew. Chem. Int. Ed. 51:4128. -BrCH=CHSiMe ) rather than arylation, see:; [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [62].p. 927.
- [62a].Dongol KG, Tay BY. Tetrahedron Lett. 47 [Google Scholar]
- [62b].Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J. Org. Chem. 2006;74 doi: 10.1021/jo8027399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Neukom JD, Perch NC, Wolfe JP. J. Am. Chem. Soc. 2010;132:6276. doi: 10.1021/ja9102259. [DOI] [PubMed] [Google Scholar]
- [64].Hanley PC, Marković D, Hartwig JF. J. Am. Chem. Soc. 2010;132:6302. doi: 10.1021/ja102172m. [DOI] [PubMed] [Google Scholar]
- [65].Hopkins BA, Wolfe JP. Angew. Chem. Int. Ed. 51:9886. doi: 10.1002/anie.201205233. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [66].Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J. Am. Chem. Soc. 2009;131:15945. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]
- [67].Zavesky BP, Babij NR, Fritz JA, Wolfe JP. Org. Lett. 2013;15:5420. doi: 10.1021/ol402377y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Babij NR, Wolfe JP. Angew. Chem. Int. Ed. 52:9247. doi: 10.1002/anie.201302720. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- [69].Alicea J, Wolfe JP. J. Org. Chem. 2014;79:4212. doi: 10.1021/jo500470m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schultz DM, Wolfe JP. Synthesis. 2012:351. doi: 10.1055/s-0031-1289668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].p. 14586.
- [71a].Streuff J, Hövelmann CH, Nieger M, Muñiz K. J. Am. Chem. Soc. 127 doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]
- [71b].Muñiz K, Hövelmann CH, Campos-Gómez E, Barluenga J, González JM, Streuff J, Nieger M. Chem. Asian J. 2005;3 doi: 10.1002/asia.200700373. [DOI] [PubMed] [Google Scholar]
- [71c].Muñiz K, Streuff J, Chávez P, Hövelmann CH. Chem. Asian J. 2008;3 doi: 10.1002/asia.200800148. [DOI] [PubMed] [Google Scholar]
- [71d].Muñiz K, Martínez C. J. Org. Chem. 2008;78 doi: 10.1021/jo302472w. [DOI] [PubMed] [Google Scholar]
- [71e].McDonald RI, Stahl SS. Angew. Chem. Int. Ed. 2013;49 [Google Scholar]; Angew. Chem. 2010;122 [Google Scholar]
- [72].A similar displacement of [Pd] by a neighboring group has been mentioned in ref. [19]
- [73].Muñiz K. J. Am. Chem. Soc. 2007;129:14542. doi: 10.1021/ja075655f. [DOI] [PubMed] [Google Scholar]
- [74].Note that a synendo -addition in the 5- process would be difficult.
- [75].p. 3994.
- [75a].Tamaru Y, Hojo M, Higashimura H, Yoshida Z. J. Am. Chem. Soc. 110 ; for an overview, see: [Google Scholar]
- [75b].Tamaru Y, Yoshida Z. J. Organomet. Chem. 1988;334 [Google Scholar]
- [76].p. 5731.
- [76a].Tamaru Y, Hojo M, Yoshida Z. J. Org. Chem. 53 [Google Scholar]
- [76b].Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J. Org. Chem. 1988;62 doi: 10.1021/jo961988b. [DOI] [PubMed] [Google Scholar]
- [77].Malkov AV, Barłóg M, Miller-Potucká L, Kabeshov MA, Farrugia L, Kočovský P. Chem. Eur. J. 2012;18:6873. doi: 10.1002/chem.201102716. [DOI] [PubMed] [Google Scholar]
- [78].The earlier studies on this type of Pd-catalyzed reactions did not address the stereochemistry issue: p. 4225.
- [78a].Bates RW, Sa-Ei K. Org. Lett. 4 doi: 10.1021/ol026701d. [DOI] [PubMed] [Google Scholar]
- [78b].Bates RW, Boonsombat J. Org. Biomol. Chem. 2002;3 doi: 10.1039/b415297b. [DOI] [PubMed] [Google Scholar]
- [79].Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]
- [80].Martínez C, Wu Y, Weinstein AB, Stahl SS, Liu G, Muñiz K. J. Org. Chem. 2013;78:6309. doi: 10.1021/jo400671q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Fles D, Majhofer B, Kovac M. Tetrahedron. 1968;24:3053. [Google Scholar]
- [82].For the stereochemistry of Pd displacement with nucleophiles, see ref. [16, 19 a, 33, 35], and the following: p. 200.
- [82a].Coulson DR. J. Am. Chem. Soc. 91 [Google Scholar]
- [82b].Wong PK, Stille JK. J. Organomet. Chem. 1969;64–83 [Google Scholar]
- [82c].Zhu G, Ma S, Lu X, Huang Q. J. Chem. Soc. Chem. Commun. 1974 [Google Scholar]
- [82d].Kočovský P, Dunn V, Gogoll A, Langer V. J. Org. Chem. 1995;64 doi: 10.1021/jo9812882. For the palladium(IV) intermediates, the S 2 inversion mechanism has been widely accepted; see ref. [66, 79] an the following: [DOI] [PubMed] [Google Scholar]
- [82e].Muñiz K. Angew. Chem. Int. Ed. 1999;48 [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- [82f].Sibbald PA, Michael FE. Org. Lett. 2009;11 doi: 10.1021/ol9000087. [DOI] [PubMed] [Google Scholar]
- [82g].Qiu S, Xu T, Zhou J, Guo Y, Liu G. J. Am. Chem. Soc. 2009;132 doi: 10.1021/ja909716k. [DOI] [PubMed] [Google Scholar]
- [83].The story is further complicated by the use of the erythrothreosynanti. p. 557. nomenclature, which is in conflict with the original Fischer notation. To avoid further confusion, we adhere to the symbolism:
- [83a].Masamune S, Ali SA, Snitman DL, Garvey DS. Angew. Chem. Int. Ed. Engl. 19 [Google Scholar]; Angew. Chem. 1980;92 For an extended discussion, see: [Google Scholar]
- [83b].Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. New York: Wiley; 1980. p. . [Google Scholar]
- [84].For this effect, see: p. 1166.
- [84a].Nettekoven U, Hartwig JF. J. Am. Chem. Soc. 124 doi: 10.1021/ja017521m. [DOI] [PubMed] [Google Scholar]
- [84b].Johns AM, Utsunomiya M, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2002;128 doi: 10.1021/ja056003z. [DOI] [PubMed] [Google Scholar]
- [85].Martínez C, Muñiz K. Angew. Chem. Int. Ed. 51:7031. doi: 10.1002/anie.201201719. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]