

Abstract
L-type amino acid transporter 1 (LAT1), overexpressed on the membrane of various tumor cells, is a potential target for tumor-targeting therapy. This study aimed to develop a LAT1-mediated chemotherapeutic agent. We screened doxorubicin modified by seven different large neutral amino acids. The aspartate-modified doxorubicin (Asp-DOX) showed the highest affinity (Km = 41.423 μmol/L) to LAT1. Aspartate was attached to the N-terminal of DOX by the amide bond with a free carboxyl and a free amino group on the α-carbon atom of the Asp residue. The product Asp-DOX was characterized by HPLC/MS. In vitro, Asp-DOX exerted stronger inhibition on the cancer cells overexpressing LAT1 and the uptake of Asp-DOX was approximately 3.5-fold higher than that of DOX in HepG2 cells. Pharmacokinetic data also showed that Asp-DOX was expressed over a longer circulation time (t1/2 = 49.14 min) in the blood compared to DOX alone (t1/2 = 15.12 min). In HepG2 and HCT116 tumor-bearing mice, Asp-DOX achieved 3.1-fold and 6.4-fold accumulation of drugs in tumor tissue, respectively, than those of the unmodified DOX. More importantly, treatment of tumor-bearing mice with Asp-DOX showed a significantly stronger inhibition of tumor growth than mice treated with free DOX in HepG2 tumor models. Furthermore, after Asp modification, Asp-DOX avoided MDR mediated by P-glycoprotein. These results suggested that the Asp-DOX modified drug may provide a new treatment strategy for tumors that overexpress LAT1 and MDR1.
Keywords: Anticancer drugs, Asp-DOX, HepG2, LAT1, P-glycoprotein
Amino acid transport across the plasma membrane is mediated by amino acid transporters located on the plasma membrane. L-type amino acid transporter 1 is an Na-independent neutral amino acid transporter. It preferentially transports large neutral amino acids such as leucine, isoleucine, valine, phenylalanine, tyrosine, tryptophan, methionine, and histidine in a Na-independent manner.1–3 Because of its broad substrate selectivity, LAT1 transports not only naturally occurring amino acids but also amino acid-related compounds such as L-dopa, melphalan, triiodothyronine, thyroxine, gabapentin, and S-(1,2-dichlorovinyl)-L-cysteine.4 Recently, it was determined that LAT1 was overexpressed primarily in various cancer cells, suggesting that LAT1 has important prognostic significance and could be a potential target in tumor therapy.5,6 Doxorubicin is a highly potent chemotherapeutic agent used to treat various cancers. However, its short half-life and low selective cytotoxicity limit its application in tumor therapy.7,8 In addition, many cancer cells express P-gp, encoded by MDR1 and localized to chromosome 7q21.P-g, possessing resistance to DOX.7,9 These defects decreased the therapeutic effect of DOX.10–12 A promising solution would be to improve the selective cytotoxicity of DOX to cancer cells for ligand-targeted therapeutics.13,14
We modified DOX on its free amidogen, −NH3, with different amino acids (as shown in Table1) to synthesize new small-molecule compounds to be transported by LAT1 into cancer cells. After screening in the LAT1 overexpressing cell line from mice with renal proximal convoluted tubules (S2-LAT1), Asp-DOX, with a free α-carboxyl group and a free α-amino group characterized by HPLC/MS, was determined to be an active and efficient formulation.
Table 1.
Different amino acid modified formations and their inhibition of the uptake of L-[H3] leucine by L-type amino acid transporter 1
| Doxorubicin | Amino acid | X | IC50 |
|---|---|---|---|
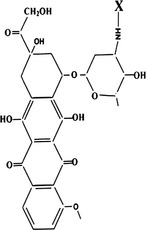 |
Leu- | 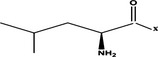 |
>100 μM |
| Val- | 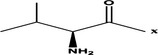 |
>100 μM | |
| Asp- |  |
30.12 μM | |
| Met- | 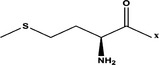 |
>100 μM | |
| Phe- | 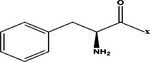 |
>100 μM | |
| Ile- | 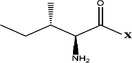 |
>100 μM | |
| Tyr- | 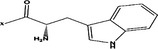 |
>100 μM |
In this study, we examined the selective cytotoxicity of Asp-DOX in vitro and in vivo. We detected the excretion rate of Asp-Dox in the MDR1-overexpressed MDCK cell line. The pharmacokinetics and biodistribution of Asp-DOX were also evaluated.
Materials and Methods
Materials
Hydroxybenzotriazole, 2-(7-Aza-1H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium hexafluorophosphate, and diisopropylethylamine were purchased from Sigma (St. Louis, MO, USA). PdCl2(PPh3)2 and Bu3SnH were provided by J&K Scientific (Beijing, China); the MTT one-step assay kit based on MTT reduction assay was provided by Hope Biotechnology (Tianjin, China). HepG2, HCT116, MCF7, and H1299 cell lines were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China) and identified by the National Cell Resource Center (Shanghai, China). Parent and MDR1 transfected MDCK cells were obtained from the State Key Laboratories of Drug Delivery Technology and Pharmacokinetics (Tianjin, China). Doxorubicin was purchased from Lark Technology (China, Beijing). Fmoc-Asp-O-allyl, Fmoc-Leu-OH, Fmoc-Met-OH, Fmoc-Phe-OH, Fmoc-Tyr-OH, Fmoc-Val-OH, and Fmoc-Ile-OH were all obtained from GL Biochem (Shanghai, China). All chemicals used for tissue culture were purchased from Gibco BRL (Grand Island, NY, USA).
Synthesis of amino acid-modified DOX
The modification of different amino acids is shown in Table1. First, 1 mmol Fmoc-Asp-O-allyl, Fmoc-Leu-OH, Fmoc-Met-OH, Fmoc-Phe-OH, Fmoc-Tyr-OH, Fmoc-Ile-OH, and Foc-Val-OH was added to 5 mL DMF (Dimethyl Formamide) in a reaction vessel placed in an ice/salt bath. Then 1.2 mmol 2-(7-Aza-1H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium hexafluorophosphate, 1.2 mmol hydroxybenzotriazole, and 1.2 mmol diisopropylethylamine were successively added into the reaction vessel. The reaction was allowed to proceed for 10 h with stirring. At the end of the reaction, the reaction mix was stopped, purified, and determined by HPLC/MS.
The procedure for deprotection of the -allyl group on the molecules with the presence of PdCl2 (PPh3)2, AcOH, and Bu3SnH in dichloromethane was described previously.15 The 20% hexahydropyridine in methanol was used for the deprotection of the Fmoc group.16 Methanol and hexahydropyridine were removed by the rotary evaporation method. The synthesis procedure of Asp-DOX is shown in Figure S1. The final product was purified by the HPLC method as previously described, and characterized and determined by MS as shown in Figures S2 and S3.
Inhibition experiment
For the inhibition of L-leucine, the uptake of 2 μmol/L L-[3H] leucine by the S2-LAT1 cell lines expressing LAT1 and 4F2hc was measured in the presence or absence of 100 μmol/L non-labeled test compounds, unless otherwise indicated. The IC50 values of the tested compounds were determined based on the LAT1-mediated 2 μmol/L L-[3H] leucine uptake measured at 1, 3, 10, 30, 100, and 300 μmol/L Asp-DOX. LAT1-mediated amino acid uptake was calculated as the difference between the mean of uptake into the S2-LAT1 cells that overexpressed LAT1 and 4F2hc. The procedure of the uptake of L-[3H] leucine was carried out as previously described.4 The S2-LAT1 cell line was pre-incubated for 10 min in DPBS and then incubated in the uptake solution (DPBS) containing 2 μmol/L L-[3H] leucine (2 μCi/mL) with or without the tested compounds for 2 min. The uptake solution was removed and the S2-LAT1 cells were then washed three times with ice-cold DPBS and were treated with 0.1 mol/L sodium hydroxide solution. After that, 400 μL cell lysis was transferred to scintillation vials and solubilized with scintillation solution. The radioactivity in the cells was measured by a liquid scintillation counter (PerkinElmer, Waltham, Mass. USA). For the inhibition of DOX or Asp-DOX, the uptake of 20 μmol/L DOX or Asp-DOX by S2-LAT1 cells was determined with or without the absence of 100 μmol/L of different amino acids (Leu, Met, Phe, Asp, Glu, and Gly) for 2 min. The uptake of DOX or Asp-DOX was stopped by washing in cold DPBS three times. Doxorubicin or Asp-DOX internalized into the cells was extracted by methanol and was determined by HPLC method as follows. The values were expressed as the percentage increase in radioactivity compared to the controls (mean values of measurements).
For the determination of Km of Asp-DOX, S2-LAT1 cells were incubated with different concentrations of Asp-DOX (1, 3, 10, 30, 100, and 300 μmol/L). The method of Eadie–Hofstee linear regression was used to draw the Michaelis–Menten equation to obtain the Km.
Cytotoxicity of Asp-DOX
The cytotoxicity of Asp-DOX or the control drug DOX to H1299 (human non-small cell lung carcinoma cell line, LAT1- and MDR1-negative), HCT116 (human colon cancer) expressing LAT1 but not MDR1, and MCF7 (human breast cancer) and HepG2 (human hepatic cancer) cells expressing both LAT1 and MDR1 were assessed using the one-step MTT assay kit. Briefly, all cancer cells were cultured in the cell culture discs (10 cm diameter) with either DMEM or RPMI-1640 mediums supplemented with 10% FBS, 100 IU/mL penicillin, and 0.05 mg/mL streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. After one or two generations, the cells were inoculated in 96-well plates. Each well of the 96-well plates was seeded with 2000–4000 cells and incubated for 48 h. The cells were then exposed to either DOX or Asp-DOX at different concentrations (10, 20, or 50 μg/mL). After being cultured for 48 h, 20 μL MTT solution per 100 μL (5 mg/mL) was added to each well, followed by incubation for another 2 h. Eventually, the absorbance of the reaction liquid was measured on a Sunrise Absorbance Microplate Reader (Tecan, Grödig, Austria) at wavelength of 492 nm. The experiment was repeated six times. The data are presented as mean values of measurements. The inhibition of DOX and Asp-DOX on the growth of cancer cells was described by the IR as follows:17
| 1 |
Cellular uptake of free DOX and Asp-DOX in vitro
Cellular uptake of free DOX and Asp-DOX was investigated using a fluorescence microscope (Nikon, Tokyo, Japan) with an excitation wavelength of 475 nm and absorption wavelength of 580 nm to observe the accumulation in the intracellular distribution of DOX as previously described.10 Cancer cell lines HepG2 and H1299 were selected as two different models and seeded into 24-well plates at 1.5 × 105/well. After 48 h, either free DOX or Asp-DOX at concentrations of 0, 10, 20, or 50 μg/mL, dissolved in serum-free medium, were added (500 μL/well). The treated cells were then incubated for 2 h at 37°C in 5% CO2. The medium was then removed and cells were rinsed three times with cold DPBS. Intrinsic fluorescence of drugs was observed using a fluorescence microscope. Four hundred microliters of methanol was added to the cells treated by different drugs. The mixture was then treated by sonication for 10 min to determine the uptake of drugs into cancer cells. Fifty microliters of supernatant was analyzed by HPLC/MS methods as follows.
Bidirectional transport assay
The MDCK-MDR1 cells were cultured in high glucose DMEM supplemented with 10% FBS, penicillin (100 IU/mL) and streptomycin (0.05 mg/mL). Cells were cultured at 37°C in an atmosphere of 5% CO2 and 95% relative humidity. For the transport assay, the cells were seeded at a density of 2 × 105 cells per 1.12 cm2 on Costar Transwell inserts (polycarbonate, 12-mm diameter, 0.4-μm pore size) and used for assays after approximately 7 days' incubation, during which the medium was changed every 3 days.
The permeability of the test compounds was measured by a Transwell assay as previously described.18,19 For permeability measurements, inserts with the endothelial monolayer were placed into 12-well plates containing HBSS with 25 mM HEPES. The efflux rate (PDR) of the test drugs was determined in the A–B and B–A directions in a shaker at 37°C with gentle shaking (60 round/min). For the A–B direction, the mediums in both compartments of the Transwell were replaced by HBSS and incubated at 37°C and 5% CO2 for 20 min. At time zero, the HBSS solution of the upper compartment was removed and replaced by 0.5 mL the same buffer containing either 50 μg/mL DOX or Asp-DOX; 100 μL solution in the lower compartment was sampled after 60 min for determining concentration. For the B–A direction, a similar procedure was carried out, except that HBSS in the bottom compartment was removed and replaced by 1.5 mL same buffer with corresponding drugs. Before transport studies, transepithelial electric resistance was measured (EVOM-2; WPI, Sarasota, FL, USA). The concentration of test compounds in the samples was determined by a Shimadzu HPLC system with a fluorescence detector (Shimadzu, Kyoto, Japan). Apparent permeability was calculated with the following equation.
| 2 |
where dQ /dt was the rate of permeability, C0 was the initial concentration in the donor compartment, and A was the surface area of the filter (1.12 cm2). The efflux ratio (PDR) was calculated, as the ratio Papp (B – A) : Papp (A – B).18 A compound with a ratio >2.0 was qualified as a substrate of the efflux mechanism.
Pharmacokinetic and biodistribution experiments
This study was carried out in strict accordance with the recommendations in the Directive 2010/63/EU revising Directive 86/609/EEC on the protection of animals used for scientific purposes. The protocol was approved by the Committee on the Ethics of Animal Experiments of Tianjin City (Permit Number: 27-2956). All surgery was carried out under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
The experiment was carried out in healthy mice. The compounds were injected through the tail vein at a dose of 5 mg/kg body weight. Blood samples were collected over a timespan of 4 h post-injection (0, 2 min, 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, and 4 h) by retrobulbar puncture.20 The blood samples were immediately mixed with 250 μL 0.5 mM EDTA-PBS and then centrifuged at 1750 g for 15 min. Supernatants (200 μL) were sampled and drugs were extracted from supernatants by incubation with 1 mL methanol, vortexed for 1 min and centrifuged at 1750 g. The extraction (approximately 1 mL) was dried by nitrogen and redissolved by 200 μL mobile phase. The concentrations of either DOX or Asp-DOX in extraction were analyzed by HPLC. Plasma drug concentration versus time was plotted and calculation of AUC0−t was carried out in the WinNonLin 4.0.1 program (Pharsight, Mountain View, CA, USA) by a non-compartment model 201 (i.v. bolus administration). Pharmacokinetic data are expressed as the concentration (μg/mL) remaining in blood at the various time points post-injection.21
For the biodistribution study, eight groups of ALB/c-nu male nude mice weighing in the range 18–22 g (Vital River Laboratory Animal Technology, Beijing, China) were injected i.p. with cancer cell lines CT116, HepG2, and H1299 to establish cancer models as previously described.18 In brief, nude ALB/c-nu mice (4 weeks old) were housed in barrier facilities in a 12:12-h light : dark cycle. Food and water were supplied ad libitum. On day 0, 1.0 × 107 of H1299, HepG2, and HCT116 cells in 0.1 mL PBS were implanted s.c. just above the right femoral joint armpit (3–5 animals for each tumor cell line). On day 20 after tumor cell injection, when the mean tumor volume reached approximately 100 mm3, free DOX and Asp-DOX were injected i.v. at a dose of 5 mg/kg. One hour after injection, the mice were killed by diethyl ether and cervical dislocation and tissue samples were collected and frozen in liquid nitrogen. All samples were kept frozen until extraction. Biodistribution was determined as previously described.21,22 Data are expressed as the percentage of injected dose/g tissue.
Animal models for therapy study
Animal models used for therapy study were carried out as previously described in a pharmacokinetics and biodistribution study. Animals bearing tumor cell lines HepG2 (LAT1-positive) or H1299 (LAT1-negative control) were divided into three groups. After the volume of tumors reached 100 mm3, the three groups were treated with saline, DOX, or Asp-DOX at a dose of 5 mg/kg by i.v. injection twice a week. The tumor size was measured twice a week. The volume of tumor was calculated by the formula: [length × (width)2/2].23
Extraction of drugs from tissues for HPLC/MS analysis
The drug extraction procedure was modified from those previously described.21 The tissue samples (100 mg each) were cut into small pieces. Saline (500 μL) and 100 μL of 2 mg/mL 1-naphthol (Sigma), as well as the internal standard, were added to all samples. Samples were then homogenized by a probe homogenizer (Sonifier 450; Branson Ultrasonics, Branson, Missouri USA). To extract DOX and Asp-DOX from the tissues, 1 mL methanol was added to 200 μL tissue homogenate and the mixture was vortexed for 1 min. Thereafter, the treated samples were centrifuged at 3000 g for 10 min at 4°C. The organic phase was separated and methanol treatment was repeated for the precipitate. The supernatant was collected and dried under a nitrogen stream. The dried samples were reconstituted with 200 μL mobile phase, vortexed for 1 min, and centrifuged for 5 min at 12 000 g at 4°C. After centrifugation, 40 μL supernatant was analyzed by HPLC/MS. The samples were kept at −80°C before HPLC analysis.
Analysis by HPLC and MS
Samples were analyzed by an HPLC/MS instrument and the products were collected at the retention time. The elution solutions were analyzed by a TSQ Quantum triple quadrupole tandem MS (Thermo Fisher Scientific, San Jose, California, USA) and an electrospray ionization source. A mixture of methanol/ammonium acetate/phosphoric acid (145/155/0.16, v/v) was used as mobile phase with a flow rate of 1 mL/min at 40°C. The Agilent C18 column (200 × 4.6 mm, 0.45 μm) was used. The injection volume was 40 μL. All measurements were carried out using the positive ion mode and MS conditions were optimized to obtain maximum sensitivity for DOX and Asp-DOX. The ion spray voltage was set at 4000 V, and the capillary temperature was set at 280°C. The sheath and auxiliary gas was nitrogen, with the pressure and flow rate of 35 psi and 5 L/min, respectively.
Statistical analysis
The independent samples t-test was used for comparisons by SPSS 15.0 software (SPSS, Chicago, IL, USA). Values of P < 0.05 were considered statistically significant.
Results
Inhibition of LAT1-mediated uptake by amino acid-related compounds
LAT1-mediated L-[3H] leucine uptake (2 μmol/L) was measured in the presence of 100 μmol/L concentrations of non-labeled compounds (Fig.1). As shown in Figure1(a), L-[3H] leucine uptake was markedly inhibited by Asp-DOX and Tyr-DOX, especially Asp-DOX, whereas the other compounds (Leu-DOX, Val-DOX, Met-DOX, Phe-DOX, and Ile-DOX) barely inhibited the uptake of L-[3H] leucine by S2-LAT1. Furthermore, in order to check the inhibition of Asp-DOX to the uptake of L-leucine, we inspected its IC50 (Fig.1b). Asp-DOX exerted high inhibition on the uptake of L-leucine and its IC50 to L-leucine was determined to be approximately 30.12 μmol/L. In order to check whether the internalization of Asp-DOX was mediated by LAT1, the inhibition to the uptake of Asp-DOX by different amino acids (Leu, Met, and Phe, substrates of LAT1; Ala, Glu, and Gly, non-substrates of LAT1) was determined with or without 100 μmol/L amino acids. The results showed that 100 μmol/L Leu, Met, and Phe strongly inhibited the uptake of Asp-DOX, whereas 100 μmol/L Ala, Glu, and Gly had no inhibition (Fig.1c). However, neither 100 μmol/L Leu, Met, or Phe, nor 100 μmol/L Ala, Glu, or Gly inhibited the uptake of DOX by S2-LAT1 cells (Fig.1d). In order to check the affinity of Asp-DOX to LAT1, the Michaelis–Menten curve was plotted. The results showed that the Km of Asp-DOX with LAT1 was determined as 41.423 μmol/L (Fig.1e). These results suggested that the internalization of Asp-DOX into S2-LAT1 cells was mainly mediated by LAT1.
Figure 1.

Inhibitory effects of different amino acid-modified compounds on the uptake of L-[H3] leucine by the S2-LAT1 transgene cell line. (a) Uptake of L-[3H] Leu (2 μmol/L) in S2-LAT1 with or without tested compounds at a concentration of 100 μmol/L (*P < 0.05; **P < 0.01). (b) Inhibition of aspartate-modified doxorubicin (Asp-DOX) at different concentrations on the uptake of L-[3H] Leu (2 μmol/L). (c, d) Inhibition of different amino acids (100 μmol/L) on the uptake of Asp-DOX (20 μmol/L) (c) or DOX (20 μmol/L) (d) by S2-LAT1 cells. (e) Curve of the Michaelis–Menten equation through Eadie–Hofstee linear regression to obtain the Michaelis constant (Km).
Cytotoxicity of Asp-DOX
The cytotoxic effects of free DOX and Asp-DOX against the LAT1-positive cell lines (HepG2, MCF7, and HCT116) and the LAT1-negative cell line (H1299) were evaluated using the MTT assay. The results are shown in Figure2 and Table2. The cytotoxicity of Asp-DOX to LAT1-positive cancer cells such as HCT116 (Fig.2a), HepG2 (Fig.2b), and MCF7 (Fig.2c) were apparently higher (P < 0.01) than that of free DOX, which suggested that LAT1 may have played an important role in the internalization of Asp-DOX into cancer cells, thus enhancing cytotoxicity by increasing the uptake of Asp-DOX into the overexpressed LAT1 cell lines. In contrast, the cytotoxicity of free DOX toward H1299 cells was much higher than that of Asp-DOX (Fig.2d). It can be inferred that the internalization of Asp-DOX was mainly mediated by the LAT1 transporter, and this inference was also supported by the inhibition results (Fig.1c,d).
Figure 2.

Cytotoxicity of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) in different cancer cell lines in vitro. Cytotoxicity levels of DOX and Asp-DOX to the LAT1-positive cell lines HCT116 (a), HepG2 (b), and MCF7 (c), and the LAT1-negative cell line H1299 (d) were determined by their inhibition of cancer cells through the MTT method.
Table 2.
Half maximal inhibitory concentration values of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) in cancer cell lines
| Drug | Cell line | IC50 |
|---|---|---|
| DOX | HCT116 | 12.54 μg/mL |
| HepG2 | >50.00 μg/mL | |
| MCF7 | >50.00 μmol/mL | |
| H1299 | 36.51 μg/ml | |
| Asp-DOX | HCT116 | <10.00 μg/mL |
| HepG2 | <10.00 μg/mL | |
| MCF7 | 17.50 μg/mL | |
| H1299 | >50 μg/mL |
Uptake of Asp-DOX in vitro
In order to inspect the role of LAT1 in the transport of Asp-DOX into cancer cells, the uptake of Asp-DOX and free DOX by a LAT1-positive cell line (HepG2) and LAT1-negative cell line (H1299 cells) was investigated using fluorescence microscopy and HPLC. The results are shown in Figure3 and Table3. It can be seen from Table3 that, after 2 h of treatment at different drug doses, the accumulation of Asp-DOX in HepG2 cells was significantly higher than that of DOX (Table3). However, the accumulation of Asp-DOX in the LAT1-negative cell line H1299 was apparently lower compared to free DOX. In addition, the fluorescence intensity in the HepG2 cells treated with Asp-DOX was stronger than that in HepG2 cells treated with DOX (Fig.3a), whereas the fluorescence strength in H1299 cells (Fig.3b) was reversed compared to HepG2. The results of HPLC analysis and fluorescence microscopy suggested that, after Asp modification, the accumulation of Asp-DOX in the LAT1-positive cell line (HepG2) increased compared to the LAT1-negative cell line H1299 (the expression of LAT1 in the four cancer cell lines is shown in Fig.3c).
Figure 3.
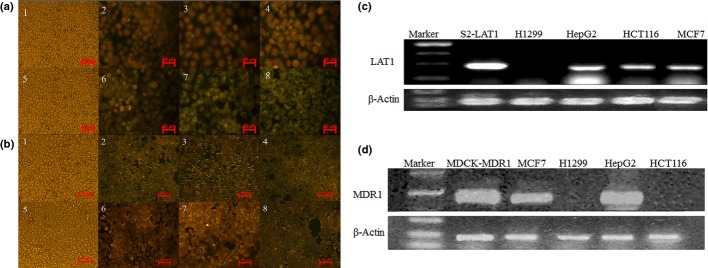
Uptake of doxorubicin (DOX) or aspartate-modified DOX (Asp-DOX) in cancer cells measured by fluorescence microscopy and the expression of LAT1 and MDR1 in cancer cell lines. The uptake of DOX and Asp-DOX was determined by the levels of fluorescence (green) at λex = 475 nm and λem = 580 nm. The LAT1-positive cell line HepG2 (a) and LAT1-negative cell line H1299 (b) treated by DOX (a1–4 and b1–4) and Asp-DOX (a5–8 and b5–8) at different doses (0, 10, 20, and 50 μg/mL). (c, d) Expression of LAT1 (c) and MDR1 (d) in H1299, HepG2, HCT116, and MCF7 cancer cell lines; β-actin, housekeeping gene.
Table 3.
Uptake of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) in HepG2 (LAT1-positive) and H1299 (LAT1-negative) cell lines at different doses (10, 20, and 50 μg/mL) in vitro (n = 3)
| Uptake of DOX, μg/mL | Uptake of Asp-DOX, μg/mL | |||
|---|---|---|---|---|
| HepG2 | H1299 | HepG2 | H1299 | |
| 10 μg/mL | 0.21 ± 0.030 | 0.20 ± 0.059 | 0.48 ± 0.019** | ND** |
| 20 μg/mL | 0.44 ± 0.032 | 0.33 ± 0.019 | 1.45 ± 0.046** | 0.016 ± 0.0012** |
| 50 μg/mL | 0.70 ± 0.079 | 0.69 ± 0.055 | 2.63 ± 0.171** | 0.018 ± 0.0022** |
P < 0.05
P < 0.01.
Results of bidirectional transport assay
Drug resistance was attributable to the decreased accumulation of drugs within cancer cells because of increased drug efflux.24 As one substrate of P-gp, DOX showed weak cytotoxicity to the MDR1-overexpressing cancer lines HepG2 and MCF7 (Fig.3d). In contrast, Asp-DOX showed a higher inhibition of HepG2 and MCF7.25 In order to check the resistance to Asp-DOX, we constructed the models of bidirectional transport assay based on the MDR1-overexpressing MDCK cells through the Transwell method (Table4). The results showed that the Papp(B–A) of DOX (6.63 ± 0.328 × 10−6 cm/s) was significantly higher (P < 0.01) than that of Asp-DOX (0.87 ± 0.073 × 10−6 cm/s), whereas the Papp(A–B) of DOX (0.53 ± 0.093 ×10−6 cm/s) was lower than that of Asp-DOX (0.67 ± 0.045 × 10−6 cm/s). The PDR of DOX was determined as 13.01 ± 1.425 whereas the PDR of Asp-DOX was only 1.30 ± 0.110. This suggested that the Asp-modified DOX was not yet a substrate of P-gp.26 These findings also suggested that Asp-DOX was better retained within the P-gp overexpressing cells than free DOX.
Table 4.
Bidirectional transport of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) in MDR1-overexpressing MDCK cells, assessed by the Transwell method (n = 3)
| Papp (10−6 cm/s) | PDR | ||
|---|---|---|---|
| A–B | B–A | ||
| DOX (50 μg/mL) | 0.53 ± 0.093 | 6.63 ± 0.328 | 13.01 ± 1.425 |
| Asp-DOX (50 μg/mL) | 0.67 ± 0.045 | 0.87 ± 0.073 | 1.30 ± 0.110* |
P < 0.01. A–B, Direction of transport from apical to basolateral profile; B–A, direction of transport from basolateral to apical profile; Papp, apparent permeability; PDR, the efflux rate.
Pharmacokinetics and biodistribution of Asp-DOX
To quantify the fate of the Asp-modified DOX, we carried out pharmacokinetic studies with Asp-DOX and DOX as previously described.27 The results are shown in Figure4. As previously reported, DOX was rapidly eliminated from the blood circulation (t1/2, 15.12 min), whereas Asp-DOX had a significantly longer circulation time (t1/2, 49.14 min). Additionally, the AUC0−t of Asp-DOX (1293.236 min μg/mL) was approximately 8.14-fold larger than that of the unmodified DOX (158.894 min μg/mL). These results suggested that, after Asp modification, the retention time of Asp-DOX in blood was prolonged and the exposure dose was also increased at the same dose.
Figure 4.
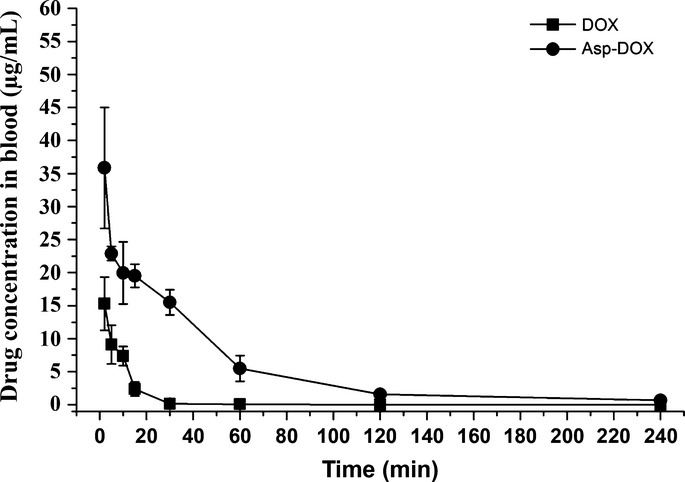
Blood drug–time curves of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) at a dose of 5 mg/kg in healthy mice (n = 3–5).
The biodistribution of DOX and Asp-DOX were investigated in mice bearing HepG2, HCT116, and H1299 tumors (Fig.5). The results showed that in the HCT116 models, with the exception that the uptake of Asp-DOX was 6.4-fold higher than that of DOX in tumor tissues, the drug levels were more or less higher in other tissues of free DOX-treated mice than in that of Asp-DOX-treated groups (Fig.5a). In HepG2 models (Fig.5b), the drug levels were significantly higher in heart, liver, and lung of DOX-treated mice than in Asp-DOX-treated mice, while the accumulation of Asp-DOX in tumor tissues was approximately 3.1-fold higher than that of free DOX-treated mice. Although there was no statistical difference, the drug levels of DOX was higher than that of Asp-DOX in other tissues (Fig.5b). In the LAT1-negative cancer cell line H1299 models (Fig.5c), the drug levels of DOX were higher than that of Asp-DOX in all tissues, especially in tumor tissues. These results suggested that LAT1 plays an important role in the uptake of Asp-DOX by tumor tissues.
Figure 5.
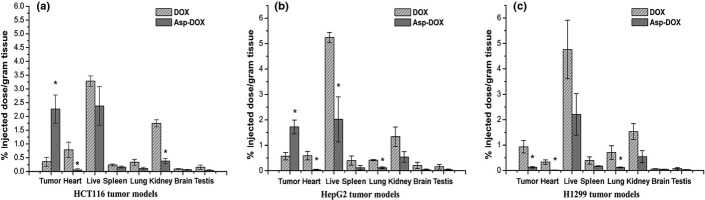
Drug biodistribution (percentage of injected dose/g of tissue) of doxorubicin (DOX) or aspartate-modified DOX (Asp-DOX) in mice bearing the LAT1-positive cell lines HCT116 (a) and HepG2 (b), and the LAT1-negative cell line H1299 (c) (n = 3–5 per group) (*P < 0.05).
Therapeutic effects of Asp-DOX in vivo
The therapeutic effect of Asp-DOX was investigated in the HepG2 (LAT1-positive) and H1299 (LAT1-negative) tumor models (representative of the four tested cancer cell lines in vitro), as previously described (Fig.6). The pharmacodynamic study revealed that, in HepG2 tumor models, tumor volumes increased rapidly in an exponential way when animals were treated with the saline and DOX, whereas treatment with Asp-DOX slowed the tumor growth rate significantly. At the end of the experiment, tumors in the animals treated with saline and free DOX continued to grow rapidly to 2490.33 ± 294.03 mm3 and 1790.334 ± 294.03 mm3, approximately 24- and 13.6-fold larger than the initial tumors; in contrast, the tumor volume of the group treated with Asp-DOX was only 709.93 ± 105.19 mm3, approximately 5-fold larger than the initial tumors (Fig.6a,b). More importantly, after 17 days of treatment with Asp-DOX, the tumor volumes showed shrinkage, which indicated a good prognosis in LAT1-positive HepG2 tumors. However, the antitumor effect exerted by DOX was stronger than that of Asp-DOX and saline in LAT1-negative H1299 tumor models (Fig.6c,d). These results suggested that Asp-DOX showed stronger inhibition of the growth of LAT1-positive HepG2 tumors in vivo than that in LAT1-negative H1299 tumors. From these results it can be inferred that Asp-DOX expressed higher selective cytotoxicity and antitumor effects to LAT1-positive tumor cell lines such as HepG2.
Figure 6.

Therapeutic responses of nude mice bearing HepG2 and H1299 tumors (n = 3–5). (a) Volume (V) changes of HepG2 tumors in three groups treated with saline, doxorubicin (DOX), or aspartate-modified DOX (Asp-DOX). (b) Volumes of HepG2 tumors in different groups after 21 days of treatment. (c) Volume changes of H1299 tumors in groups treated with saline, DOX, and Asp-DOX. (d) Volumes of H1299 tumors in different groups after 21 days of treatment.
Discussion
LAT1 is overexpressed on various human tumor cells and is a potential molecular target for tumor therapy.5,28 It was proposed that it must have a free carboxyl and an amino group and that the hydrophobic interaction between the substrate side chain and the substrate binding site of LAT1 seems to be crucial for the substrate binding, which was supported by the results of inhibition of different amino acid-modified formations to the uptake of L-[3H] leucine (Fig.1). Amino acids, the substrates of LAT1, provided ideal ligands for a targeted delivery system of antitumor drugs. This was supported by our results in vitro and in vivo (Figs5, Table3). The higher accumulation of Asp-DOX in LAT1-overexpressing tumor tissues and the better antitumor activity than free DOX in our pharmacodynamics studies proved that Asp-DOX was an efficient formulation that exerted stronger antitumor effects and higher selective cytotoxicity to LAT1-positive tumor cells in vitro and in vivo. (Fig.6a,b).
Overexpression of P-gp associated with MDR is a prominent mechanism in tumor therapy,29 which decreases effective drug accumulation within tumors. Doxorubicin is a widely used anticancer drug, and a proven substrate of P-gp.26 Therefore, it was practicable to decrease the resistance of cancer cells to DOX through reconstruction on the molecule. In this study, after Asp modification, Asp-DOX showed a lower PDR and a lower affinity to P-gp (Table4) and it showed stronger growth inhibition in cancer cell lines HCT116, MCF7, and HepG2 in vitro and in vivo (Figs6). It can be inferred that this formulation, Asp-DOX, successfully avoided the recognition and elimination of P-gp in tumors for an efficient antitumor therapy.
Doxorubicin had a shorter retention time than Asp-DOX and it was quickly cleared during the blood circulation (t1/2, 15.12 min; AUC0−t, 158.89 min * mg/mL).30,31 The t1/2 and AUC0−t of Asp-DOX (49.14 min and 1293.236 min * mg/mL, respectively) were both greatly enhanced compared to DOX (Fig.4). These results suggested that the internalization of Asp-DOX was different from DOX and that the clearance of Asp-DOX became slower and the exposure dose increased greatly after modification. The lower clearance rate and higher exposure dose of Asp-DOX were necessary for its accumulation in tumor sites to exert its potent effect.
In conclusion, Asp-DOX efficiently avoided the MDR effect induced by P-gp and expressed better properties in pharmacodynamics and pharmacokinetics. It is suggested that Asp-DOX deserves consideration as a candidate for the anticancer drug arsenal and provides another choice for LAT1-positive cancer treatment because of its long circulation, high affinity to LAT1, and efficient avoidance of the MDR effect induced by P-gp. More importantly, the present study not only provided support for the transport properties of LAT1 but also revealed a new clue for the conventional antitumor agents to overcome MDR.
Acknowledgments
This work was supported by the Chinese Ministry of Sciences and Technology Research project (Grant No. 2009ZX09304-002), the State 973 plan project (Grant No. 2010CB735602), and the Major National Technology Programs in the “Twelfth Five-Year” Plan period (Grant No. 2012ZX09304002).
Glossary
- A–B
apical-to-basolateral
- AUC0−t
area under the curve of concentration versus time
- B–A
basolateral-to-apical
- DOX
doxorubicin
- DPBS
Dulbecco's PBS
- IR
inhibition rate
- Km
Michaelis constant
- LAT1
L-type amino acid transporter 1
- MDR1
multiple drug resistance gene 1
- Papp
apparent permeability
- PDR
the efflux rate
- P-gp
P-glycoprotein
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Fig. S1. Procedure of the synthesis of aspartate-modified doxorubicin (Asp-DOX).
Fig. S2. Characterizations of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) by HPLC.
Fig. S3. Mass spectrometry results of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX).
References
- Kanai Y, Segawa H, Miyamoto K-i, Uchino H, Takeda E, Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98) J Biol Chem. 1998;273:23629–32. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- Christensen HN. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev. 1990;70:43. doi: 10.1152/physrev.1990.70.1.43. [DOI] [PubMed] [Google Scholar]
- Segawa H, Fukasawa Y, Miyamoto K-i, Takeda E, Endou H, Kanai Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem. 1999;274:19745–51. doi: 10.1074/jbc.274.28.19745. [DOI] [PubMed] [Google Scholar]
- Uchino H, Kanai Y, Kim DK, et al. Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): insights into the mechanisms of substrate recognition. Mol Pharmacol. 2002;61:729–37. doi: 10.1124/mol.61.4.729. [DOI] [PubMed] [Google Scholar]
- Nawashiro H, Otani N, Shinomiya N, et al. L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int J Cancer. 2006;119:484–92. doi: 10.1002/ijc.21866. [DOI] [PubMed] [Google Scholar]
- Kaira K, Oriuchi N, Imai H, et al. Prognostic significance of L-type amino acid transporter 1 expression in resectable stage I–III nonsmall cell lung cancer. Br J Cancer. 2008;98:742–8. doi: 10.1038/sj.bjc.6604235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko T, Kopečková P, Kopeček J. Chronic exposure to HPMA copolymer-bound adriamycin does not induce multidrug resistance in a human ovarian carcinoma cell line. J Controlled Release. 1999;59:133–48. doi: 10.1016/s0168-3659(98)00186-2. [DOI] [PubMed] [Google Scholar]
- O'brien M, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX™/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–9. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- Peterson CM, Shiah J-G, Sun Y, et al. HPMA Copolymer Delivery of Chemotherapy and Photodynamic Therapy in Ovarian Cancer. Polymer Drugs in the Clinical Stage: Springer; 2003. pp. 101–23. [DOI] [PubMed] [Google Scholar]
- Ye WL, Teng ZH, Liu DZ, et al. Synthesis of a new pH-sensitive folate–doxorubicin conjugate and its antitumor activity in vitro. J Pharm Sci. 2013;102:530–40. doi: 10.1002/jps.23381. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–85. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci. 1987;84:3004–8. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–63. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Orbán E, Mező G, Schlage P, et al. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin–GnRH-III bioconjugates and DNA-binding properties of daunorubicin–amino acid metabolites. Amino Acids. 2011;41:469–83. doi: 10.1007/s00726-010-0766-1. [DOI] [PubMed] [Google Scholar]
- Loffet A, Zhang H. Allyl-based groups for side-chain protection of amino-acids. Int J Pept Protein Res. 1993;42:346–51. doi: 10.1111/j.1399-3011.1993.tb00504.x. [DOI] [PubMed] [Google Scholar]
- Mellor SLMC, Chan WC. Fmoc-aminooxy-2-chlorotrityl polystyrene resin: a facile solid-phase methodology for the synthesis of hydroxamic acids. Tetrahedron Lett. 1997;38:3311–4. [Google Scholar]
- Shmeeda H, Amitay Y, Gorin J, et al. Delivery of zoledronic acid encapsulated in folate-targeted liposome results in potent in vitro cytotoxic activity on tumor cells. J Controlled Release. 2010;146:76–83. doi: 10.1016/j.jconrel.2010.04.028. [DOI] [PubMed] [Google Scholar]
- Hellinger É, Veszelka S, Tóth AE, et al. Comparison of brain capillary endothelial cell-based and epithelial (MDCK-MDR1, Caco-2, and VB-Caco-2) cell-based surrogate blood–brain barrier penetration models. Eur J Pharm Biopharm. 2012;82:340–51. doi: 10.1016/j.ejpb.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Perrière N, Yousif S, Cazaubon S, et al. A functional in vitro model of rat blood–brain barrier for molecular analysis of efflux transporters. Brain Res. 2007;1150:1–13. doi: 10.1016/j.brainres.2007.02.091. [DOI] [PubMed] [Google Scholar]
- Takara K, Hatakeyama H, Kibria G, Ohga N, Hida K, Harashima H. Size-controlled, dual-ligand modified liposomes that target the tumor vasculature show promise for use in drug-resistant cancer therapy. J Controlled Release. 2012;162:225–32. doi: 10.1016/j.jconrel.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Lehtinen J, Raki M, Bergström KA, et al. Pre-targeting and direct immunotargeting of liposomal drug carriers to ovarian carcinoma. PLoS ONE. 2012;7:e41410. doi: 10.1371/journal.pone.0041410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino F, Brignole C, Marimpietri D, et al. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003;63:7400–9. [PubMed] [Google Scholar]
- Gana-Weisz M, Halaschek-Wiener J, Jansen B, Elad G, Haklai R, Kloog Y. The Ras inhibitor S-trans, trans-farnesylthiosalicylic acid chemosensitizes human tumor cells without causing resistance. Clin Cancer Res. 2002;8:555–65. [PubMed] [Google Scholar]
- Moshtaghian J, Ghaedi K, Moafi A, et al. MDR1 gene expression in acute lymphoblastic leukemia; Implications in pharmacokinetics and relapse. Res Pharm Sci. 2012;7:S690. [Google Scholar]
- Zhang FY, Du GJ, Zhang L, Zhang CL, Lu WL, Liang W. Naringenin enhances the anti-tumor effect of doxorubicin through selectively inhibiting the activity of multidrug resistance-associated proteins but not P-glycoprotein. Pharm Res. 2009;26:914–25. doi: 10.1007/s11095-008-9793-y. [DOI] [PubMed] [Google Scholar]
- Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. J Pharmacol Exp Ther. 2006;317:1372–81. doi: 10.1124/jpet.106.101154. [DOI] [PubMed] [Google Scholar]
- Loi M, Marchiò S, Becherini P, et al. Combined targeting of perivascular and endothelial tumor cells enhances anti-tumor efficacy of liposomal chemotherapy in neuroblastoma. J Controlled Release. 2010;145:66–73. doi: 10.1016/j.jconrel.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Yanagida O, Kanai Y, Chairoungdua A, et al. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochim Biophys Acta. 2001;1514:291–302. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- Longley D, Johnston P. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–92. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- Tseng YL, Hong RL, Tao MH, Chang FH. Sterically stabilized anti-idiotype immunoliposomes improve the therapeutic efficacy of doxorubicin in a murine B-cell lymphoma model. Int J Cancer. 1999;80:723–30. doi: 10.1002/(sici)1097-0215(19990301)80:5<723::aid-ijc16>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Peer D, Margalit R. Tumor-targeted hyaluronan nanoliposomes increase the antitumor activity of liposomal doxorubicin in syngeneic and human xenograft mouse tumor models. Neoplasia. 2004;6:343. doi: 10.1593/neo.03460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Procedure of the synthesis of aspartate-modified doxorubicin (Asp-DOX).
Fig. S2. Characterizations of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX) by HPLC.
Fig. S3. Mass spectrometry results of doxorubicin (DOX) and aspartate-modified DOX (Asp-DOX).