

Abstract
We have previously reported that an adaptor protein CRK, including CRK-I and CRK-II, plays essential roles in the malignant potential of various aggressive human cancers, suggesting the validity of targeting CRK in molecular targeted therapy of a wide range of cancers. Nevertheless, the role of CRK in human bladder cancer with marked invasion, characterized by distant metastasis and poor prognosis, remains obscure. In the present study, immunohistochemistry indicated a striking enhancement of CRK-I/-II, but not CRK-like, in human bladder cancer tissues compared to normal urothelium. We established CRK-knockdown bladder cancer cells using 5637 and UM-UC-3, which showed a significant decline in cell migration, invasion, and proliferation. It is noteworthy that an elimination of CRK conferred suppressed phosphorylation of c-Met and the downstream scaffold protein Gab1 in a hepatocyte growth factor-dependent and -independent manner. In epithelial–mesenchymal transition-related molecules, E-cadherin was upregulated by CRK elimination, whereas N-cadherin, vimentin, and Zeb1 were downregulated. A similar effect was observed following treatment with c-Met inhibitor SU11274. Depletion of CRK significantly decreased cell proliferation of 5637 and UM-UC-3, consistent with reduced activity of ERK. An orthotopic xenograft model with bioluminescent imaging revealed that CRK knockdown significantly attenuated not only tumor volume but also the number of circulating tumor cells, resulted in a complete abrogation of metastasis. Taken together, this evidence uncovered essential roles of CRK in invasive bladder cancer through the hepatocyte growth factor/c-Met/CRK feedback loop for epithelial–mesenchymal transition induction. Thus, CRK might be a potent molecular target in bladder cancer, particularly for preventing metastasis, leading to the resolution of clinically longstanding critical issues.
Keywords: Bladder cancer, c-Met, CRK, EMT, metastasis
Bladder cancer (BC) is the fourth most common cancer, occupying the eighth position among the estimated cancer death causes in the world.1 BC is one of the most aggressive human genitourinary cancers. Overall, the 5-year survival rate for BC is approximately 80%, falling to only 6% for metastatic disease. After BC progresses to metastatic disease, the residual treatment option is cisplatin-based combination chemotherapy.2 Therefore, an elucidation of the molecular mechanisms underlying local invasion linked to metastasis is obligatory for an identification of novel therapeutic targets, ultimately improving the overall outcome.
Increasing evidence indicates that epithelial–mesenchymal transition (EMT) has a critical role in tumor invasion and metastasis. In the process of EMT, expression levels of epithelial markers such as E-cadherin are reduced, whereas those of mesenchymal markers such as Snail, Slug, Zeb1, N-cadherin, and vimentin are increased, leading to a morphological change from epithelial to mesenchymal-like appearance, such as spindle shape. EMT also plays important roles in therapeutic resistance, tumor recurrence, and metastatic progression in various cancers.3,4 In BC, the EMT expression profile is associated with disease progression.5
The signaling adaptor protein CRK was originally isolated as an oncogene fusion product of an avian sarcoma CT10 retrovirus.6 Its mammalian homologs include CRK-I, CRK-II, and CRK-L (CRK-like). CRK-I consists of an SH2 and an SH3 domain, whereas CRK-II contains an additional SH3 domain. Elevated levels of CRK-I/-II mRNAs and the protein are associated with malignant potential of various human tumors, including ovarian cancer, synovial sarcoma, glioblastoma, and breast cancer.7–10 CRK promotes EMT and is required for hepatocyte growth factor (HGF)-mediated cell spreading in kidney epithelial cells.11 We have previously shown that in synovial sarcoma cells, CRK is required for sustained phosphorylation of the SH2 domain binding protein Gab1 in response to HGF stimulation, and the consequent downstream Rac1 activation.12 These reports suggest that CRK has the potential to modulate EMT through growth factors including HGF.
The involvement of CRK in the invasiveness and metastasis of human BC remains unclear. In the present study, we investigated whether CRK is highly expressed in BC and its contribution to malignant properties in vitro and in vivo. We uncovered novel mechanisms underlying CRK-induced EMT through the HGF/c-Met/CRK-mediated positive feedback loop. Moreover, we elucidated the contribution of CRK on metastasis of BC in an orthotopic mouse xenograft model that we originally established.
Materials and Methods
Cell lines and antibodies
Four BC cell lines, T24, UM-UC-3, 5637, and RT4, were purchased from the ATCC (Manassas, VA, USA). The cells were cultured in McCoy's 5A (T24 and RT4), DMEM (UM-UC-3), and RPMI-1640 (5637) containing 10% FBS. Antibodies used are described in Data S1.
Establishment of Crk knockdown cells
BC cell lines were cotransfected with a plasmid producing siRNAs for CRK (pSUPER-i-Crk) and pBabe-puro using FuGENE HD transfection reagent (Promega, Madison, WI, USA) as described previously.7,9 Following selection with 3–10 μg/mL puromycin, colonies were isolated, and expression levels of CRK were analyzed by immunoblotting.
Establishment of UM-UC-3 cells stably expressing tdTomato-Luc2
UM-UC-3 cells were stably transfected with pCSII-CMV-tdTomato-Luc2, followed by selection using 0.5 mg/mL bleomycin. The resultant survival cells were sorted by FACS Aria II (Becton Dickinson, Tokyo, Japan), and the cells with excessive expression of tdTomato were isolated. Luciferase activity was examined by luciferase assay (Promega).
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblot analyses were carried out as described previously.7–9
RNA extraction and RT-PCR
RNA extraction and RT-PCR were carried out as described previously.4,9 Bladder tumor specimens from patients at Hokkaido University Hospital (Sapporo, Japan), obtained after informed consent, were used for semiquantitative RT-PCR. Sequences of the primers used are listed in Table S1. Details are described in Data S1.
Wound healing assay, invasion assay, and cell proliferation assay
The wound healing assay assessing cell motility, Matrigel invasion assay, and cell proliferation assay were carried out as described previously.7,8 Details are described in Data S1.
Xenograft model
Six- to 8-week-old female nude mice, Balb/cA Jcl nu/nu (Clea Japan, Tokyo, Japan) were used as the orthotopic xenograft model. After the mice were anesthetized with either 1.75% isoflurane or sodium pentobarbital (60 mg/kg) by i.p. injection, an incision of approximately 6 mm was made in the lower abdomen, followed by direct injection of 5 × 105 tumor cells into the bladder wall using a 30G needle under magnification. All animal experiments were carried out in accordance with the protocol approved by the Institutional Animal Care and Use Committee at the Hokkaido University Graduate School of Medicine.
Bioluminescent imaging was carried out using the IVIS Spectrum imaging system (Caliper Life-Sciences, Hopkinton, MA, USA) post-i.p. injection of VivoGlo Luciferin in vivo Grade (Promega). Values are expressed as relative light units per second at 15 min after injection.
Histological analysis and immunohistochemistry
Formalin-fixed paraffin-embedded tissues, including human BC specimens (surgically resected in the Department Urology, Hokkaido University Graduate School of Medicine) and UM-UC-3-derived xenografts, were sectioned and stained with H&E. Immunohistochemistry was carried out using antibodies to CRK, CRK-L, Ki67 (MIB1; Dako, Glostrup, Denmark), and MMP-9 (Santa Cruz Biotechnology, Dallas, TX, USA) antibodies.
Statistical analysis
The data represent the average means of triplicate experiments and are presented as mean and SEM or mean and SD where indicated. Student's t-test or the Mann–Whitney U-test was chosen to analyze the statistical difference. P < 0.05 was considered significant and P < 0.01 was considered highly significant.
Results
CRK-I/CRK-II highly expressed in human BC tissues and cell lines
The signaling adaptor protein CRK has been reported to be overexpressed in various cancers,13 and was shown to be linked to various aspects of tumor development, including cell migration, invasion, adhesion, and growth.7–10 However, its role in malignant features of BC, particularly in aggressive invasion/metastasis causing poor prognosis, remains unknown. In the present study, we first investigated the expression levels of CRK family adaptors in human BC tissues. Immunohistochemistry indicated a striking enhancement of CRK-I and CRK-II in all of the BC tissues tested, including non-invasive (n = 5) and invasive cancers (n = 5), compared to that of normal urothelium (n = 5) (Fig.1a,b). The nuclear staining of CRK-I/II was prominent in non-invasive BC, but not in invasive BC, suggesting a pro-apoptotic function of nuclear CRK mediated by interactions with cell cycle protein Wee1 and Crm1/Exportin as a cellular defense mechanism in early-stage cancer (Fig.1b).14,15 The expression level of CRK-L was low. Reverse transcription PCR revealed that the expression levels of CRK-I and CRK-II mRNAs were increased in BCs with low (n = 4) and high (n = 12) grade, relative to normal urothelium (n = 3) (Fig.1c).
Figure 1.
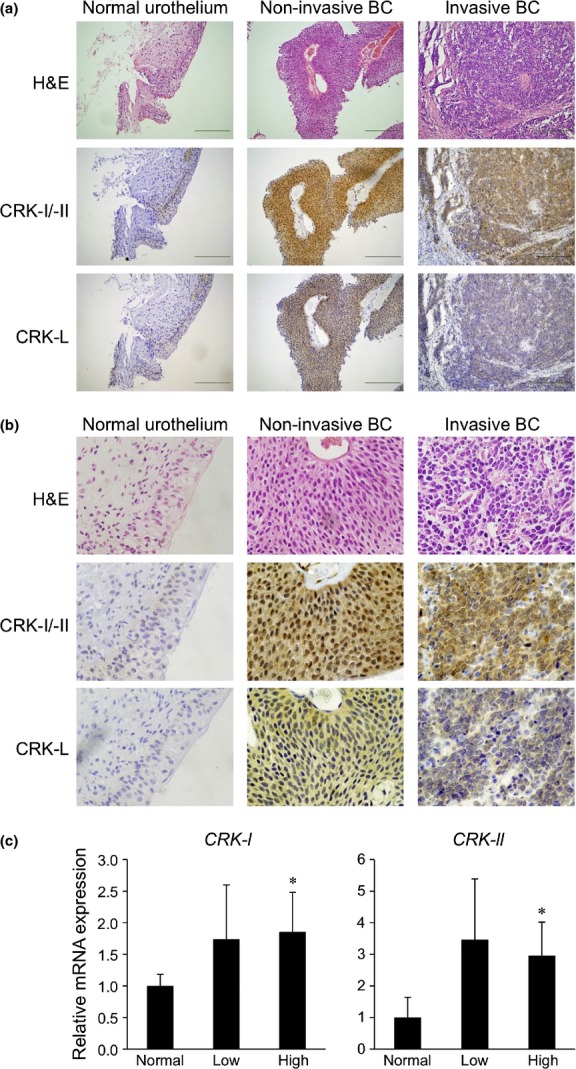
Adaptor proteins CRK-I and CRK-II are overexpressed in human BC tissues. (a,b) Clinical specimens of normal urothelium tissues (n = 5) and non-invasive (n = 5) and invasive BC (n = 5) were subjected to H&E staining and immunostaining for CRK-I/-II and CRK-L. (a) Scale bar = 100 μm. (b) Higher magnification images of part in FIg. 1(a) are displayed. (c) Expression levels of CRK-I and CRK-II mRNAs in surgical specimens, including normal urothelium (n = 3) and low- (n = 4) and high-grade (n = 12) BC, were examined by semiquantitative RT-PCR. *P < 0.05 versus normal urothelium. CRK-L, CRK-like.
In four BC cell lines (T24, UM-UC-3, and 5637 were originated from invasive type of cancer, and RT4 from non-invasive type), CRK-II expression was significantly higher than CRK-I, and CRK-L was also considerably detected (Fig.2a). Among the three invasive cell lines, CRK-related focal adhesion molecules, p130Cas and paxillin, were tyrosine-phosphorylated, and were especially high in invasive T24 and 5637 cells (Fig.2b). Of note, CRK-I and CRK-II possessed higher binding abilities to the downstream targets, guanine nucleotide exchange factors such as Dock180 and C3G, than that of CRK-L (Fig.2c), suggesting distinct functional roles of CRK-I and CRK-II in regulating cell motility and adhesion of invasive BC.
Figure 2.
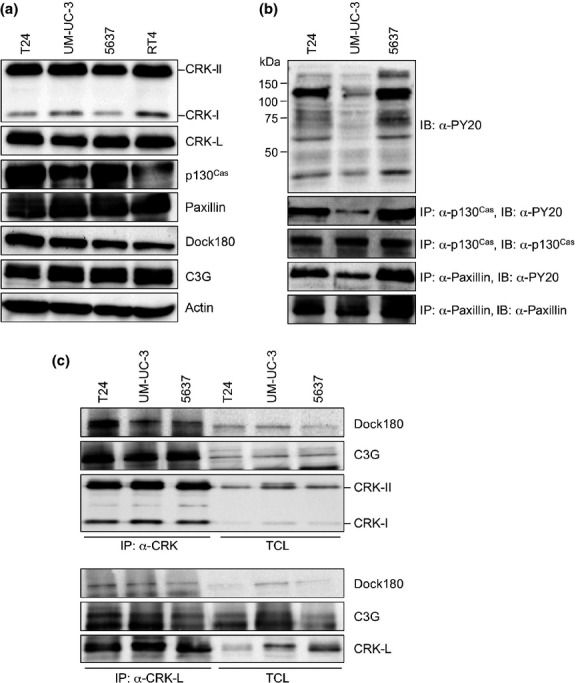
Expression levels of CRK and its related proteins in BC cell lines. (a) In BC cell lines (invasive, T24, UM-UC-3, 5637; non-invasive, RT4), expression levels of CRK adaptors and the indicated proteins were investigated by immunoblotting (IB). (b) In invasive BC cell lines, tyrosine phosphorylation levels of p130Cas and paxillin were examined by immunoprecipitation (IP), followed by IB with anti-tyrosine phosphorylation Ab (PY20). (c) Binding between CRK adaptors and downstream Dock180 and C3G. Lysates were immunoprecipitated with anti-CRK (for CRK-I/-II) or anti-CRK-L Abs, followed by IB using anti-Dock or anti-C3G Abs. TCL, total cell lysate; CRK-L, CRK-like.
CRK knockdown decreased motility and invasion in invasive BC cells
To clarify the significance of CRK-I and CRK-II in the malignant features of invasive BC, we undertook the stable knockdown of CRK-I/-II in three invasive BC cell lines, and succeeded in establishing 5637 and UM-UC-3 cells with the marked depletion (CRKi; Fig.3a). Elimination of CRK impaired cell spreading, leading to morphological changes, in contrast to their parental (WT) and control (empty) cells with an elongated mesenchymal-like shape (Fig.3b). Wound healing and Matrigel invasion assays revealed that cell motility and invasion were significantly decreased by CRK depletion in parallel to the knockdown efficiencies (Fig.3c,d). The phospho-p130Cas/CRK complex formation in focal adhesion has been shown to modulate the migration and invasion of cancer cells,16,17 and here we found that CRK elimination induced a decline in phosphorylation of p130Cas in 5637 and UM-UC-3 cells (Fig.3e).
Figure 3.
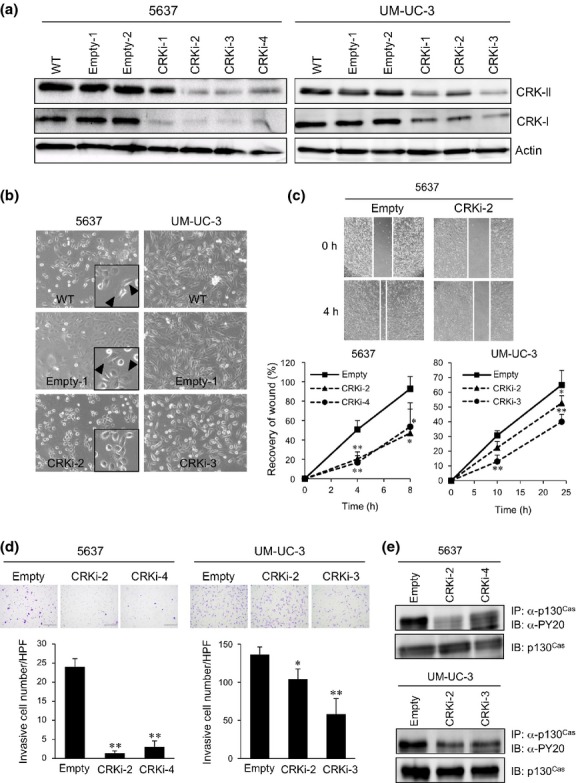
Knockdown of CRK suppresses cell spreading, motility, and invasion of BC cells. (a) Establishment of CRK knockdown bladder cancer cells. The 5637 and UM-UC-3 cells were stably transfected with expression plasmids producing shRNA targeting CRK (CRKi) or its control vector (empty). Cell lysate of WT, control (empty), and CRK knockdown cells (CRKi) were subjected to immunoblotting (IB) with anti-CRK Ab. Actin was used as a loading control. (b) Photomicrographs of 5637 and UM-UC-3 cells as indicated were taken under bright-field illumination. Arrowheads indicate elongated mesenchymal-like cells. (c) Wound healing assay. Extent of cell movement was calculated at the indicated time points, and displayed as mean ± SE of three independent experiments. Representative photomicrographs of 5637 cells are shown. (d) Matrigel invasion assay. Cells were seeded on Matrigel-coated Transwell chambers. After 24 h, the invading cells under the filter were counted and graphed as means ± SD. Representative photographs are shown. (e) In 5637 and UM-UC-3 cells with or without CRK, tyrosine phosphorylation levels of p130Cas were examined. *P < 0.05, **P < 0.01. IP, immunoprecipitation.
Tyrosine phosphorylation of c-Met was reduced by depletion of CRK in invasive BC cell lines
CRK is involved in diverse signaling transduction derived from various receptor tyrosine kinases such as epidermal growth factor receptor, fibroblast growth factor receptor, c-Met, nerve growth factor receptor, and platelet-derived growth factor receptor, cooperating with adaptor molecules such as IRS-1, Gab1, and Cbl.18–20 To identify CRK-associated signaling in invasive BC cells, the phosphorylation status of several receptor tyrosine kinases and downstream molecules were examined in the presence or absence of CRK-I/-II. The phosphorylation level of c-Met was definitely decreased by CRK elimination, whereas those of epidermal growth factor receptor and platelet-derived growth factor receptor-α were constant (Fig.4a). Src, FAK, and Gab1 have been shown to play a role in integrating signals from c-Met, coordinating with CRK.7,11,21 Indeed, the reduction of phosphorylation levels of Src and Gab1 was detected in Crk-depleted cells (Fig.4b). It is noteworthy that Crk knockdown significantly decreased expression levels of HGF mRNA, a ligand of the c-Met receptor (Fig.4c). Taken together, in invasive BC, CRK contributes to promote c-Met signaling by facilitating Gab1/p130Cas/Src complex assembly in focal adhesion and, ultimately, HGF expression.
Figure 4.
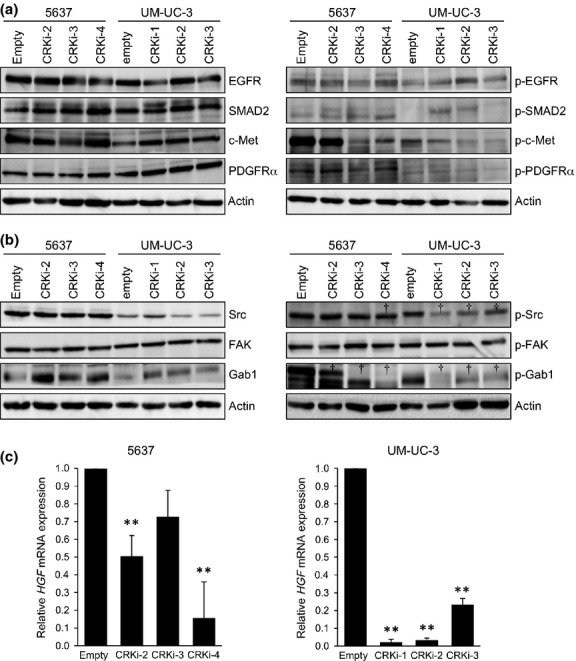
Knockdown of CRK reduces c-Met activation through impaired hepatocyte growth factor (HGF) production and a decline in Gab1 phosphorylation. (a,b) In 5637 and UM-UC-3 BC cells with or without CRK depletion, levels of the expression and phosphorylation (p-) of indicated proteins were analyzed by immuno-blotting. †Suppression of phosphorylation levels in CRK knockdown cells. (c) Total RNA was isolated from indicated cell lines, and endogenous expression level of HGF mRNA was examined by real-time RT-PCR. **P < 0.01 versus control cells (empty). EGFR, epidermal growth factor receptor; PDGFRα, platelet-derived growth factor receptor-α; SMAD2,
Knockdown of CRK suppresses EMT in invasive BC cells
CRK promotes an epithelial–mesenchymal-like transition and is required for HGF-mediated cell proliferation in kidney epithelial cells.11 Given that CRK-depleted BC cells markedly decreased the motility and invasion of BC cells (Fig.3c,d), we next examined the effect of CRK on EMT regulation. Four BC cell lines showed a variety of expression levels of EMT markers (Fig. S1). Quantitative RT-PCR indicated that CRK depletion significantly suppressed N-cadherin mRNA expression, accompanied with elevated E-cadherin (Fig.5a). Transcription factor Zeb1, which was highly expressed in UM-UC-3 cells (Fig. S1), was downregulated by Crk abrogation, as well as vimentin (Fig.5a). These events were also verified by immunoblotting (Fig.5b). MMP-2 and MMP-9 showed decreased expression in some Crk-depleted cells (Fig.5c). In UM-UC-3 cells, Met inhibitor SU11274 significantly reduced expression of N-cadherin, Zeb1, and vimentin, but conversely induced E-cadherin (Fig.5d), similar to the alterations produced by CRK depletion (Fig.5a).
Figure 5.
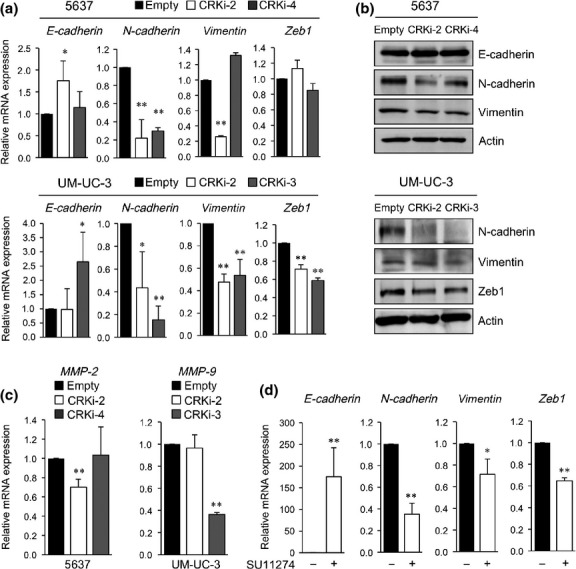
Depletion of CRK suppresses expression of epithelial–mesenchymal transition markers in bladder cancer cells. (a,b) Expression levels of mRNAs and proteins of epithelial–mesenchymal transition markers were examined by real-time RT-PCR (a) and immunoblotting (b). (c) Expression levels of MMP-2 and MMP-9mRNAs were examined by real-time RT-PCR in 5637 and UM-UC-3 cells, respectively. (d) UM-UC-3 cells were incubated in the presence of c-Met inhibitor SU11274 (1 μM), or DMSO as a control, for 4 h. Expression levels of E-cadherin, N-cadherin, vimentin, and Zeb1 were analyzed by real-time RT-PCR. *P < 0.05, **P < 0.01.
Knockdown of CRK reduced growth of invasive BC cells
Given that CRK has been shown to upregulate cell growth,7–9 we investigated the role of CRK in cell proliferation. Compared to control cells, the growth rates of CRK-knockdown cells were significantly lower in vitro (Fig.6a). Consistent with this, a decline in phosphorylation level of ERK, but not Akt, was observed with CRK depletion (Fig.6b). To further analyze the contribution of CRK to tumor development and progression of BC, we established UM-UC-3 cells labeled with tdTomato-Luc2 in the presence or absence of CRK, enabling us to monitor non-invasively the growth of primary tumor and metastasis using the IVIS imaging system.22 Cells (5 × 105) were s.c. injected into nude mice (Data S1). Tumors formed by the CRK-knockdown cells developed slowly (Fig. S2a,b); after 28 days, the weight of tumors was significantly lower than the controls (Fig. S2c,d). Staining of MIB-1 and MMP-9 was lower in the CRK-depleted tumors (Fig. S2e).
Figure 6.
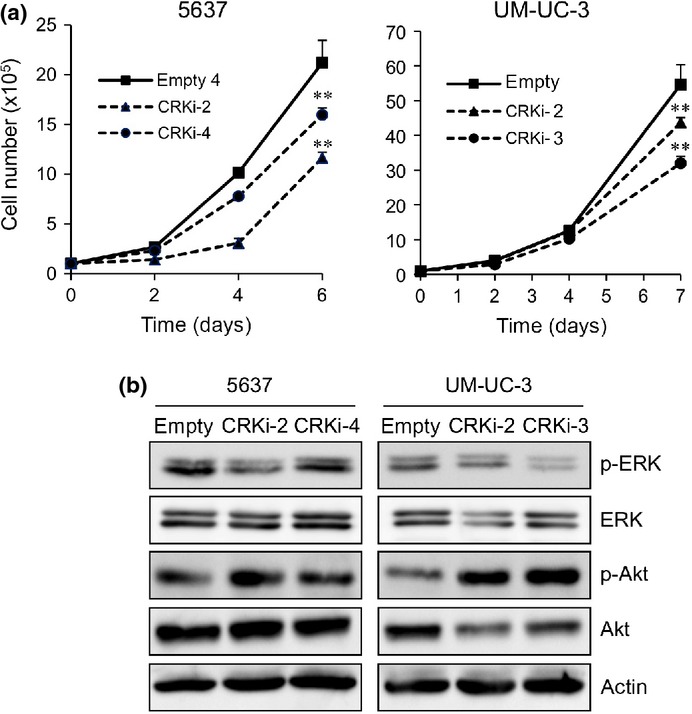
Elimination of CRK inhibits proliferation of BC cells through a decline in phosphorylation of ERK. (a) Proliferation of 5637 and UM-UC-3 cells in the presence or absence of CRK. The cells were counted under a microscope at the indicated time points, and expressed as means ± SE of three independent experiments. **P < 0.01. (b) Levels of expression and phosphorylation of ERK and Akt were examined by immunoblotting.
Elimination of CRK decreases primary tumor growth, circulating tumor cells, and metastasis in orthotopic xenograft model
To mimic human BC, we used orthotopic xenograft model. UM-UC-3 cells (5 × 105) with or without CRK were injected under the bladder muscle layer, and tumor growth and metastasis were monitored weekly using the IVIS imaging system. Tumor growth was significantly reduced in the CRK-knockdown group (Fig.7a,b). Forty-eight days after implantation, the weight of CRK-depleted tumors was significantly decreased compared to those of controls (Fig.7c). It is noteworthy that CRK-knockdown significantly declined the number of circulating tumor cells, and almost completely abolished metastases to the liver and lung (Fig.7d). The primary bladder tumor and the metastatic region were subjected to immunohistochemical analysis (Data S1, Fig.7e), which indicated an abrogation of invasive features with declines in MMP-9 expression (Fig.7f) and c-Met phosphorylation (Fig. S3) in CRK-depleted tumors.
Figure 7.

Knockdown of CRK inhibits tumor growth and circulating tumor cells, and completely abolishes BC metastasis in vivo. UM-UC-3 cells labeled with tdTomato-luc2 (control, empty; CRK depletion, CRKi-3) were orthotopically injected under the bladder muscle layer in athymic mice (n = 5 each group). (a,b) Tumor growth was measured weekly by a bioluminescence imaging system, and graphed as mean ± SE from five mice in each group (b). (c) After 48 days, the mice were killed and the weight of the excised primary bladder tumors was measured. (d) After 48 days, bioluminescent imaging of photons from blood (100 μL), and liver and lung metastatic tumor burden was taken ex vivo (left) and plotted (right). (e) Excised tumors were fixed and stained with H&E. In mice injected with CRKi cells, the metastasis to the liver and lung was absent. (f) Primary bladder tumors were subjected to immunostaining for CRK, MIB-1, and MMP9. *P < 0.05, **P < 0.01.
Discussion
New molecular target drugs have been emerging in cancer therapy; however, in progressive BC, the sole treatment option is cisplatin-based combination chemotherapy, ultimately linked to poor prognosis. Therefore, an identification of novel therapeutic targets to control the proliferation, invasion, and metastasis of tumor cells is urgently needed. Previous studies reported that CRK overexpression in various aggressive cancers is associated with tumor progression.7,10,13,23 Based on the evidence from the present study, we propose that CRK is a potential therapeutic molecular target in invasive BC. Indeed, CRK-I and CRK-II were excessively expressed in BC tissue specimens compared to normal urothelium. In two independent invasive BC cell lines, simultaneous knockdown of CRK-I and CRK-II reduced cell proliferation, motility, and invasiveness through an impaired HGF/c-Met feedback loop (Fig. S4). These findings were also verified in an orthotopic xenograft model, which revealed a decline in primary tumor growth, circulating tumor cells, and metastatic potential by CRK depletion. To the best of our knowledge, the present study provides the first functional and mechanistic role of CRK in human BC cells.
Adaptor protein CRK has been shown to play a role in HGF/c-Met-mediated biological responses involved in morphogenesis, EMT, cell spreading, and breakdown of epithelial adherence junctions.11,24,25 Overexpression of c-Met and enhanced phosphorylation have been reported in human BC, associated with metastatic progression and poor prognosis;26,27 however, the underlying molecular mechanisms triggering c-Met activation were obscure. In the present study, using specific shRNA targeting both CRK-I and CRK-II, we clarified a crucial role of CRK in c-Met-mediated EMT induction, through the sustained phosphorylation of Gab1 and enhancement of HGF production. We have previously shown that the elimination of CRK in synovial sarcoma cells induced the disorganization of actin cytoskeleton and complete abolition of HGF-mediated Rac1 activation and cell motility.12 In addition, CRK facilitates tyrosine phosphorylation of Gab1-Y307 through Src, contributing to the organization of focal adhesions and enhanced cell migration.21 These findings suggest that CRK plays an essential role in the long-term activation of c-Met signaling, by promoting the assembly of Gab1/p130Cas/Src/CRK. In addition, we uncovered the novel mechanism underlying the constitutive activation of c-Met signaling through the HGF/c-Met/CRK positive feedback loop, which effectively induces EMT, ultimately leading to a facilitation of BC progression (Fig. S4).
EMT is a process in which tumor cells change their epithelial properties to mesenchymal features and acquires metastatic potential in various tumor cells, including BC.3,5,28 Indeed, several studies have shown an association between HGF/c-MET signaling and EMT.29,30 In the present study, a depletion of CRK protein suppressed the expression levels of EMT markers such as N-cadherin and vimentin, and conversely increased E-cadherin (Fig.5a,b). In UM-UC-3 cells, the expression levels of Zeb1 and MMP-9 were suppressed by CRK elimination, whereas that of MMP-2 was unaltered (Fig.5a,c). In contrast, CRK knockdown in 5637 cells led to a decline in MMP-2, but not in Zeb1 or MMP-9 (Fig.5a,c, data not shown). These results suggest that c-Met/CRK signaling definitely provokes EMT in BC, but the underlying molecular mechanisms seem to be partly different depending on the cellular lineage. Since CRK is a pivotal regulator in c-Met-mediated EMT, albeit differences in the downstream signaling, raising CRK as an effective therapeutic molecular target to depress EMT phenomena.
The Ras-ERK-MAPK signaling pathway is well known to play a critical role in the regulation of cell proliferation, survival, and differentiation.31 In fact, previous studies have shown that Src, Grb2, Gab1, CRK, and p130Cas act as downstream mediators leading to the activation of ERK and MAPK,8,32 and that ERK activation was necessary for Ras-induced EMT.33 All of these results suggest that c-Met/CRK signaling might synergistically regulate EMT with the Ras-ERK-MAPK pathway. Further investigation is therefore necessary to identify the precise mechanisms responsible for CRK-mediated EMT in BC.
In conclusion, our results clearly indicated that CRK promoted the proliferation, migration, invasion, and progression of BC. CRK overexpression in human BC specimens was detected by RT-PCR and immunohistochemistry. Depletion of CRK by shRNA effectively inhibited BC cell proliferation and migration in an in vitro setting. Of note, in the orthotopic xenograft model, CRK elimination significantly decreased BC invasion and metastasis by suppressing EMT. Taken together, we propose here that CRK as a potent therapeutic target for BC, especially in invasive cancer.
Acknowledgments
We thank all members of our laboratory for helpful discussions. This work was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Japan Society for the Promotion of Science, the Ministry of Health, Labor, and Welfare of Japan, and the Japan Science and Technology Agency.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Fig. S1. Four BC cell lines showed a variety of expression levels of epithelial–mesenchymal transition markers.
Fig. S2. UM-UC-3 cells were s.c. injected into nude mice, and CRK knockdown attenuated the tumor growth.
Fig. S3. UM-UC-3 cells were orthotopically injected into nude mice, and the phosphorylation levels of c-Met in the formed tumors declined with CRK elimination.
Fig. S4. An essential role for CRK was evident in promoting proliferation, invasion, and metastasis in BC through the c-Met-mediated positive feedback loop for inducing epithelial–mesenchymal transition.
Table S1. Sequences of primers for quantitative real-time RT-PCR.
Data S1. Supplementary materials and methods.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Bellmunt J, Albiol S, Suarez C, Albanell J. Optimizing therapeutic strategies in advanced bladder cancer: update on chemotherapy and the role of targeted agents. Crit Rev Oncol Hematol. 2009;69:211–22. doi: 10.1016/j.critrevonc.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Mahabir R, Tanino M, Elmansuri A, et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014;16:671–85. doi: 10.1093/neuonc/not239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart E, Cohen MS, Neto BS, et al. Identification and prognostic significance of an epithelial-mesenchymal transition expression profile in human bladder tumors. Clin Cancer Res. 2007;13:1685–94. doi: 10.1158/1078-0432.CCR-06-2330. [DOI] [PubMed] [Google Scholar]
- Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–5. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- Linghu H, Tsuda M, Makino Y, et al. Involvement of adaptor protein Crk in malignant feature of human ovarian cancer cell line MCAS. Oncogene. 2006;25:3547–56. doi: 10.1038/sj.onc.1209398. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tsuda M, Tanaka S, et al. Adaptor protein Crk induces Src-dependent activation of p38 MAPK in regulation of synovial sarcoma cell proliferation. Mol Cancer Res. 2009;7:1582–92. doi: 10.1158/1541-7786.MCR-09-0064. [DOI] [PubMed] [Google Scholar]
- Wang L, Tabu K, Kimura T, et al. Signaling adaptor protein Crk is indispensable for malignant feature of glioblastoma cell line KMG4. Biochem Biophys Res Commun. 2007;362:976–81. doi: 10.1016/j.bbrc.2007.08.106. [DOI] [PubMed] [Google Scholar]
- Fathers KE, Bell ES, Rajadurai CV, et al. Crk adaptor proteins act as key signaling integrators for breast tumorigenesis. Breast Cancer Res. 2012;14:R74. doi: 10.1186/bcr3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte L, Royal I, Naujokas M, et al. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell. 2002;13:1449–61. doi: 10.1091/mbc.01-10-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Tsuda M, Makino Y, et al. Adaptor molecule Crk is required for sustained phosphorylation of Grb2-associated binder 1 and hepatocyte growth factor-induced cell motility of human synovial sarcoma cell lines. Mol Cancer Res. 2006;4:499–510. doi: 10.1158/1541-7786.MCR-05-0141. [DOI] [PubMed] [Google Scholar]
- Nishihara H, Tanaka S, Tsuda M, et al. Molecular and immunohistochemical analysis of signaling adaptor protein Crk in human cancers. Cancer Lett. 2002;180:55–61. doi: 10.1016/s0304-3835(01)00763-7. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Richardson DA, Kopf J, Yoshida M, Hollingsworth RE, Kornbluth S. Apoptotic regulation by the Crk adapter protein mediated by interactions with Wee1 and Crm1/exportin. Mol Cell Biol. 2002;22:1412–23. doi: 10.1128/mcb.22.5.1412-1423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar B, Reichman CT, Singh S, O'Connor JP, Birge RB. Proapoptotic function of the nuclear Crk II adaptor protein. Biochemistry. 2007;46:10828–40. doi: 10.1021/bi700537e. [DOI] [PubMed] [Google Scholar]
- Takino T, Nakada M, Miyamori H, Yamashita J, Yamada KM, Sato H. CrkI adapter protein modulates cell migration and invasion in glioblastoma. Cancer Res. 2003;63:2335–7. [PubMed] [Google Scholar]
- Coon M, Herrera R. Modulation of HeLa cells spreading by the non-receptor tyrosine kinase ACK-2. J Cell Biochem. 2002;84:655–65. doi: 10.1002/jcb.10078. [DOI] [PubMed] [Google Scholar]
- Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–71. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- Feller SM, Lewitzky M. Potential disease targets for drugs that disrupt protein-protein interactions of Grb2 and Crk family adaptors. Curr Pharm Des. 2006;12:529–48. doi: 10.2174/138161206775474369. [DOI] [PubMed] [Google Scholar]
- Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–9. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tsuda M, Makino Y, et al. Crk adaptor protein induced-phosphorylation of Gab1 on tyrosine 307 via Src is important for organization of focal adhesions and enhanced cell migration. Cell Res. 2009;19:638–50. doi: 10.1038/cr.2009.40. [DOI] [PubMed] [Google Scholar]
- van der Horst G, van Asten JJ, Figdor A, et al. Real-time cancer cell tracking by bioluminescence in a preclinical model of human bladder cancer growth and metastasis. Eur Urol. 2011;60:337–43. doi: 10.1016/j.eururo.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Yamada S, Yanamoto G, Kawasaki S, et al. Overexpression of CRKII increases migration and invasive potential in oral squamous cell carcinoma. Cancer Lett. 2011;303:84–91. doi: 10.1016/j.canlet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Lamorte L, Rodrigues S, Naujokas M, Park M. Crk synergizes with epidermal growth factor for epithelial invasion and morphogenesis and is required for the met morphogenic program. J Biol Chem. 2002;277:37904–11. doi: 10.1074/jbc.M201743200. [DOI] [PubMed] [Google Scholar]
- Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream form the met receptor tyrosine kinase. Mol Cell Biol. 2000;20:8513–25. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Trink B, Tzai TS, et al. Overexpression of c-met as a prognostic indicator for transitional cell carcinoma of the urinary bladder: a comparison with p53 nuclear accumulation. J Clin Oncol. 2002;20:1544–50. doi: 10.1200/JCO.2002.20.6.1544. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Sagara Y, Kanda S, Hayashi T, Kanetake H. Phosphorylated hepatocyte growth factor receptor/c-Met is associated with tumor growth and prognosis in patients with bladder cancer: correlation with matrix metalloproteinase-2 and -7 and E-cadherin. Hum Pathol. 2009;40:496–504. doi: 10.1016/j.humpath.2008.09.011. [DOI] [PubMed] [Google Scholar]
- McConkey DJ, Choi W, Marquis L, et al. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009;28:335–44. doi: 10.1007/s10555-009-9194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzo MG, Lesurf R, Petkiewicz S, et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106:12903–8. doi: 10.1073/pnas.0810402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM. MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene. 2014;33:4069–76. doi: 10.1038/onc.2013.369. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Blaukat A, Ivankovic-Dikic I, Grönroos E, et al. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J Biol Chem. 1999;274:14893–901. doi: 10.1074/jbc.274.21.14893. [DOI] [PubMed] [Google Scholar]
- Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. 2010;38:114–27. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Four BC cell lines showed a variety of expression levels of epithelial–mesenchymal transition markers.
Fig. S2. UM-UC-3 cells were s.c. injected into nude mice, and CRK knockdown attenuated the tumor growth.
Fig. S3. UM-UC-3 cells were orthotopically injected into nude mice, and the phosphorylation levels of c-Met in the formed tumors declined with CRK elimination.
Fig. S4. An essential role for CRK was evident in promoting proliferation, invasion, and metastasis in BC through the c-Met-mediated positive feedback loop for inducing epithelial–mesenchymal transition.
Table S1. Sequences of primers for quantitative real-time RT-PCR.
Data S1. Supplementary materials and methods.