

Abstract
We conducted a phase I study of a weekly nab-paclitaxel and S-1 combination therapy in patients with human epidermal growth factor receptor type 2-negative metastatic breast cancer. The primary objective was to estimate the maximum tolerated and recommended doses. Each treatment was repeated every 21 days. Levels 1, 2a, 2b, and 3 were set depending on the S-1 dose (65 or 80 mg/m2) and nab-paclitaxel infusion schedule (days 1 and 8 or days 1, 8, and 15). Fifteen patients were enrolled. Dose-limiting toxicity was observed in one patient at Level 3 (100 mg/m2 nab-paclitaxel on days 1, 8, and 15 with 80 mg/m2 S-1 daily for 14 days, followed by 7 days of rest). Although the maximum tolerated dose was not reached, the recommended dose was determined to be Level 3. Neutropenia was the most frequent grade 3–4 treatment-related adverse event. For patients with measurable lesions, the response rate was 50.0% and the median time to treatment failure and median progression-free survival was 13.2 and 21.0 months, respectively. The present results show the feasibility and potential for long-term administration of this combination therapy.
Keywords: Combination chemotherapy, metastatic breast cancer, nab-paclitaxel, phase I study, S-1
Chemotherapies for breast cancer, including molecular targeted therapies, have undergone remarkable development in recent years; conventional anthracycline and taxane-containing regimens continue to play a key role in this treatment. For cases of human epidermal growth factor receptor type 2 (HER2)-negative breast cancer, the treatment options are limited compare to those for HER2-positive cases and the development of highly efficacious therapy is warranted.
Combination chemotherapy represents a treatment choice that has been prescribed for increased efficacy. The selection of a combination of cytotoxic chemotherapies versus sequential single-agent treatment is controversial.1 In phase III clinical trials involving metastatic breast cancer (MBC), O'Shaughnessy et al. evaluated a combination therapy with docetaxel and capecitabine, whereas Albain et al. prescribed a combination therapy with paclitaxel and gemcitabine; both research groups reported the superiority of the combined regimens over monotherapies.2,3 Combination therapies have also been reported to correlate with a high incidence of toxicity and high efficacies, therefore, the development of a well-tolerated, highly efficacious therapy is anticipated.
Nab-paclitaxel is a 130-nm nanoparticulate drug preparation comprising paclitaxel bound to human serum albumin particles and is widely used as a key drug for the treatment of breast cancer.4 In a pivotal phase III clinical study, treatment with nab-paclitaxel showed a significantly better response rate (RR; a primary endpoint) of 24.0%, as compared with an RR of 11.1% for treatment with the standard solvent-based paclitaxel.5 Furthermore, in a randomized phase II clinical study, the median progression-free survival (PFS) and RR of weekly nab-paclitaxel was 12.9 months and 49%, respectively, which suggested that weekly nab-paclitaxel might be superior to tri-weekly administration.6,7 In that study, the major toxicities associated with weekly nab-paclitaxel were myelosuppression and peripheral neuropathy.
The oral, fixed-dose combination agent S-1 comprises tegafur (FT), a fluoropyrimidine prodrug of 5-fluorouracil (5-FU), and the 5-FU metabolism modulating agents 5-chloro-2.4-dihydrooxypyridine (CDHP) and oteracil potassium (Oxo). S-1 is designed to orally deliver 5-FU, a pyrimidine analog antimetabolite and antineoplastic agent while reducing the rate of 5-FU degradation and conversion in the gastrointestinal tract to a toxic phosphorylated metabolite.8 The results of a phase II clinical study revealed an RR of 41.7% for patients with MBC who received S-1 monotherapy, indicating the efficacy of this regimen.9 The major adverse events associated with S-1 treatment in that study were myelosuppression and gastrointestinal toxicity. A phase III study (SELECT BC) carried out in chemotherapy-naïve patients with HER2-negative MBC, which investigated overall survival as a primary endpoint, confirmed the non-inferiority of S-1 to taxanes.10,11
Thymidine phosphorylase is an enzyme that converts 5-FU to its active form, fluorodeoxyuridylate, and taxanes have been reported to induce the upregulation of thymidine phosphorylase in tumor tissues.12 Nukatsuka et al.13 reported a synergistic reduction in tumor size following treatment with paclitaxel combined with S-1 in a mouse model of human breast cancer.
The mechanisms of cytotoxic action differ between nab-paclitaxel and S-1. A major toxicity of both nab-paclitaxel and S-1 is myelosuppression; otherwise, these two drugs have no other overlapping toxicity profiles that would affect the continuation of treatment. Given this information and the assumption from the results of basic studies that the combined use of these two drugs might yield synergistically enhanced efficacy, we carried out a phase I study of weekly nab-paclitaxel in combination with S-1 in patients with HER2-negative MBC.
Materials and Methods
This phase I dose-escalation study to evaluate treatment with weekly nab-paclitaxel in combination with S-1 in patients with HER2-negative MBC was carried out in conformance with the Good Clinical Practice guidelines and the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board of each participating medical institution prior to initiation of the study. Written informed consent was obtained from every patient prior to participation in the study.
Patient population
Patients who met the following major criteria were considered eligible to participate in the study: women with cytologically or histologically confirmed breast cancer who were aged 20–74 years; patients with clinically confirmed MBC; patients with demonstrated HER2-negativity through immunohistochemical analysis or FISH; patients previously treated with single-regimen or no chemotherapy for MBC; a survival expectancy of ≥60 days; an Eastern Cooperative Oncology Group performance status of 0 or 1; and an absolute neutrophil count (ANC) of ≥2000/mm3, hemoglobin concentration of ≥9.0 g/dL, platelet count of ≥10.0 × 104/mm3, total bilirubin concentration of ≤1.5 mg/dL, albumin concentration of ≥3.5 g/dL, serum aspartate aminotransferase concentration of <100 IU/L, serum alanine aminotransferase concentration of <100 IU/L, and creatinine clearance of ≥60 mL/min as determined from a 24-h urine collection or predicted creatinine clearance calculated using the Cockcroft–Gault formula.14
However, patients with tumor progression during or within 12 months after the last dose of pre- or post-operative taxane chemotherapy were excluded from the study. Patients with a history of taxane or S-1 chemotherapy for MBC and those who had experienced grade ≥2 peripheral neuropathy before or since enrolment were also excluded. Measurable disease using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 was not required.
Study design and treatment
The dosage schedules at each dose level are shown in Figure1. Nab-paclitaxel (100 mg/m2) was given by i.v. drip infusion over a 30-min period in doses based on the body surface area (BSA) and calculated using the Mosteller formula; doses were given on days 1 and 8 for Levels 1 and 2b, and on days 1, 8, and 15 for Levels 2a and 3.15 S-1 was given orally twice daily for 14 consecutive days, followed by a 7-day rest. The S-1 dosage was set at 65 mg/m2 for Levels 1 and 2a, and at 80 mg/m2 for Levels 2b and 3. The following daily S-1 dose levels were based on the BSA and calculated using the Fujimoto formula:16 Level 1 and 2a cohorts (65 mg/m2), the dose was 50, 80, or 100 mg at a BSA of <1.25, 1.25–1.5, or ≥1.5 m2, respectively; Level 2b and 3 cohorts (80 mg/m2), the dose was 80, 100, or 120 mg at a BSA of <1.25, 1.25–1.5, or ≥1.5 m2, respectively. Administration of the combination chemotherapy was repeated in 21-day cycles until the occurrence of disease progression or development of intolerable toxicities. Although the rule was to avoid corticosteroid or anti-allergic pretreatments, such treatments were allowed in cases with signs of hypersensitivity.
Figure 1.
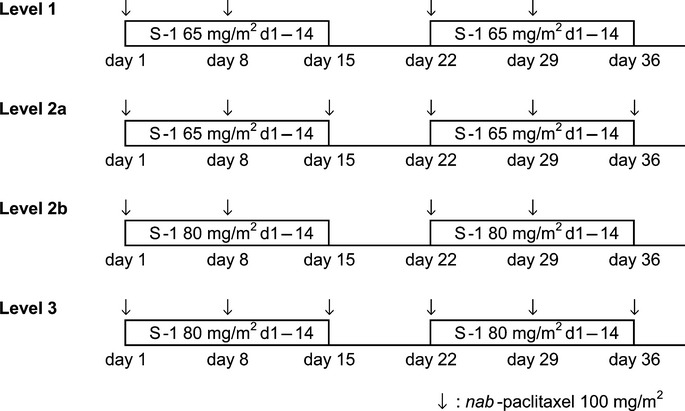
Therapeutic experimental regimens for nab-paclitaxel and S-1 combination therapy in patients with human epidermal growth factor receptor type 2-negative metastatic breast cancer.
Dose modification was carried out in accordance with the protocol. Before commencement each cycle, patients were required to have an ANC ≥1500/mm3, platelet count ≥75 000/mm3, total bilirubin ≤1.5 mg/dL, liver transaminase <100 IU/L, serum creatinine ≤1.5 mg/dL, ≤grade 2 peripheral sensory neuropathy, ≤grade 2 eye disorders, ≤grade 3 diarrhea, and ≤grade 3 stomatitis. If any toxicity applicable to Table S1 occurred during the administration period in a cycle, the study treatment was to be interrupted. If any toxicity applicable to Table S2 occurred, the dose of each drug in the next administration was to be decreased according to Table S3.
Level escalation plan
This study was carried out with a unique 3 + 3 design in a sequential order of Levels 1, 2a, 2b, and 3. Whether to proceed to the next Level was determined by deliberation between the investigator, medical officer, and study sponsor by considering the dose-limiting toxicity (DLT) and administration conditions during the first and second cycles. For cases in which DLT was observed in one or two of three patients at any Level, three additional patients were to be recruited for the same Level. When DLT was observed in three or more of six patients at any Level, that Level was considered a maximum tolerated dose (MTD) and the dose level immediately below that Level was defined as a recommended dose (RD). In the case that the DLT incidence was <50% at Level 3, the study sponsor was entrusted with the final judgment of an RD after deliberation with the medical officer and at the suggestion of the Data and Safety Monitoring Committee. Dose-limiting toxicity was defined as the occurrence of any of the following during cycle 1: grade 4 platelet count decrease; grade 3 platelet count decrease requiring blood transfusion; febrile neutropenia with a neutrophil count of <500/mm3 and pyrexia at ≥38.5°C; grade 4 neutrophil count decrease persisting for ≥8 days; grade ≥3 nausea, vomiting, and diarrhea refractory to symptomatic treatment; and the postponement of cycle 2 initiation for ≥15 days from the scheduled time point because of adverse reaction(s). For ≥grade 3 non-hematological toxicities, cases of all other abnormal clinical laboratory test values, and/or transient non-hematological toxicities, the principle investigators, medical officer, and sponsor would confer to determine the presence or absence of a DLT.
Study objectives
The primary objective of this study was to estimate the MTD and RD of the combination therapy including weekly nab-paclitaxel and S-1 in patients with MBC. The secondary objective comprised evaluations of safety, antitumor responses, administration conditions, and pharmacokinetic profiles.
Safety and efficacy assessments
Adverse events were evaluated for severity in accordance with the Common Terminology Criteria for Adverse Events version 4.0. Antitumor response evaluations were carried out every two cycles according to RECIST version 1.1.
Pharmacokinetics
The paclitaxel and S-1 component plasma pharmacokinetics were investigated in this study. Blood samples were collected at 0, 0.5, 1, 2, 4, 10, 24, 48, and 72 h after nab-paclitaxel dosing and at 0, 0.5, 1, 2, 4, 6, 8, and 10 h after S-1 dosing during the first cycle only. The plasma concentrations of paclitaxel, FT, 5-FU, CDHP, and Oxo were determined through validated analytical procedures that incorporated HPLC with tandem mass spectrometry at the Shin Nippon Biochemical Laboratories (Wakayama, Japan). The lower limit of quantitation for paclitaxel in human plasma was 1 ng/mL, and the reliable response range was 1–1000 ng/mL. The lower limit of quantitation values for FT, 5-FU, CDHP, and Oxo in human plasma were 20, 2, 4, and 4 ng/mL, respectively, and the reliable response ranges were 20–4000, 2–400, 4–800, and 4–400 ng/mL, respectively.
The pharmacokinetic parameters were calculated according to non-compartmental techniques using the WinNonlin software program (Pharsight, Mountain View, CA, USA). The maximum observed concentration (Cmax) and the time to Cmax (tmax) were determined directly from the observed plasma concentration–time profiles over the 72-h sampling interval. The apparent terminal elimination rate constant (λz) was estimated by linear regression of the individual plasma concentration–time data. The terminal elimination half-life (t1/2) was calculated as t1/2 = ln (2)/λz for each individual. Individual areas under the concentration–time curves (AUCs) from time 0 to the last measurable time point (AUC0–t) were calculated according to the trapezoidal rule. Individual AUCs extrapolated to infinity (AUCinf) were calculated using the last measurable concentration (Clast) according to the formula AUCinf = AUC0–t + Clast/λz.
Results
Fifteen patients were enrolled at two medical institutions in Japan between July 2010 and December 2012. A follow-up to the study treatment continued until December 2013.
Patient characteristics
The patient characteristics are summarized in Table1. All 15 patients were subjected to the safety analysis. Eleven and four patients had histologically positive and negative hormone receptor statuses, respectively. Nine patients had chemotherapy-naïve MBC and the other six had undergone chemotherapy for MBC with anthracycline-containing regimens.
Table 1.
Characteristics of patients with human epidermal growth factor receptor type 2-negative metastatic breast cancer (MBC) treated with nab-paclitaxel and S-1 combination therapy (n = 15)
| Characteristic | No. of patients (%) |
|---|---|
| Age, years | |
| Median (range) | 63.0 (41–67) |
| ECOG PS | |
| 0 | 9 (60.0) |
| 1 | 6 (40.0) |
| Hormonal status | |
| ER-positive and/or PgR-positive | 11 (73.3) |
| ER-negative and PgR-negative | 4 (26.7) |
| Metastatic site | |
| Lung | 6 (40.0) |
| Bone | 9 (60.0) |
| Liver | 4 (26.7) |
| Distant lymph nodes | 6 (40.0) |
| Other | 3 (20.0) |
| Prior chemotherapy for MBC | |
| 0 | 9 (60.0) |
| 1 | 6 (40.0) |
ECOG, Eastern Cooperative Oncology Group; ER, estrogen receptor; PgR, progesterone receptor; PS, performance status.
Dose-limiting toxicity, MTD, and RD
The dosage level was escalated up to Level 3, the highest level specified in the protocol; however, no DLT was observed in three patients per group through Levels 1 to 3. Three additional patients were enrolled for Level 3 treatment with the intent to evaluate tolerability at that dosage level in six patients. As a result, a DLT (neutropenia leading to a delay in the start of cycle 2 for ≥15 days beyond the scheduled day) occurred in one patient, so MTD was not reached. However, at Level 3 dose reductions were required in three of the six patients in cycle 2 (grade 1 diarrhea in one patient, grade 2 diarrhea and grade 1 vomiting in one patient, and a prolonged neutropenia in one patient); therefore, it was determined that the dosage should not be increased further, and the RD was determined to be Level 3.
Drug administration and safety profile
Fifteen patients received a total of 206 cycles of combination chemotherapy. The median number of cycles administered per patient was 14.0 (range, 1–35). The overall relative dose intensity (RDI) was 62.5% for nab-paclitaxel and 70.5% for S-1. The overall RDIs up to cycle 2, as required to determine whether to proceed to the next Level, were 84.0% and 81.0% for nab-paclitaxel and S-1, respectively. The RDIs up to cycle 2 at Level 3 were 68.5% and 76.3% for nab-paclitaxel and S-1, respectively. The major reasons for requiring a nab-paclitaxel dose reduction were peripheral sensory neuropathy (33.3%; n = 5) and fatigue (20.0%; n = 3); S-1 dose reductions were mainly because of fatigue (26.7%; n = 4) or diarrhea (20.0%; n = 3). Skipping of nab-paclitaxel administration was most often because of fatigue (33.3%; n = 5) or peripheral sensory neuropathy (20.0%; n = 3), whereas neutropenia (26.7%; n = 4), decreased appetite (13.3%; n = 2), and diarrhea (13.3%; n = 2) were the main reasons for skipping S-1 treatment. Neutropenia was a major reason for delaying the initiation of the next cycle (86.7%; n = 13). The following factors accounted for the discontinuation of treatment: disease progression in six patients; adverse events (psoriasis, keratitis, cheilitis, and diarrhea) in four patients; refusal of further treatment in three patients; and end of study in two patients.
The treatment-related adverse events that occurred in ≥30% (≥5 patients) of all patients are listed in Table2. The hematological toxicities with high incidence were neutropenia (100%; n = 15), leukopenia (100%; n = 15), and anemia (80%; n = 12). The non-hematological toxicities with high incidence included alopecia (93%; n = 14), peripheral sensory neuropathy (87%; n = 13), diarrhea (80%; n = 12), and decreased appetite (80%; n = 12). Most of the treatment-related adverse events, although high in incidence, were grade ≤2 and clinically manageable. Grade ≥3 treatment-related adverse events that occurred in two or more patients included neutropenia (93%; n = 14), leukopenia (67%; n = 10), lymphopenia (20%; n = 3), fatigue (20%; n = 3), and peripheral sensory neuropathy (13%; n = 2). Grade 3 peripheral sensory neuropathy improved to grade 2 rapidly after skipping the administration.
Table 2.
Treatment-related adverse events at each level
| Adverse Events / CTCAE Grade | Level 1 | Level 2a | Level 2b | Level 3 | Total | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 3 | n = 3 | n = 3 | n = 6 | n = 15 | ||||||||||||||||
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| Neutropenia | 0 | 0 | 2 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 2 | 1 | 0 | 0 | 4 | 2 | 0 | 1 | 9 | 5 |
| Leukopenia | 0 | 0 | 3 | 0 | 0 | 1 | 2 | 0 | 0 | 2 | 1 | 0 | 1 | 1 | 3 | 1 | 1 | 4 | 9 | 1 |
| Alopecia | 3 | 0 | NA | NA | 1 | 2 | NA | NA | 0 | 2 | NA | NA | 2 | 4 | NA | NA | 6 | 8 | NA | NA |
| Peripheral sensory neuropathy | 1 | 0 | 1 | 0 | 0 | 2 | 1 | 0 | 0 | 2 | 0 | 0 | 3 | 3 | 0 | 0 | 4 | 7 | 2 | 0 |
| Anemia | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 2 | 0 | 0 | 2 | 4 | 0 | 0 | 3 | 8 | 1 | 0 |
| Diarrhea | 1 | 1 | 0 | 0 | 2 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 3 | 2 | 0 | 0 | 7 | 4 | 1 | 0 |
| Decreased appetite | 2 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 4 | 1 | 0 | 0 | 8 | 3 | 1 | 0 |
| Nausea | 2 | 0 | 0 | NA | 2 | 0 | 0 | NA | 2 | 1 | 0 | 0 | 2 | 1 | 0 | 0 | 8 | 2 | 0 | 0 |
| Stomatitis | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 4 | 0 | 0 | 0 | 8 | 2 | 0 | 0 |
| Fatigue | 1 | 0 | 0 | NA | 0 | 2 | 0 | NA | 1 | 1 | 1 | NA | 1 | 1 | 2 | NA | 3 | 4 | 3 | NA |
| Dysgeusia | 1 | 0 | NA | NA | 3 | 0 | NA | NA | 0 | 1 | NA | NA | 4 | 0 | NA | NA | 8 | 1 | NA | NA |
| Skin hyperpigmentation | 1 | 0 | NA | NA | 1 | 0 | NA | NA | 2 | 0 | NA | NA | 4 | 1 | NA | NA | 8 | 1 | NA | NA |
| Dry skin | 0 | 1 | 0 | NA | 0 | 1 | 0 | NA | 1 | 1 | 0 | NA | 4 | 0 | 0 | NA | 5 | 3 | 0 | NA |
| ALT level increased | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 1 | 0 | 5 | 0 | 1 | 0 |
| AST level increased | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 1 | 0 | 5 | 0 | 1 | 0 |
| Myalgia | 1 | 1 | 0 | NA | 1 | 0 | 0 | NA | 2 | 0 | 0 | NA | 1 | 0 | 0 | NA | 5 | 1 | 0 | NA |
| Abdominal pain | 0 | 0 | 0 | NA | 1 | 1 | 0 | NA | 0 | 2 | 0 | NA | 2 | 0 | 0 | NA | 3 | 3 | 0 | NA |
| Peripheral edema | 0 | 0 | 0 | NA | 1 | 1 | 0 | NA | 0 | 0 | 0 | NA | 2 | 2 | 0 | NA | 3 | 3 | 0 | NA |
| Lymphopenia | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 3 | 3 | 0 |
| Constipation | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 5 | 0 | 0 | 0 |
| Thrombocytopenia | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 5 | 0 | 0 | 0 |
| Watering of eyes increased | 0 | 0 | 0 | NA | 0 | 0 | 0 | NA | 1 | 0 | 0 | NA | 4 | 0 | 0 | NA | 5 | 0 | 0 | NA |
ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; CTCAE, Common Terminology Criteria for Adverse Events; NA, not applicable
Efficacy
The RRs and disease control rates (complete response [CR] + partial response [PR] + stable disease [SD] for ≥16 weeks) are shown in Table3. Twelve of the 15 patients had measurable lesion(s) as defined by RECIST version 1.1. The responses in the 12 patients included CR in one patient, PR in five patients, SD in five patients, progressive disease in one patient, and not evaluable in one patient, with a RR of 50.0% (95% confidence interval [CI], 21.1–78.9). Among the triple-negative cases, the responses were CR in one patient, PR in one patient, and progressive disease in one patient. Among patients with hepatic metastasis, the responses were PR in three patients and SD in one patient. The disease control rate was 83.3% (95% CI, 51.6–97.9), and the median time to treatment failure (TTF) and median PFS were 13.2 months (95% CI, 6.9–16.2) and 21.0 months (95% CI, 14.9–not reached), respectively.
Table 3.
Efficacy of combination therapy according to RECIST
| Response | Level 1 (n = 2) | Level 2a (n = 3) | Level 2b (n = 3) | Level 3 (n = 4) | Total (n = 12) |
|---|---|---|---|---|---|
| CR | 1 | 0 | 0 | 0 | 1 |
| PR | 0 | 2 | 0 | 3 | 5 |
| SD | 0 | 1 | 2 | 1 | 4 |
| PD | 1 | 0 | 0 | 0 | 1 |
| NE | 0 | 0 | 1 | 0 | 1 |
| Response rate (CR+PR) | 50.0% | 66.7% | 0.0% | 75.0% | 50.0% (95% CI, 21.1–78.9) |
| Disease control rate (CR+PR+SD for ≥16 weeks) | 50.0% | 100.0% | 66.7% | 100.0% | 83.3% (95% CI, 51.6–97.9) |
CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; NE, not evaluable
Pharmacokinetics
Twelve patients (six patients at Levels 1 and 2a, and six patients at Levels 2b and 3) underwent pharmacokinetic evaluations. The pharmacokinetic parameters for the S-1 components and paclitaxel are summarized in Table4. The plasma concentrations of FT, 5-FU, CDHP, and Oxo increased in a dose-dependent manner at a dose of 65 or 80 mg/m2 S-1 with the co-administration of 100 mg/m2 nab-paclitaxel. The pharmacokinetic parameters of paclitaxel following the concomitant administration of nab-paclitaxel and S-1 were similar regardless of the S-1 dose level.
Table 4.
Plasma pharmacokinetic parameters for S-1 components and paclitaxel
| Dose level | FT | 5-FU | CDHP | Oxo | Paclitaxel | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Level 1 and 2a (n = 6) | ||||||||||
| tmax, h | 1.3 | 0.5 | 2.3 | 0.8 | 1.5 | 0.5 | 1.7 | 0.5 | 0.5 | 0.0 |
| Cmax, ng/mL | 1901 | 310 | 148.9 | 65.0 | 417.0 | 69.9 | 53.51 | 77.41 | 4778 | 343 |
| AUC0–t, ng/h/mL | 12 201 | 3119 | 695.4 | 276.3 | 1463 | 360 | 230.5 | 409.8 | 4583 | 479 |
| AUCinf, ng/h/mL | 21 998 | 8800 | 718.6 | 271.7 | 1567 | 422 | 1217 | NA | 4806 | 490 |
| t1/2, h | 8 | 1.8 | 1.6 | 0.5 | 2.5 | 0.2 | 3.4 | NA | 26.2 | 3.9 |
| Level 2b and 3 (n = 6) | ||||||||||
| tmax, h | 1.3 | 0.5 | 2.3 | 0.8 | 1.2 | 0.4 | 1.7 | 0.5 | 0.5 | 0.0 |
| Cmax, ng/mL | 2768 | 432 | 193.9 | 83.1 | 452.6 | 97.5 | 80.19 | 63.11 | 5689 | 1056 |
| AUC0–t, ng/h/mL | 16 057 | 3697 | 903.5 | 310.4 | 1648 | 175 | 275.8 | 201.7 | 4876 | 1007 |
| AUCinf, ng/h/mL | 29 732 | 12 675 | 933.7 | 300.9 | 1772 | 119 | 411.5 | 258.6 | 5087 | 1066 |
| t1/2, h | 8.3 | 2.4 | 1.7 | 0.4 | 2.7 | 0.7 | 2.7 | 2.0 | 25.2 | 2.6 |
AUC, area under the curve; AUCinf, AUC from time zero extrapolated to infinity; AUC0–t, AUC from time zero to t; CDHP, 5-chloro-2.4-dihydrooxypyridine; Cmax, maximum plasma concentration; FT, tegafur; 5-FU, 5-fluorouracil; NA, not applicable; Oxo, oteracil potassium; t1/2, half-life time; tmax, maximum concentration time.
Discussion
To our knowledge, this phase I study represents the first clinical trial carried out to evaluate a combination treatment of weekly nab-paclitaxel and S-1 in patients with HER2-negative MBC.
Because the attempt to estimated MTD failed under the 3 + 3 design in this study, we explored the possibility of further nab-paclitaxel dose escalation in order to estimate MTD. However, this dose escalation was determined inappropriate upon deliberation with the medical officer and the Data and Safety Monitoring Committee on the grounds of the RDI at Level 3 as well as the adverse reaction occurrence status, which included <grade 3 adverse reactions. Therefore, Level 3 was determined as the RD (100 mg/m2 nab-paclitaxel on days 1, 8, and 15 with an 80 mg/m2 S-1 dose for 14 days, followed by 7 days of rest).
One of the most important factors for evaluating combination chemotherapies is the balance of efficacy and toxicity. In previously reported phase III clinical studies of docetaxel in combination with capecitabine and of paclitaxel in combination with gemcitabine, the median time to disease progression was 6.1 months for both combination therapy groups, a significantly longer duration than those in the corresponding monotherapy groups.2,3 The incidence of hand–foot syndrome, oral mucositis, and diarrhea in the docetaxel–capecitabine combination therapy group and of neutropenia and febrile neutropenia in the paclitaxel–gemcitabine combination therapy group, nevertheless, tended to be higher when compared with the corresponding monotherapy groups.
Regarding the efficacy and safety in our clinical trial, the PFS of the nab-paclitaxel with S-1 combination therapy was longer than the nab-paclitaxel monotherapy (21.0 and 12.9 months, respectively), and the therapy was feasible, with manageable toxicities. Neutropenia was seen as a notable adverse event with the present combination therapy when compared with nab-paclitaxel or S-1 monotherapy.6,9 Grade 4 neutropenia occurred in 33.3% (n = 5) of patients in the present study. Delayed initiation of the next cycle because of neutropenia was rather common; however, no patients were discontinued from the study because of neutropenia, none developed febrile neutropenia, and only two patients required granulocyte colony-stimulating factor administration. Therefore, the adverse reactions were clinically controllable. None of the other noted toxicities constituted any noticeable add-on when compared with the toxicities in either corresponding monotherapy group, although grade 1 or 2 mild toxicities occurred relatively frequently. Grade ≥3 toxicities were uncommon and the TTF and PFS were also longer when compared with those in either corresponding historical monotherapy group, thus allowing the continuation of long-term treatment. Discrepancy between PFS and TTF was seen in this study; one of the reasons is considered to be that nine patients were censored in PFS because post-discontinuation treatment was initiated before disease progression had occurred.
The appropriateness of combination versus sequential monotherapy was discussed in the 1st International Consensus Guidelines for Advanced Breast Cancer and developed at the Consensus Conference on Metastatic/Recurrent Breast Cancer.17 Although sequential single-agent chemotherapy was recommended for the treatment of MBC, it was agreed that combination chemotherapy might also be included among the options in cases requiring the urgent control of disease progression, such as a life-threatening case of visceral metastasis. Likewise, combination chemotherapy should be considered as an option when a rapid and significant response is required, according to the European Society for Medical Oncology guidelines.18 Inasmuch as gratifying results were obtained with the combination chemotherapy in our present study in a case with multiple hepatic metastases and another with triple-negative disease and an otherwise poor prognosis, the applicability of this regimen in similar subpopulations would be anticipated. It is also important to identify populations in which this combination therapy is effective. Regarding nab-paclitaxel, SPARC (secreted protein, acidic and rich in cysteine, also known as osteonectin, BM40) is expected to be a valuable biomarker, and further investigation into this protein as a therapeutic response-predicting factor is needed.19
Although Level 3 was set as the RD in this study based on the importance of the patients’ quality of life during chemotherapy for MBC, it may be appropriate to adjust the medicinal dosage and administration schedule according to each patient's condition in the clinical practice setting. As the non-inferiority of S-1 to taxanes in terms of overall survival was verified in the SELECT BC study, a flexible approach that begins with combined nab-paclitaxel and S-1 therapy and then shifts to maintenance therapy with S-1 monotherapy after attaining control of the tumor size and symptoms might be proven valid.11
We also evaluated the pharmacokinetics of combination therapy with nab-paclitaxel and S-1. To compare our findings with previously reported data, we reanalyzed the AUC0–10 h from a phase I study.20 There was no significant difference between the administration of S-1 alone and in combination with 100 mg/m2 nab-paclitaxel in terms of the Cmax and AUC0–10 h of 5-FU, which is considered a relevant compound with respect to the efficacy and safety of S-1. However, the Cmax of FT or CDHP was significantly increased in comparison with data from a previous report.20 An additional pharmacokinetic study should be carried out to evaluate the pharmacokinetic parameters of combination therapy with nab-paclitaxel and S-1. When compared with the mean total clearance (18.6–24.8 L/h/m2) and mean volume of the terminal phase (527–935 L/m2) for paclitaxel in Japanese patients following the administration of nab-paclitaxel alone (80–300 mg/m2), there were no obvious differences between those results and the results of this study.21,22
In conclusion, the present data shows the feasibility of a combination therapy with weekly nab-paclitaxel and S-1 and the possibility of long-term administration of this regimen, suggesting that this combination may be a promising therapy for HER2-negative MBC. Further investigation regarding the long-term safety and efficacy in phase II and ensuing studies is needed.
Acknowledgments
We thank all of the participating patients and their families, as well as the investigators and clinical research coordinators. We are grateful to Yutaka Ariyoshi, Kazuo Tamura, and Hironobu Minami, who served as members of the Data and Safety Monitoring Committee. This study was sponsored by Taiho Pharmaceutical Co., Ltd.
Disclosure Statement
The study was designed under the responsibility of Taiho Pharmaceutical Co., Ltd. Katsumasa Kuroi, Toshinari Yamashita, and Kazuhiko Nakagawa received honoraria from Taiho. Toshiaki Saeki received honoraria and research funding from Taiho. The other authors declare no conflict of interest.
Supporting Information
Table S1. Dose interruption criteria within treatment cycle.
Table S2. Dose reduction criteria for nab-paclitaxel and S-1.
Table S3. Dose reduction schema for nab-paclitaxel and S-1.
References
- Cardoso F, Bedard PL, Winer EP, et al. International guidelines for management of metastatic breast cancer: combination vs sequential single-agent chemotherapy. J Natl Cancer Inst. 2009;101:1174–81. doi: 10.1093/jnci/djp235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shaughnessy J, Miles D, Vukelja S, et al. Superior survival with capecitabine plus docetaxel combination therapy in anthracycline-pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol. 2002;20:2812–23. doi: 10.1200/JCO.2002.09.002. [DOI] [PubMed] [Google Scholar]
- Albain KS, Nag SM, Calderillo-Ruiz G, et al. Gemcitabine plus paclitaxel versus paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26:3950–7. doi: 10.1200/JCO.2007.11.9362. [DOI] [PubMed] [Google Scholar]
- Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7:1041–53. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil–based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- Gradishar WJ, Krasnojon D, Cheporov S, et al. Significantly longer progression-free survival with nab-paclitaxel compared with docetaxel as first-line therapy for metastatic breast cancer. J Clin Oncol. 2009;27:3611–9. doi: 10.1200/JCO.2008.18.5397. [DOI] [PubMed] [Google Scholar]
- Gradishar WJ, Krasnojon D, Cheporov S, et al. Phase II trial of nab-paclitaxel compared with docetaxel as first-line chemotherapy in patients with metastatic breast cancer: final analysis of overall survival. Clin Breast Cancer. 2012;12:313–21. doi: 10.1016/j.clbc.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Shirasaka T, Shimamato Y, Ohshimo H, et al. Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs. 1996;7:548–57. doi: 10.1097/00001813-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Saeki T, Takashima S, Sano M, et al. A phase II study of S-1 in patients with metastatic breast cancer–a Japanese trial by the S-1 Cooperative Study Group, Breast Cancer Working Group. Breast Cancer. 2004;11:194–202. doi: 10.1007/BF02968301. [DOI] [PubMed] [Google Scholar]
- Mukai H, Takashima T, Hozumi Y, et al. Randomized study of taxane versus TS-1 in women with metastatic or recurrent breast cancer (SELECT BC) Jpn J Clin Oncol. 2010;40:811–4. doi: 10.1093/jjco/hyq054. [DOI] [PubMed] [Google Scholar]
- Hara F, Matsubara N, Saito T, et al. Randomized phase III study of taxane versus TS-1 as first-line treatment for metastatic breast cancer (SELECT BC) J Clin Oncol. 2014;32:1012. [Google Scholar]
- Puglisi F, Andreetta C, Valent F, et al. Anthracyclines and taxanes induce the upregulation of thymidine phosphorylase in breast cancer cells. Anticancer Drugs. 2007;18:883–8. doi: 10.1097/CAD.0b013e32816ebede. [DOI] [PubMed] [Google Scholar]
- Nukatsuka M, Fujioka A, Nakagawa F, et al. Antimetastatic and anticancer activity of S-1, a new oraldihydropyrimidine-dehydrogenase-inhibiting fluoropyrimidine, alone and in combination with paclitaxel in an orthotopically implanted human breast cancer model. Int J Oncol. 2004;25:1531–6. [PubMed] [Google Scholar]
- Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- Mosteller RD. Simplified calculation of body-surface area. N Engl J Med. 1987;317:1098. doi: 10.1056/NEJM198710223171717. [DOI] [PubMed] [Google Scholar]
- Fujimoto S, Watanabe T, Sakamoto A, Yukawa K, Morimoto K. Studies on the physical surface area of Japanese. 18. Calculation formulae in three stages over all ages. Nippon Eiseigaku Zasshi. 1968;23:443–50. doi: 10.1265/jjh.23.443. [DOI] [PubMed] [Google Scholar]
- Cardoso F, Costa A, Norton L, et al. 1st International consensus guidelines for advanced breast cancer (ABC 1) Breast. 2012;21:242–52. doi: 10.1016/j.breast.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Cardoso F, Harbeck N, Fallowfield L, Kyriakides S, Senkus E ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23:vii11–9. doi: 10.1093/annonc/mds232. [DOI] [PubMed] [Google Scholar]
- Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of Cremophor-free, albumin-bound paclitaxel, ABI-007, compared with Cremophor-based paclitaxel. Clin Cancer Res. 2006;12:1317–24. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- Hirata K, Horikoshi N, Aiba K, et al. Pharmacokinetic study of S-1, a novel oral fluorouracil antitumor drug. Clin Cancer Res. 1999;5:2000–5. [PubMed] [Google Scholar]
- Yamada K, Yamamoto N, Yamada Y, Mukohara T, Minami H, Tamura T. Phase I and pharmacokinetic study of ABI-007, albumin-bound paclitaxel, administered every 3 weeks in Japanese patients with solid tumors. Jpn J Clin Oncol. 2010;40:404–11. doi: 10.1093/jjco/hyp192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando M, Yonemori K, Katsumata N, et al. Phase I and pharmacokinetic study of nab-paclitaxel, nanoparticle albumin-bound paclitaxel, administered weekly to Japanese patients with solid tumors and metastatic breast cancer. Cancer Chemother Pharmacol. 2012;69:457–65. doi: 10.1007/s00280-011-1726-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Dose interruption criteria within treatment cycle.
Table S2. Dose reduction criteria for nab-paclitaxel and S-1.
Table S3. Dose reduction schema for nab-paclitaxel and S-1.