

Abstract
The aim of the present study was to determine whether chlorogenic acid (CA) is able to modulate cadmium (Cd)-induced oxidative brain damage. Cd-treated rats displayed numerous pathological effects, including the inhibition of acetylcholinesterase, elevated lipid peroxidation, the depletion of enzymatic and non-enzymatic antioxidants, the reduction of membrane-bound ATPase activity, mitochondrial dysfunction and DNA fragmentation. Pretreatment of the rats with CA significantly attenuated these effects. These results lead to the hypothesis that the mechanisms by which CA attenuates the effects of Cd-induced oxidative brain damage include the maintenance of antioxidant homeostasis, inhibition of the membrane effects and the perpetuation of mitochondrial dysfunction. These data support the potential of CA as a beneficial intervention in the prevention of heavy metal poisoning due to Cd exposure.
Keywords: neurotoxicity, cadmium, chlorogenic acid, oxidative stress
Introduction
Heavy metal pollution due to cadmium (Cd), arsenic and lead is under vital scrutiny as a result of potential neurotoxicological hazards to the human population (1,2). Cd is a universal ecological toxicant frequently encountered through metal industries, in aerosolized form in cigarette smoke and in certain industrial workplaces, including those where battery manufacture and smelting take place (3). In addition to the noxious effect of Cd on vital organs such as the kidneys, liver and testes, Cd also elicits damaging effects on the central nervous system (CNS) due to a variety of factors, such as its toxicity at low concentration, extended half-life (15–30 years in humans) and low clearance rate (4). Although the blood-brain barrier (BBB) is impermeable to chronic low levels of Cd exposure, Cd may affect the permeability of the barrier and reach the brain (5). Preclinical studies have indicated that Cd induces behavioral disorders, as well as morphological and biochemical changes in the CNS (6,7). The brain region displays a high potential for oxidation and a low antioxidant defense capacity relative to the hepatic system. The neurotoxic potential of Cd has been attributed to the changes it induces in the brain enzyme network involved in counteracting oxidative stress and the disturbance of brain metabolism. Further, Cd increases the generation of free radicals, in particular superoxide ions, in the brain and hinders the antioxidant defence system, thus increasing lipid peroxidation. Moreover, free radicals generated by Cd interact with mitochondrial sites, leading to the breakdown of mitochondrial potentials, a consequence of reductions in intracellular glutathione levels (8). The mechanism by which Cd induces oxidative stress appears to be based mainly on the disruption of the prooxidant/antioxidant balance, which may lead to brain injury via oxidative damage to critical biomolecules, such as thiols, lipids, proteins and DNA (9).
Dietary polyphenols are proposed to be major antioxidants that provide health benefits due to their free radical-scavenging and metal-chelating properties, and the modulation of enzymatic activity and signal transduction pathways (10). Chlorogenic acid (CA) is a potent polyphenolic antioxidant abundantly found in apples, coffee beans, tomatoes, potatoes and apricots (11,12). CA is an ester formed between caffeic acid and quinic acid that is hydrolyzed by intestinal bacteria to various aromatic acid metabolites (13). The antioxidant potential of CA is due to the presence of vicinal hydroxyl groups on an aromatic residue, which enable it to scavenge free radicals in the aqueous phase (14) and prevent oxidative DNA damage (15). Previous studies have reported the antioxidant potential (16,17) and widespread bioactive effects of CA, such as antimutagenic, antiviral (18), anticarcinogenic (19) and tissue protection (20) effects. The neuroprotective potential of CA against oxidative insult to the brain has been well documented (21–23). The present study was carried out to investigate the neuroprotective efficacy of CA against the changes to the brain provoked by cadmium.
Materials and methods
Chemicals
CA, cadmium chloride (CdCl2) and other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were of the highest available commercial grade.
Animals
Male Wistar rats weighing 170–200 g were obtained from the animal facility at Shandong University (Jinan, China). The animals were maintained under standard laboratory conditions of relative humidity (55±5%), temperature (25±2°C) and light (12 h light/12 h dark). They were fed standard diet pellets and water was provided ad libitum.
Experimental design
Twenty four rats were divided into four groups (n=6 per group) and subjected to treatment as follows: Group 1: Rats received distilled water 1 ml/100 g body weight once daily by oral gavage for 30 days and served as a control group. Group 2: Rats orally received Cd as CdCl2 in saline (5 mg/kg body weight) each day for 30 days. Group 3: Rats received CA alone (60 mg/kg body weight) dissolved in distilled water, intragastric administration once daily for 30 days. Group 4: Rats orally received Cd (5 mg/kg body weight) followed by intragastric administration of CA (60 mg/kg body weight) in distilled water once daily for 30 days.
At the end of the experimental period, rats were fasted overnight and sacrificed by decapitation. Blood was collected in a heparinized BD vacutainer (BD Biosciences, Franklin Lakes, NJ, USA) and plasma samples were collected by centrifugation at 2,000 × g for 20 min. The whole brain was immediately excised and rinsed in ice-cold saline to remove the blood. The brain tissues were homogenized (25%, w/v) in 25 mmol/l phosphate buffer (pH 7.0) containing 0.25 mol/l sucrose (w/v) and centrifuged at 1,000 × g for 10 min. The clear supernatant was used for various biochemical assays to assess oxidative stress.
Preparation of mitochondrial fraction
This was conducted as previously described (24). Brain homogenate (25%, w/v) was prepared and the supernatant was subject to centrifugation at 12,000 g for 10 min at 4°C (Remi cooling centrifuge, C-24 BL; Remi Laboratory Instruments, Mumbai, India). The mitochondrial pellets thus obtained were washed twice with phosphate buffer (pH 7.4) to remove sucrose and homogenized in phosphate buffer containing 0.5% Tween-80 (v/v). The supernatant was collected and used for enzyme analysis, then washed and resuspended in the same buffer.
Biochemical assays
Estimation of acetylcholinesterase (AChE) activity
The level of AChE in the brain homogenate was measured according to a previously described method (25) using acetylthiocholine iodide (ATCI) as a substrate. In this method, AChE in the sample hydrolyzed ATCI into thiocholine and butyric acid. The thiocholine reacted with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) to form 5-thio-2-nitrobenzoic acid. The yellow color that developed was measured spectrophotometrically at 412 nm. The values are expressed as µmol of ATCI hydrolyzed/min/mg protein.
Assessment of enzymatic antioxidants
The brain tissue level of superoxide dismutase (SOD) was estimated by the method of Kakkar et al (26). The reaction was based on the inhibition of the reduction of the nitro blue tetrazolium chromogen by NADH and phenazine methosulfate. One unit of SOD activity corresponds to the amount of enzyme that causes a 50% suppression of the reduction of nitro blue tetrazolium/min/mg of protein.
The catalase (CAT) activity in the brain homogenate was analyzed by the method of Sinha et al (27) wherein the breakdown of H2O2 was evaluated by measuring the UV absorption of H2O2 at 240 nm. The CAT activity is expressed as µmol H2O2 consumed/min/mg of protein.
The brain levels of glutathione peroxidase (GPx) were assayed by the method of Rotruck et al (28). The assay system was based on the reaction between a measured amount of enzyme preparation H2O2 and reduced glutathione (GSH) for a specified time period. The unreacted GSH was then measured by reaction with DTNB. The GPx activity is expressed as nmol GSH oxidized/min/mg of protein.
Glutathione S-transferase (GST) was estimated by the method of Habig et al (29). The increase in absorbance was measured at 340 nm using 1-chloro-2,4-dinitrochlorobenzene (CDNB) as substrate. GST activity is expressed in units of nmol of GSH-CDNB conjugate formed/min/mg of protein.
Estimation of protein content
The protein content of the brain homogenate was estimated by the method of Lowry et al (30) using bovine serum albumin as a standard.
Estimation of non-enzymatic antioxidants
The GSH levels in the brain were determined by the method of Moron et al (31). The nonprotein sulfhydryl content of cells is in the form of GSH. DTNB is a disulfide compound that is readily reduced by sulfhydryl compounds to form a highly colored yellow anion. The optical density of this yellow substance was measured at 412 nm. Results are expressed as µg/mg of protein.
The concentration of ascorbic acid (vitamin C) in the brain was determined by the method of Omaye et al (32). In this method dehydroascorbic acid, the oxidized product of ascorbic acid, reacts with 2,4-dinitrophenylhydrazine to form bis-2,4-dinitrophenylhydrazone. This undergoes further rearrangement to form a product with an absorption maximum at 520 nm. Thiourea provides the reducing medium to prevent interference from non-ascorbic acid chromogens. Results are expressed in µg/mg of protein.
Vitamin E was analyzed by the method of Desai (33). Ferric ions were reduced to ferrous ions in the presence of vitamin E, and a pink-colored complex was formed. The chelating agent orthophosphoric acid was added to minimize carotene interference in the assay. Results are expressed as µg/mg of protein.
Measurement of tissue lipid peroxides (LPOs)
The LPO levels in the brain homogenate were measured according to the method of Ohkawa et al (34). The final colored end-product was assayed spectrophotometrically at 532 nm. The LPO level was expressed as nmol malondialdehyde (MDA)/mg protein.
Determination of membrane-bound ATPase activities
Na+/K+-ATPase activity in the brain homogenate was assayed by the method of Bonting (35). In this method, 0.2 ml brain homogenate was added to a reaction mixture containing Tris HCl buffer (pH 7.5), 50 mM MgSO4, 50 mM KCl, 600 mM NaCl, 1 mm EDTA, 40 mM ATP and 2.5% ammonium molybdate and incubated for 15 min at 37°C. The reaction was then arrested by the addition of 1 ml ice cold 10% trichloroacetic acid. Then the amount of inorganic phosphorus (Pi) liberated in the protein-free supernatant was estimated. The activity of Mg2+-ATPase in the tissue homogenate was assayed by the method of Ohinishi (36). Brain levels of Ca2+-ATPase level were estimated as described by the method of Hjertan and Pan (37). The activities of these ATPase enzymes in tissue homogenate were expressed as µmol of Pi liberated/min/mg protein.
Determination of mitochondrial citric acid cycle enzymes
The activity of α-ketoglutarate dehydrogenase (KDH) was estimated from the rate of reduction of NAD+ in the presence of α-KDH at 340 nm (38). The activity of isocitrate dehydrogenase (ICDH) was assayed by the method of King (39). Succinate dehydrogenase (SDH) was assayed by the method of Nulton-Persson and Szweda (40) and the activity was estimated from the rate of reduction of dichloroindophenol (DCIP) in the presence of sodium succinate at 600 nm. The levels of malate dehydrogenase (MDH) were estimated by the method of Mehler et al (41). In this reaction the rate of oxidation of NADH was measured in the presence of oxaloacetate at 340 nm.
DNA fragmentation analysis
The DNA fragmentation pattern obtained using agarose gel electrophoresis was analyzed by the method of Watabe et al (42). Briefly, the brain samples were washed twice with phosphate-buffered saline and were lysed in a solution of 10 mM Tris-HCl (pH 8.0), 10 mM EDTA, 0.5% (w/v) sodium dodecyl sulfate and 0.1% (w/v) RNase A, with incubation for 60 min at 50°C. The lysate was then further incubated for 60 min at 50°C with 1 mg/ml proteinase K and subjected to electrophoresis on 1% agarose gel for 60 min at 50 V using 40 mM Tris acetate (pH 7.5) containing 1 mM EDTA. Subsequent to electrophoresis, DNA was visualized by staining with ethidium bromide.
Statistical analysis
The results are expressed as mean ± standard deviation for 6 rats per group. Statistical analysis was performed by one-way analysis of variance using the SPSS software package for Windows (version 10.0; SPSS, Inc., Chicago, IL, USA. Post hoc testing was performed for inter-group comparisons using the least significance difference (LSD) test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of CA and Cd on brain AChE status
Fig. 1 displays the activity of AChE in the brains of control and experimental rats. In the present study, the levels of AChE in the brain were significantly (P<0.05) decreased in the Cd-treated rats whereas the administration of CA (60 mg/kg body weight) significantly (P<0.05) attenuated the Cd-induced reduction in AChE activity, resulting in normal AChE levels.
Figure 1.
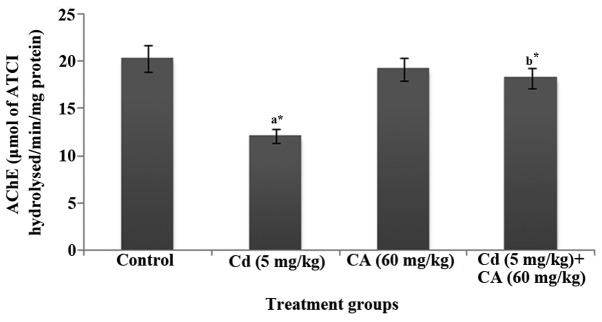
Effect of Cd and CA on the level of AChE in rat brain homogenate. Values are mean ± standard deviation (n=6 per group). a*P<0.05 vs. the control group; b*P<0.05 vs. the Cd group. Cd, cadmium; CA, chlorogenic acid; AChE, acetylcholinesterase; ATCI, acetylthiocholine iodide.
Effects of CA and Cd on brain enzymatic antioxidant levels
The activities of the enzymatic antioxidants SOD, CAT, GPx and GST in the brains of control and experimental rats are displayed in Table I. Cd-treated rats displayed significant (P<0.05) reductions in the activities of SOD, CAT, GPx and GST when compared with the control rats. The intragastric administration of CA reversed the noxious effect of Cd and elicited a significant (P<0.05) restoration of the levels of antioxidant enzymes to normal values.
Table I.
Effects of CD and CA on the activities of enzymatic antioxidants in the brain.
| Groups | SOD (U/mg protein) | CAT (U/mg protein) | GPx (nmol/min/mg protein) | GST (nmol/min/mg protein) |
|---|---|---|---|---|
| Control | 7.22±0.19 | 4.35±0.1 | 2.87±0.13 | 5.59±0.45 |
| Cd (5 mg/kg) | 4.11±0.13a | 1.56±0.07a | 1.38±0.06a | 3.18±0.21a |
| CA (60 mg/kg) | 7.91±0.17a,b | 3.70±0.08a,b | 3.95±0.18a,b | 4.62±0.17b |
| Cd (5 mg/kg) + CA (60 mg/kg) | 7.10±0.09b | 4.20±0.14b | 4.10±0.15a,b | 5.43±0.10b |
Values are expressed as mean ± standard deviation for 6 rats in each group.
P<0.05 compared with the control group
P<0.05 compared with the Cd group. Units of measurement are: for SOD, U/mg protein where 1 unit is equal to the amount of enzyme that causes a 50% suppression of the reduction of nitro blue tetrazolium/min/mg of protein; for CAT, U/mg protein where 1 unit corresponds to 1 nmol H2 consumed/min/mg protein; for GPx, nmol GSH oxidized/min/mg protein; and for GST, nmol GSH-CDNB conjugate formed/min/mg protein. Cd, cadmium; CA, chlorogenic acid; SOD, superoxide dismutase; CAT, catalase; GPx, glutathione peroxidase; GST, glutathione S-transferase; GSH, reduced glutathione; CDNB, 1-chloro-2,4-dinitrochlorobenzene.
Effects of CA and Cd on brain non-enzymatic antioxidant levels
Table II displays the levels of the non-enzymatic antioxidants GSH, vitamin C and vitamin E in the brains of control and experimental rats. The levels of GSH and of vitamins C and E were significantly (P<0.05) diminished in the brain tissues of Cd-treated rats when compared with control rats. Treatment with CA significantly (P<0.05) restored the depleted levels of GSH, vitamins C and E to near normal levels.
Table II.
Effect of Cd and CA on the levels of non-enzymatic antioxidants (µg/mg protein).
| Groups | GSH | Vitamin C | Vitamin E |
|---|---|---|---|
| Control | 5.82±0.48 | 2.7±0.19 | 2.8±0.24 |
| Cd (5 mg/kg) | 2.16±0.29a | 1.2±0.07a | 1.6±0.18a |
| CA (60 mg/kg) | 5.64±0.35b | 2.6±0.12 | 2.9±0.31 |
| Cd (5 mg/kg) + CA (60 mg/kg) | 6.12±0.51b | 2.1±0.12b | 2.3±0.21b |
Values are expressed as mean ± standard deviation for 6 rats in each group.
P<0.05 compared with the control group
P<0.05 compared with the Cd group. Cd, cadmium; CA, chlorogenic acid; GSH, reduced glutathione.
Effects of CA and Cd on brain lipid peroxidation
Fig. 2 depicts the level of MDA, an index of lipid peroxidation, in the brains of control and experimental rats. Cd-treated rats displayed a significant (P<0.05) increase in the level of MDA in comparison with control rats. However, treatment with CA significantly (P<0.05) diminished the degree of elevation of the MDA level in the brain when compared with that in the rats treated with Cd alone.
Figure 2.
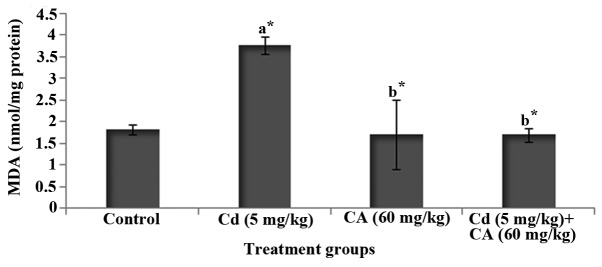
Levels of MDA in the brains of experimental rats. Values are expressed as mean ± standard deviation for 6 rats in each group. a*P<0.05 compared with the control group; b* P<0.05 compared with the Cd group. Cd, cadmium; CA, chlorogenic acid; MDA, malondialdehyde.
Effect of CA and Cd on membrane-bound ATPases
The results displayed in Table III are the changes in the activities of the ATPase enzymes Na+/K+-ATPase, Ca2+-ATPase and Mg2+-ATPase in the brains of control and experimental rats. The Cd-treated rats displayed significant (P<0.05) reductions in the activities of these ATPase enzymes on comparison with the control rats. However, treatment with CA significantly (P<0.05) increased the levels of the three ATPase enzymes in the brain when compared with those in the rats treated with Cd alone.
Table III.
Effect of Cd and CA on the levels of brain membrane ATPases (µmol phosphorus/min/mg protein).
| Groups | Na+/K+-ATPase | Mg2+-ATPase | Ca2+-ATPase |
|---|---|---|---|
| Control | 2.27±0.06 | 1.18±0.05 | 1.23±0.07 |
| Cd (5 mg/kg) | 1.18±0.05a | 0.38±0.07a | 0.53±0.07a |
| CA (60 mg/kg) | 2.21±0.04 | 1.07±0.05 | 1.11±0.05 |
| Cd (5 mg/kg) + CA (60 mg/kg) | 2.15±0.04b | 0.80±0.11b | 0.90±0.07b |
Values are expressed as mean ± standard deviation for 6 rats in each group.
P<0.05 compared with the control group
P<0.05 compared with the Cd group. Cd, cadmium; CA, chlorogenic acid.
Modulation of mitochondrial membrane integrity by CA and Cd
Table IV shows the activities of the citric cycle enzymes ICDH, α-KDH, SDH and MDH in the brain mitochondria of control and experimental rats. Significant reductions (P<0.05) in enzyme activities were brought about by Cd exposure in comparison with the control. However, treatment with CA restored the levels of mitochondrial enzymes to normal.
Table IV.
Effect of Cd and CA on the activities of brain mitochondrial citric acid cycle enzymes.
| Groups | SDH | MDH | α-KDH | ICDH |
|---|---|---|---|---|
| Control | 43.57±2.35 | 875.85±32.65 | 54.27±3.97 | 155.57±3.88 |
| Cd (5 mg/kg) | 12.65±4.55a | 553.43±26.38a | 23.43±1.83a | 99.41±6.25a |
| CA (60 mg/kg) | 43.94±1.47 | 870.05±31.41 | 52.84±2.73 | 160.64±5.58 |
| Cd (5 mg/kg) + CA (60 mg/kg) | 26.25±1.35b | 711.59±26.26b | 38.46±1.52b | 134.02±3.57b |
Units of measurement are: for SDH, nmol succinate oxidized/min/mg protein; for MDH, nmol NADH oxidized/min/mg protein; for KDH, nmol ferricyanide formed/h/mg protein; and for ICDH, nmol α-ketoglutarate formed/h/mg protein. Values are expressed as mean ± standard deviation for 6 rats in each group.
P<0.05 compared with the control group
P<0.05 compared with the Cd group. CD, cadmium; CA, chlorogenic acid; SDH, succinate dehydrogenase; MDH, malate dehydrogenase; α-KDH, α-ketoglutarate dehydrogenase; ICDH, isocitrate dehydrogenase.
Effects of CA and Cd on brain DNA fragmentation
Cd provoked oxidative DNA damage as demonstrated by agarose gel electrophoresis, as shown in Fig. 3. The results indicate that there was significant DNA fragmentation in the Cd-treated group compared with the control group. However, CA supplement restored the DNA integrity in comparison with that in the Cd-treated rats.
Figure 3.
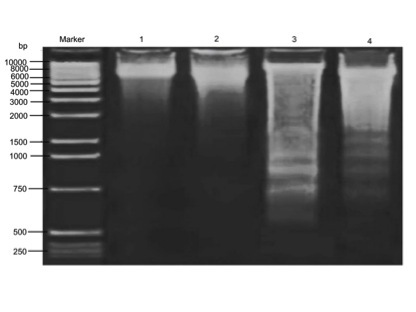
Effect of Cd and CA on brain DNA fragmentation. Marker, DNA molecular weight marker; lane 1, control; lane 2, Cd (5 mg/kg); lane 3, CA (60 mg/kg); lane 4: Cd (5 mg/kg) + CA (60 mg/kg). Cd, cadmium; CA, chlorogenic acid.
Discussion
The brain is a highly complex tissue that regulates an array of biological metabolic events and utilizes 20% of cardiac output. However, high concentrations of peroxidizable unsaturated lipids and minimal antioxidant defence render the brain more prone to noxious oxidative stress attack than the visceral vital tissues are (43). Mounting evidence substantiates the hypothesis that oxidative stress plays a pivotal role in the etiology of neurodegenerative diseases (44,45). The neurotoxic effects of Cd are mediated through the depletion of enzymatic antioxidants and a resultant increase in lipid peroxidation (5). Further, Cd exposure elicits modulation of thiol status, alteration of ion transport and ultimately DNA damage (46).
Research substantiates that alteration of the cholinergic transmission system is a hallmark of Cd neurotoxicity (47). AChE is a key biomarker detected in order to evaluate the neurotoxicity of heavy metals. There is evidence to suggest that free radicals provoked by heavy metals are responsible for diminished AChE activity in the brain (48). In the brain, low levels of AChE could lead to noxious events such as the accumulation of acetylcholine, which causes the rapid firing of neurons leading to convulsions and status epilepticus (49). The modulation of AChE in the brain displayed by Cd is concentration dependent; at a low concentration (0.01 mM) activation of AChE occurs whilst at higher concentrations (>0.1 mM) AChE activity is inhibited (7). Further, Cd induces a conformational change in AChE that leads to the enzyme becoming unreactive (50). In the present study toxic doses (5 mg/kg) of Cd were reflected by diminished levels of AChE and treatment with CA restored the activity of AChE in the brain. The neuroprotective effects that CA elicited in the present study may be due to antioxidant and free radical-scavenging actions. Furthermore, regulation of the ionic homeostasis imbalance in neurons and the highly lipophilic nature of CA may be responsible to its neuroprotective efficacy. Studies suggest that CA has the ability to cross the BBB so that it may effectively block the deleterious effects of free radicals within the brains vascular and cellular compartments (51,52).
In the oxidative stress cascade, lipid peroxidation is the chief culprit and plays an imperative role in the toxicity of many xenobiotics (53,54). In the present study, the oxidative insult of Cd increased the MDA level, an oxidative product of LPO in brain tissue. Previous studies suggest that Cd-induced lipid peroxidation is due to the generation of hydroxyl radicals, superoxide anions, nitric oxide and H2O2 (55–57). Indeed, in the present study, treatment with CA effectively reduced the MDA level and this may be due to the ability of CA to transfer electrons, scavenge free radicals, chelate metals and activate antioxidant enzymes. GSH, the first line of defense against reactive oxygen species (ROS), is a readily available source of endogenous sulfhydryl (-SH) groups. It has been demonstrated that Cd exposure causes a marked decline in GSH levels, which may be ascribed to direct conjugation with free or protein-bound -SH groups (58). Further, GSH depletion may be due to the inhibition of glutathione reductase, the enzyme responsible for the catalytic conversion of glutathione disulfide (GSSG) to GSH (59). Cd interferes with the -SH group of metallothionein, a metal chelating protein (60) and thus reduces the GSH level. Vitamin C is a vital antioxidant that also acts as an anti-stress factor. Vitamin E, a lipophilic chain-breaking antioxidant, also plays a critical role in the detoxification of Cd. The present study exemplifies the depletion of non-enzymatic antioxidant levels in the brain, which may be due to increased utilization of these biomolecules to reduce the Cd-induced oxidative stress. CA directly acts as a scavenger to inhibit the Cd-mediated lipid peroxidation and thus reduces the utilization of non-enzymatic antioxidants, consequently leading to improvements of GSH and vitamin C and E levels in the brain. The findings observed in the present study are consistent with previous findings (22).
Cells are equipped with an array of antioxidant defence mechanisms to counteract the effects of free radicals. The present study shows that the Cd-induced increase in lipid peroxidation was accompanied by a concomitant decline in the activities of the cellular enzymatic antioxidants SOD, CAT, GPx and GST. The diminished levels of antioxidant enzymes may be due to the interaction of Cd with the -SH groups of enzymes and the decreased activity of GPx may be due to competition by Cd-metallothioneins (61). Further, a number of studies have indicated that Cd inactivates a wide array of enzymes and proteins involved in the anti-stress mechanism (62–64). In one study, the administration of CA significantly elevated the enzymatic antioxidant status in Cd-intoxicated rats, an effect that may be due to its anti-lipid peroxidative potential (65).
Membrane-bound ATPase enzymes are highly vulnerable to peroxidation and the actions of LPOs. They are bound to the plasma membrane and are involved in the translocation of sodium, potassium, calcium and magnesium ions. In the brain, the active transport of sodium and potassium mediated by Na+/K+-ATPase generates membrane potentials. In addition, Na+/K+-ATPase regulates the uptake and release of catecholamines (66,67) and serotonin (68). Ca2+-ATPase modulates intracellular calcium levels (69), and the role of Mg2+-ATPase is to maintain high brain levels of intracellular magnesium ions to control the rates of protein synthesis and cell growth (70). Thus, ATPases are vital for neuronal functions and to maintain the resting membrane potential and nerve conduction. Previous studies indicate that Cd toxicity disrupts the activities of ATPase in CNS (47,71–73), reflecting the changes to membrane and neurotransmitter functions. The reduction of ATPase activity may result from the formation of Cd-ATPase complexes through -SH groups of the enzyme and/or increased oxidative stress. Treatment with CA increased the tissue levels of ATPases, suggesting that it protects the brain by preserving its structural integrity against Cd challenge (74).
Research into cell death has focused on mitochondria due to their role as the arbiters of cell fate in response to stress. Cd is known to cause stress to the mitochondria by affecting the thiol status and depleting ATP, thereby leading to a cascade of pathophysiological events that ultimately result in necrotic cell death (75). The citric acid cycle is an essential metabolic process that is also an integral part of the oxidative defense machinery of the cells. When KDH, ICDH, SDH and MDH are inhibited, the permeability of the inner mitochondrial membrane is increased, which causes mitochondrial dysfunction, and subsequently leads to oxidative brain injury. Notably, in the present study, poisoning with Cd decreased the levels of these mitochondrial marker enzymes, whilst treatment with CA significantly attenuated these effects, possibly as a result of mitochondrial membrane stabilization (76).
Finally, in the present study Cd elevated the extent of DNA fragmentation, possibly as a result of increased ROS production during heavy metal exposure. Treatment with the CD-treated rats with CA profoundly attenuated the toxicity by diminishing the level of DNA fragmentation, possibly by inhibiting the oxidative reactive radicals induced by Cd. These results are consistent with a previous study (77).
In conclusion, the present study provides evidence for the cytoprotective potential of CA against Cd-provoked derangement of brain homeostasis. The neuroprotective effects of CA are mediated through the inhibition of lipid peroxidation and by supporting the endogenous antioxidant defense systems in brain with the subsequent restoration of AChE and membrane-bound ATPase enzyme activities, and prevention of mitochondrial dysfunction and DNA fragmentation. Recently, it was demonstrated that Cd-induced neuronal cell death involves the downregulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and the activation of phosphoinositide 3′-kinase/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) (78). This finding may serve as a basis for future research to explore the effect of CA on the signaling mechanisms that mediate the negative effects of Cd.
References
- 1.Brender JD, Suarez L, Felkner M, et al. Maternal exposure to arsenic, cadmium, lead, and mercury and neural tube defects in offspring. Environ Res. 2006;101:132–139. doi: 10.1016/j.envres.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Jadhav SH, Sarkar SN, Patil RD, Tripathi HC. Effects of subchronic exposure via drinking water to a mixture of eight water contaminating metals: a biochemical and histopathological study in male rats. Arch Environ Contam Toxicol. 2007;53:667–677. doi: 10.1007/s00244-007-0031-0. [DOI] [PubMed] [Google Scholar]
- 3.Nawrot T, Plusquin M, Hogervorst J, et al. Environmental exposure to cadmium and risk of cancer: a prospective population-based study. Lancet Oncol. 2006;7:119–126. doi: 10.1016/S1470-2045(06)70545-9. [DOI] [PubMed] [Google Scholar]
- 4.Fowler BA. Monitoring of human populations forearly markers of cadmium toxicity; a review. Toxicol Appl Pharmacol. 2009;238:294–300. doi: 10.1016/j.taap.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Shukla A, Shukla GS, Srimal RC. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum Exp Toxicol. 1996;15:400–405. doi: 10.1177/096032719601500507. [DOI] [PubMed] [Google Scholar]
- 6.Abu-Taweel GM, Ajarem JS, Ahmad M. Protective effect of curcumin on anxiety, learning behavior, neuromuscular activities, brain neurotransmitters and oxidative stress enzymes in cadmium intoxicated mice. Behav Brain Sci. 2013;3:74–84. doi: 10.4236/jbbs.2013.31008. [DOI] [Google Scholar]
- 7.Carageorgiou H, Tzotzes V, Pantos C, Mourouzis C, Zarros A, Tsakiris S. In vivo and in-vitro effects of cadmium on adult rat brain total antioxidant status, acetylcholinesterase, (Na+, K+)-ATPase and Mg2+-ATPase activities: protection by L-cysteine. Basic Clin Pharmacol Toxicol. 2004;94:112–118. doi: 10.1111/j.1742-7843.2004.pto940303.x. [DOI] [PubMed] [Google Scholar]
- 8.Lopez E, Arce C, Oset-Gasque MJ, Cañadas S, González MP. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radical Biol Med. 2006;40:940–951. doi: 10.1016/j.freeradbiomed.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 9.Kasprzak KS. Oxidative DNA and protein damage in metal-induced toxicity and carcinogenesis. Free Radical Biol Med. 2002;32:958–967. doi: 10.1016/S0891-5849(02)00809-2. [DOI] [PubMed] [Google Scholar]
- 10.Kasai H, Fukada S, Yamaizumi Z, Sugie S, Mori H. Action of chlorogenic acid in vegetables and fruits as an inhibitor of 8-hydroxydeoxyguanosine formation in vitro and in a rat carcinogenesis model. Food Chem Toxicol. 2000;38:467–471. doi: 10.1016/S0278-6915(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki A, Yamamoto N, Jokura H, et al. Chlorogenic acid attenuates hypertension and improves endothelial function in spontaneously hypertensive rats. J Hypertens. 2006;24:1065–1073. doi: 10.1097/01.hjh.0000226196.67052.c0. [DOI] [PubMed] [Google Scholar]
- 12.Shibata H, Sakamoto Y, Oka M, Kono Y. Natural antioxidant, chlorogenic acid, protects against DNA breakage caused by monochloramine. Biosci Biotechnol Biochem. 1999;63:1295–1297. doi: 10.1271/bbb.63.1295. [DOI] [PubMed] [Google Scholar]
- 13.Gonthier MP, Verny MA, Besson C, Rémésy C, Scalbert A. Chlorogenic acid bioavailability largely depends on its metabolism by the gut microflora in rats. J Nutr. 2003;133:1853–1859. doi: 10.1093/jn/133.6.1853. [DOI] [PubMed] [Google Scholar]
- 14.Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Biol Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 15.Cheng JC, Dai F, Zhou B, Yang L, Liu ZL. Antioxidant activity of hydroxycinnamic acid derivatives in human low density lipoprotein: mechanism and structure-activity relationship. Food Chem. 2007;104:132–139. doi: 10.1016/j.foodchem.2006.11.012. [DOI] [Google Scholar]
- 16.López-Giraldo LJ, Laguerre M, Lecomte J, et al. Kinetic and stoichiometry of the reaction of chlorogenic acid and its alkyl esters against the DPPH radical. J Agric Food Chem. 2009;57:863–870. doi: 10.1021/jf803148z. [DOI] [PubMed] [Google Scholar]
- 17.Sato Y, Itagaki S, Kurokawa T, et al. In vitro and in vivo antioxidant properties of chlorogenic acid and caffeic acid. Int J Pharm. 2011;403:136–138. doi: 10.1016/j.ijpharm.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 18.Wang GF, Shi LP, Ren YD, et al. Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antiviral Res. 2009;83:186–190. doi: 10.1016/j.antiviral.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Gordon MH, Wishart K. Effects of chlorogenic acid and bovine serum albumin on the oxidative stability of low density lipoproteins in vitro. J Agric Food Chem. 2010;58:5828–5833. doi: 10.1021/jf100106e. [DOI] [PubMed] [Google Scholar]
- 20.Tang YZ, Liu ZQ. Chemical kinetic behavior of chlorogenic acid in protecting erythrocyte and DNA against radical-induced oxidation. J Agric Food Chem. 2008;56:11025–11029. doi: 10.1021/jf802462h. [DOI] [PubMed] [Google Scholar]
- 21.Nakajima Y, Shimazawa M, Mishima S, Hara H. Water extract of propolis and its main constituents, caffeoylquinic acid derivatives, exert neuroprotective effects via antioxidant actions. Life Sci. 2007;80:370–377. doi: 10.1016/j.lfs.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Shi W, Li Y, et al. Neuroprotective effects of chlorogenic acid against apoptosis of PC12 cells induced by methylmercury. Environ Toxicol Pharmacol. 2008;26:13–21. doi: 10.1016/j.etap.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 23.Vardi N, Parlakpinar H, Ates B. Beneficial effects of chlorogenic acid on methotrexate-induced cerebellar Purkinje cell damage in rats. J Chem Neuroanat. 2012;43:43–47. doi: 10.1016/j.jchemneu.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Johnson D, Lardy H. Isolation of liver or kidney mitochondria. Methods Enzymol. 1967;10:94–96. doi: 10.1016/0076-6879(67)10018-9. [DOI] [Google Scholar]
- 25.Ellman GL, Courtney KD, Andres VJ, Jr, Feather-stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmcol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 26.Kakkar P, Das B, Viswanathan PN. A modified spectrophotometric assay of superoxide dismutase. Ind J Biochem Biophys. 1984;21:130–132. [PubMed] [Google Scholar]
- 27.Sinha AK. Colorimetric assay of catalase. Anal Biochem. 1972;47:389–394. doi: 10.1016/0003-2697(72)90132-7. [DOI] [PubMed] [Google Scholar]
- 28.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 29.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 30.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 31.Moron MS, Depierre JW, Mannervik B. Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochim Biophys Acta. 1979;582:67–78. doi: 10.1016/0304-4165(79)90289-7. [DOI] [PubMed] [Google Scholar]
- 32.Omaye ST, Turnbull JD, Sauberlich HE. Selected methods for the determination of ascorbic acid in animal cells, tissues and fluids. Methods Enzymol. 1979;62:3–8. doi: 10.1016/0076-6879(79)62181-X. [DOI] [PubMed] [Google Scholar]
- 33.Desai ID. Vitamin E analysis methods for animal tissues. Methods Enzymol. 1984;105:138–147. doi: 10.1016/S0076-6879(84)05019-9. [DOI] [PubMed] [Google Scholar]
- 34.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxidation in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 35.Bonting SL. Presence of enzyme systems in mammalian tissues. In: Bilter EE, editor. Membrane and Ion Transport. Wiley Interscience; London: 1970. pp. 257–263. [Google Scholar]
- 36.Ohinishi T, Suzuki T, Suzuki Y, Ozawa K. A comparative study of plasma membrane Mg2+ ATPase activities in normal, regenerating and malignant cells. Biochem Biophys Acta. 1982;684:67–74. doi: 10.1016/0005-2736(82)90050-5. [DOI] [PubMed] [Google Scholar]
- 37.Hjertan S, Pan H. Purification and characterization of two forms of a low affinity Ca2+-ATPase from erythrocyte membranes. Biochim Biophys Acta. 1983;728:281–288. doi: 10.1016/0005-2736(83)90480-7. [DOI] [PubMed] [Google Scholar]
- 38.Reed LJ, Mukherjee BB. α-ketoglutarate dehydrogenase complex from Escherichia coli. Methods Enzymol. 1969;113:55–61. doi: 10.1016/0076-6879(69)13016-5. [DOI] [Google Scholar]
- 39.King J, editor. Practical Clinical Enzymology. Van D Nostrand Co.; London: 1965. The hydrolases - acid and alkaline phosphatases; pp. 199–208. [Google Scholar]
- 40.Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem. 2001;276:23357–23361. doi: 10.1074/jbc.M100320200. [DOI] [PubMed] [Google Scholar]
- 41.Mehler AH, Kornberg A, Grisolia S, Ochoa S. The enzymatic mechanism of oxidation-reductions between malate or isocitrate and pyruvate. J Biol Chem. 1948;174:961–977. [PubMed] [Google Scholar]
- 42.Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J Biol Chem. 1996;271:14067–14072. doi: 10.1074/jbc.271.24.14067. [DOI] [PubMed] [Google Scholar]
- 43.Bondy SC. Free-radical-mediated toxic injury to the nervous system. In: Wallace KB, editor. In: Free Radical Toxicology. Taylor and Francis; Oxford: 1997. pp. 221–248. [Google Scholar]
- 44.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58:39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010. doi: 10.1155/2012/428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar R, Agarwal AK, Seth PK. Oxidative stress-mediated neurotoxicity of cadmium. Toxicol Lett. 1996;89:65–69. doi: 10.1016/S0378-4274(96)03780-0. [DOI] [PubMed] [Google Scholar]
- 47.Antonio MT, Corredor L, Leret ML. Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicol Lett. 2003;143:331–340. doi: 10.1016/S0378-4274(03)00194-2. [DOI] [PubMed] [Google Scholar]
- 48.Tsakiris S, Angelogianni P, Schulpis KH, Starridis JC. Protective effect of L-phenylalanine on rat brain acetylcholinesterase inhibition induced by free radicals. Clin Biochem. 2000;33:103–106. doi: 10.1016/S0009-9120(99)00090-9. [DOI] [PubMed] [Google Scholar]
- 49.Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 1986;44:857–877. [PubMed] [Google Scholar]
- 50.Tomlinson G, Mutus B, McLennan I. Activation and inactivation of acetylcholinesterase by metal ions. Can J Biochem. 1981;59:728–735. doi: 10.1139/o81-101. [DOI] [PubMed] [Google Scholar]
- 51.Chu YF, Brown PH, Lyle BJ, et al. Roasted coffees high in lipophilic antioxidants and chlorogenic acid lactones are more neuroprotective than green coffees. J Agric Food Chem. 2009;57:9801–9808. doi: 10.1021/jf902095z. [DOI] [PubMed] [Google Scholar]
- 52.Lapchak PA. The phenylpropanoid micronutrient chlorogenic acid improves clinical rating scores in rabbits following multiple infarct ischemic strokes: synergism with tissue plasminogen activator. Exp Neurol. 2007;205:407–413. doi: 10.1016/j.expneurol.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 53.Stohs SJ, Bagchi D. Oxidative Mechanisms in the toxicity of metal ions. Free Radical Biology and Medi. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-H. [DOI] [PubMed] [Google Scholar]
- 54.Anane R, Creppy EE. Lipid peroxidation as pathway of aluminium cytotoxicity in human skin fibroblast cultures: prevention by superoxide dismutase+catalase and vitamins E and C. Hum Exp Toxicol. 2001;2:477–481. doi: 10.1191/096032701682693053. [DOI] [PubMed] [Google Scholar]
- 55.O'Brien P, Salasinski HJ. Evidence that the reactions of cadmium in the presence of metallothionein can produce hydroxyl radicals. Arch Toxicol. 1998;72:690–700. doi: 10.1007/s002040050562. [DOI] [PubMed] [Google Scholar]
- 56.Koizumi T, Shirakura G, Kumagai H, Tatsumoto H, Suzuki KT. Mechanism of cadmium-induced cytotoxicity in rat hepatocytes: cadmium-induced active oxygen-related permeability changes of the plasma membrane. Toxicology. 1996;114:124–134. doi: 10.1016/S0300-483X(96)03477-4. [DOI] [PubMed] [Google Scholar]
- 57.Tandom SK, Singh S, Prasad S, et al. Reversal of cadmium induced oxidative stress by chelating agent, antioxidant or their combination in rat. Toxicol Lett. 2003;145:211–217. doi: 10.1016/S0378-4274(03)00265-0. [DOI] [PubMed] [Google Scholar]
- 58.Rana SV, Verma S. Protective effects of GSH, vitamin E and selenium on lipid peroxidation in cadmium-fed rats. Biol Trace Elem Res. 1996;51:161–168. doi: 10.1007/BF02785435. [DOI] [PubMed] [Google Scholar]
- 59.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 60.Hidalgo J, Aschner M, Zatta P, Vasák M. Roles of the metallothionein family of proteins in the central nervous system. Brain Res Bull. 2001;15:133–145. doi: 10.1016/S0361-9230(01)00452-X. [DOI] [PubMed] [Google Scholar]
- 61.Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/S0300-483X(03)00305-6. [DOI] [PubMed] [Google Scholar]
- 62.Jamall IS, Sprowls JJ. Effects of cadmium and dietary selenium on cytoplasmic and mitochondrial antioxidant defense systems in the heart of rats fed high dietary copper. Toxicol Appl Pharmacol. 1987;87:102–110. doi: 10.1016/0041-008X(87)90088-3. [DOI] [PubMed] [Google Scholar]
- 63.Sarkar S, Yadov P, Bhatnagar D. Lipid peroxidative damage on cadmium exposure and alterations in antioxidant system in rat erythrocytes: A study with relation to time. Biometals. 1998;11:153–157. doi: 10.1023/A:1009286130324. [DOI] [PubMed] [Google Scholar]
- 64.Casalino E, Calzaretti G, Sblano C, Landriscina C. Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology. 2002;179:37–50. doi: 10.1016/S0300-483X(02)00245-7. [DOI] [PubMed] [Google Scholar]
- 65.Pari L, Karthikesan K, Menon VP. Comparative and combined effect of chlorogenic acid and tetrahydrocurcumin on antioxidant disparities in chemical induced experimental diabetes. Mol Cell Biochem. 2010;341:109–117. doi: 10.1007/s11010-010-0442-5. [DOI] [PubMed] [Google Scholar]
- 66.Bogdanski DF, Tissuri A, Brodie BB. Role of sodium, potassium, ouabain and reserpine in uptake, storage and metabolism of biogenic amines in synaptosomes. Life Sci. 1968;7:419–428. doi: 10.1016/0024-3205(68)90013-1. [DOI] [PubMed] [Google Scholar]
- 67.Mata M, Fink DJ, Gainer H, et al. Activity-dependent energy metabolism in rat posterior pituitary, primarily reflects sodium pump activity. J Neurochem. 1980;34:214–215. doi: 10.1111/j.1471-4159.1980.tb04643.x. [DOI] [PubMed] [Google Scholar]
- 68.Hernandez R. Brain Na+, K+-ATPase activity possibly regulated by a specific serotonin receptor. Brain Res. 1989;408:399–402. doi: 10.1016/0006-8993(87)90414-8. [DOI] [PubMed] [Google Scholar]
- 69.Skalska J, Bednarczyk P, Piwońska M, et al. Calcium ions regulate K+ uptake into brain mitochondria: The evidence for a novel potassium channel. Int J Mol Sci. 2009;10:1104–1120. doi: 10.3390/ijms10031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanui H, Rubin H. The role of magnesium in cell proliferation and transformation. In: Boynton AL, McKochan WL, Whitfield JP, editors. Ions, Cell Proliferation and Cancer. Academic Press; New York: 1982. pp. 517–537. [Google Scholar]
- 71.Rajanna B, Hobson M, Boykin M, Chetty CS. Effects of chronic treatment with cadmium on ATPases, uptake of catecholamines and lipid peroxidation in rat brain synaptosomes. Ecotoxicol Environ Saf. 1990;20:36–41. doi: 10.1016/0147-6513(90)90044-6. [DOI] [PubMed] [Google Scholar]
- 72.Carfagna MA, Ponsler GD, Muhoberac BB. Inhibition of ATPase activity in rat synaptic plasma membranes by simultaneous exposure to metals. Chem Biol Interact. 1996;100:53–65. doi: 10.1016/0009-2797(95)03685-7. [DOI] [PubMed] [Google Scholar]
- 73.El-Missiry MA, Shalaby F. Role of beta-carotene in ameliorating the cadmium-induced oxidative stress in rat brain and testis. J Biochem Mol Toxicol. 2000;14:238–243. doi: 10.1002/1099-0461(2000)14:5<238::AID-JBT2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 74.Lee K, Lee JS, Jang HJ, et al. Chlorogenic acid ameliorates brain damage and edema by inhibiting matrix metalloproteinase-2 and 9 in a rat model of focal cerebral ischemia. Eur J Pharmacol. 2012;689:89–95. doi: 10.1016/j.ejphar.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 75.Nigam D, Shukla GS, Agarwal AK. Glutathione depletion and oxidative damage in mitochondria following exposure to cadmium in rat liver and kidney. Toxicol Lett. 1999;106:151–157. doi: 10.1016/S0378-4274(99)00059-4. [DOI] [PubMed] [Google Scholar]
- 76.Ho L, Varghese M, Wang J, et al. Dietary supplementation with decaffeinated green coffee improves diet-induced insulin resistance and brain energy metabolism in mice. Nutr Neurosci. 2012;15:37–45. doi: 10.1179/1476830511Y.0000000027. [DOI] [PubMed] [Google Scholar]
- 77.Xu JG, Hu QP, Liu Y. Antioxidant and DNA-protective activities of chlorogenic acid isomers. J Agric Food Chem. 2012;60:11625–11630. doi: 10.1021/jf303771s. [DOI] [PubMed] [Google Scholar]
- 78.Chen S, Gu C, Xu C, et al. Celastrol prevents cadmium-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. J Neurochem. 2014;128:256–266. doi: 10.1111/jnc.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]