

Summary
The development of new gene-editing tools, in particular the CRISPR/Cas system, has greatly facilitated site-specific mutagenesis in human embryonic stem cells (hESCs), including the introduction or correction of patient-specific mutations for disease modeling. However, integration of a reporter gene into an endogenous locus in hESCs still requires a lengthy and laborious two-step strategy that involves first drug selection to identify correctly targeted clones and then excision of the drug-resistance cassette. Through the use of iCRISPR, an efficient gene-editing platform we recently developed, this study demonstrates a knockin strategy without drug selection for both active and silent genes in hESCs. Lineage-specific hESC reporter lines are useful for real-time monitoring of cell-fate decisions and lineage tracing, as well as enrichment of specific cell populations during hESC differentiation. Thus, this selection-free knockin strategy is expected to greatly facilitate the use of hESCs for developmental studies, disease modeling, and cell-replacement therapy.
Highlights
-
•
A selection-free knockin strategy was established for making reporter hESC lines
-
•
OCT4-mOrange, OCT4-eGFP, and PDX1-eGFP hESC reporter lines were created
-
•
Faithful reporter activities were established
Current hESC knockin strategies typically include a drug-resistance cassette in the donor template for enrichment of correctly targeted clones. Huangfu and colleagues demonstrate a strategy for generating knockin reporter hESC lines without drug selection for both active and silent genes using the CRISPR/Cas9 system. This development may further facilitate the use of hESCs for disease modeling and cell-replacement therapy.
Introduction
Human embryonic stem cells (hESCs) are capable of unlimited self-renewal in culture while maintaining the potential to differentiate into any cell type present in the human body and thus provide researchers great opportunities for human developmental studies, disease modeling, and cell-replacement therapies (Zhu and Huangfu, 2013). All these applications benefit from lineage-specific knockin reporters that allow real-time observation of gene-expression dynamics, cell-lineage tracing, and isolation of a specific cell population of interest from a heterogeneous differentiation culture for downstream analysis. However, creating knockin alleles in hESCs is usually a lengthy and technically challenging process. Because of the low efficiency of homologous recombination, the donor vector needs to contain a drug-resistance gene for enrichment of cells with the correct integration. Due to the concern that the insertion of a drug-resistance cassette may interfere with the expression of the reporter gene or neighboring genes, it is usually necessary to remove the drug-resistance cassette through a second electroporation step followed by isolation of clonal lines and further characterization (Davis et al., 2008). Thus, substantial time and effort is needed to generate a knockin reporter hESC line through this two-step procedure.
The development of engineered “genomic scissors” that introduce site-specific DNA double-strand breaks (DSBs), including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and more recently the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system, has greatly facilitated gene targeting in hESCs (Kim and Kim, 2014). Repair of a DSB by non-homologous end joining (NHEJ) often results in insertion and/or deletions (Indels) that can be used to knock out a target gene in hESCs (Ding et al., 2013a, b; González et al., 2014). Alternatively, homology-directed repair (HDR) can be employed to efficiently incorporate exogenous sequences such as a fluorescent reporter into a specific genomic locus in hESCs (Hockemeyer et al., 2009, 2011; Hou et al., 2013; Merkert et al., 2014). Despite the significant improvement, a drug-resistance cassette is still required for generating knockin reporters of genes that are not expressed in undifferentiated hESCs.
To overcome these limitations, we made use of the CRISPR/Cas system, in which the CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) duplex or a single chimeric guide RNA (gRNA) recognizes a 20-nucleotide (nt) DNA sequence upstream of the 5′-NGG-3′ protospacer adjacent motif (PAM) and directs the DNA endonuclease Cas9 for site-specific cleavage (Cong et al., 2013; Jinek et al., 2012; Mali et al., 2013a). Based on this, we have developed an efficient genome-editing platform in hESCs, which we named iCRISPR (González et al., 2014). Through TALEN-mediated gene targeting in the AAVS1 locus, we have created hESC lines (referred to as iCas9 hESCs) that allow robust, doxycycline-inducible expression of Cas9. By transfecting iCas9 hESCs with gRNAs, the iCRISPR system enables efficient NHEJ-mediated gene disruption as well as HDR-mediated precise nucleotide modifications in the presence of short single-stranded DNA (ssDNA) donors (∼100 nt).
We reasoned that the iCRISPR system would also facilitate the generation of knockin reporter alleles using longer double-stranded (dsDNA) donors and may further enable the identification of correctly targeted hESC lines without drug selection. Thus, this work explores the utility of iCRISPR for targeting fluorescent reporters into two endogenous loci, OCT4 (POU5F1) and PDX1, and demonstrates the generation of knockin hESC lines without drug selection for both expressed and silent genes. Further characterization confirmed the creation of multiple hESC reporter lines with no undesired mutations in the targeted loci or any potential off-target sites analyzed, supporting the broad application of this approach for efficient generation of knockin alleles in hESCs.
Results
CRISPR/Cas-Mediated Targeting of the OCT4 Locus
We first evaluated the efficiency of the CRISPR/Cas system for making knockin reporter alleles by targeting the OCT4 locus using drug selection. HUES8 hESCs were co-electroporated with two plasmids: one expressing Cas9 and a crRNA/tracrRNA duplex targeting OCT4 and the other containing the fluorescent reporter and a drug-resistance cassette as the HDR template (Figures 1A, S1A, and S1B). We used a donor plasmid, 2A-eGFP-PGK-Puro (Hockemeyer et al., 2011), in which the last OCT4 coding codon is fused in frame with a 2A sequence followed by eGFP (2A-eGFP) and a loxP-flanked (floxed) puromycin-resistance gene expressed from the constitutive PGK promoter (PGK-Puro) (Figure 1A). This strategy minimizes any potential impact on the endogenous protein and is applicable to targeting both silent and expressed genes. PCR and Southern blot analysis identified eight correctly targeted clones without random transgene integration from a total of 288 puromycin-resistant clones screened (Figures 1B and S1C). The targeting efficiency (2.8%) was comparable to the efficiencies observed with TALENs and ZFNs using similar targeting strategies (Hockemeyer et al., 2009, 2011).
Figure 1.

CRISPR/Cas-Mediated Targeting of the OCT4 Locus through Drug Selection
(A) Schematics of the targeting strategy. In the presence of the donor plasmid, HDR results in the replacement of the OCT4 stop codon with 2A-eGFP-PGK-Puro. The PCR primers (F + GFP-R) used for genotyping are indicated with red arrowheads. OCT4 cr1-dp (the duplex version) targets a 30-nt sequence (indicated with a green line) upstream of the PAM sequence (indicated with a purple line). In all targeting schematics here and after, boxes are exons, filled blue boxes indicate the coding sequence (CDS), connecting lines are introns, the stop codon (TGA) is labeled in red, HL and HR indicate left and right homology arms, and the Southern blot external and internal probes are indicated with red bars.
(B) Southern blot analysis using the external probe (WT: 4,173 bp; GFP+Puro: 6,835 bp) and the internal puromycin probe (GFP+Puro: 2,415 bp) identified eight correctly targeted clones, which are labeled in red. WT, wild-type allele; GFP+Puro, correctly targeted allele with puromycin-selection cassette.
(C) Three of the correctly targeted clones (nos. 1, 4, and 7) were electroporated with Cre recombinase. Four days after electroporation, eGFP+ cells were isolated using FACS.
(D) For each clone (nos. 1, 4, and 7) electroporated with CRE recombinase, two GFP+ clones were picked (e.g., C1.1 and C1.2 for clones derived from no. 1), and Southern blot analysis using the external probe (WT: 4,173 bp; GFP+Puro: 6,835 bp; GFP only: 5,015 bp) and the internal puromycin probe (GFP+Puro: 2,415 bp) showed correct removal of the puromycin-selection cassette. A clone (C1) prior to Cre electroporation was used as the Pre-Cre control. GFP+Puro, targeted allele prior to Cre-mediated excision of the PGK-Puro cassette; GFP only, targeted allele after Cre-mediated excision.
Despite correct targeting, we failed to detect eGFP expression in any of the targeted lines. This is likely caused by the presence of the drug-resistance cassette as observed for other genes (Davis et al., 2008). Indeed, after Cre-mediated excision of the PGK-Puro cassette, all resulting OCT4-eGFP lines showed proper co-expression of eGFP with pluripotency markers OCT4, SOX2, and NANOG (Figures 1C, 1D, and S1D). These results highlight the necessity of removing the drug-resistance cassette for proper reporter gene expression.
Selection-free Targeting of the OCT4 Locus Using a Mini-vector Donor
To further explore the possibility of making knockin reporter alleles without drug selection, we designed a “mini-vector” donor plasmid, 2A-mOrange, which is similar to 2A-eGFP-PGK-Puro except that there is no PGK-Puro cassette and eGFP was replaced by mOrange (Figure 2A). We also replaced the crRNA/tracrRNA duplex cr1-dp with the single gRNA cr1 targeting the same sequence (Figures 2A and S2A), as the chimeric version works more efficiently than the original duplex design (Hsu et al., 2013; Jinek et al., 2012). Similar to the experiment with the 2A-eGFP-PGK-Puro donor, we co-electroporated HUES8 hESCs with a plasmid expressing Cas9/gRNA and the new 2A-mOrange mini-vector (Figure 2A). In contrast to the absence of fluorescence after integration of the 2A-eGFP-PGK-Puro cassette, integration of the 2A-mOrange cassette resulted in mOrange expression in ∼0.001% of cells as detected by fluorescence-activated cell sorting (FACS) (Figure S2B). One may enrich mOrange-expressing cells for establishing OCT4 reporter lines. However, this low efficiency is impractical for genes not expressed in undifferentiated hESCs, as one has to rely on randomly picking individual colonies to establish clonal lines.
Figure 2.
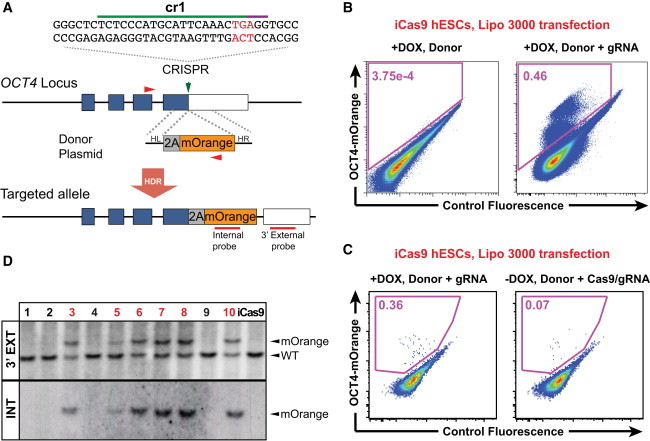
Targeting the OCT4 Locus without Drug Selection
(A) Schematics of the targeting strategy without drug selection. In the presence of the donor plasmid, HDR results in the replacement of the stop codon with 2A-mOrange. OCT4 cr1 targets a 20-nt sequence upstream of the PAM sequence. The PCR primers (F + mOr-R) used for genotyping are indicated with red arrowheads.
(B) FACS enrichment for OCT4-mOrange+ cells after transfection of the OCT4-mOrange donor plasmid and the OCT4-targeting gRNA into HUES8 iCas9 cells treated with doxycycline.
(C) FACS analysis for OCT4-mOrange fluorescence in doxycycline-treated HUES8 iCas9 cells co-transfected with the OCT4-mOrange donor plasmid and the OCT4-targeting gRNA, compared to HUES8 iCas9 cells (not treated with doxycycline) co-transfected with the Cas9/gRNA and the donor plasmids using Lipofectamine 3000.
(D) Ten colonies were randomly picked from individual FACS-enriched mOrange+ cells. Southern blot analysis using the external probe (WT: 4,173 bp; mOrange: 4,963 bp) and the internal mOrange probe (mOrange: 4,963 bp) identified six correctly targeted clones, which are labeled in red. mOrange: correctly targeted allele.
Our recent study shows that the iCRISPR platform enables efficient gene editing using short ssDNA HDR templates (González et al., 2014), prompting us to further optimize the iCRISPR platform for HDR using longer circular dsDNA donor vectors. After optimizing transfection conditions, we co-transfected doxycycline-treated HUES8 iCas9 cells twice in 2 days with the OCT4 cr1 gRNA and the 2A-mOrange mini-vector using Lipofectamine 3000 (Figures S2D and S3). FACS analysis identified ∼0.4% mOrange-expressing cells (Figure 2B), >100-fold greater than results from our electroporation experiments (Figures S2B and S2C). Similar results were observed in experiments using iCas9 cells generated from MEL-1 hESCs (Figure S2E), supporting the general utility of this new approach in diverse human pluripotent stem cell (hPSC) backgrounds. The much-improved efficiencies can be partially attributed to the integration of Cas9 in the genome as an ∼5- to 6-fold increase of mOrange+ cells was observed compared to the control condition where iCas9 hESCs (not treated with doxycycline) were transfected with Cas9/gRNA and the donor vector using Lipofectamine 3000 (Figure 2C). The use of Lipofectamine transfection also substantially increased the targeting efficiency compared to the electroporation method (Figures S2B and S2C). There may be other ways to improve the transfection efficiency (e.g., through nucleofection) to achieve similar results with or without the use of iCas9 hESCs (Byrne et al., 2015).
We picked ten colonies from individual FACS-isolated mOrange+ cells and identified six correctly targeted clones by PCR and Southern blot analysis (Figures 2B, 2D, and S2F). All six lines co-expressed mOrange with pluripotency markers such as OCT4, SOX2, and NANOG and displayed normal hESC morphology (Figure S2G). We further examined the OCT4-mOrange hESC reporter lines along with the OCT4-eGFP lines for reporter gene expression after differentiation. After 3 days of treatment with BMP4 and SB431542, a TGFβ inhibitor (Hou et al., 2013), hESCs exhibited a differentiated morphology, and eGFP and mOrange expression were downregulated in the respective OCT4-eGFP and OCT4-mOrange hESC reporter lines with concomitant loss of endogenous OCT4 expression as determined by immunostaining and FACS analysis (Figures 3A and 3B). Thus, the OCT4-eGFP and OCT4-mOrange reporters faithfully reflect endogenous gene expression during the maintenance and differentiation of hESCs.
Figure 3.
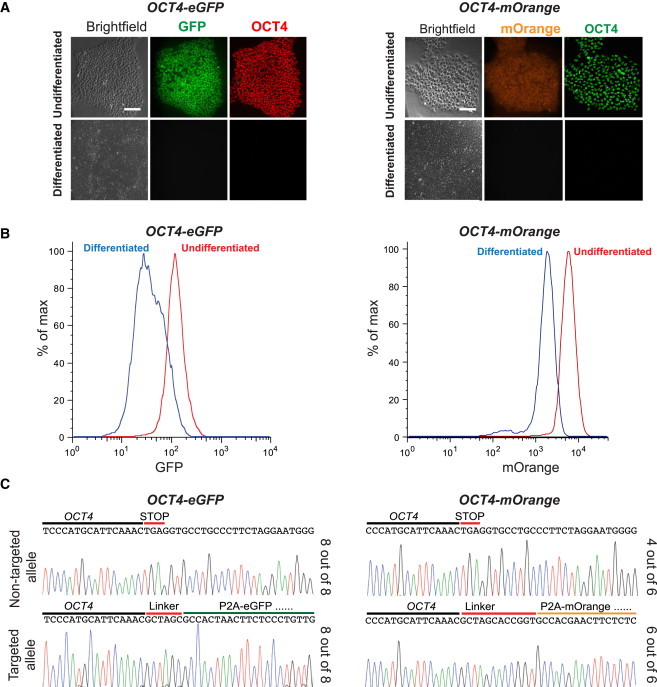
Characterization of OCT4 Reporter Lines
(A) OCT4-eGFP and OCT4-mOrange hESCs were treated with SB431542 and BMP4 to initiate differentiation. Three days after this treatment, the cells displayed concomitant loss of OCT4 protein expression with GFP or mOrange by immunostaining. An RFP antibody was used to detect mOrange expression, whereas the GFP expression was detected directly. The scale bar represents 100 μm.
(B) Three days after SB431542 and BMP4 treatment, flow cytometry analysis showed a loss of GFP and mOrange, verifying that OCT4-eGFP and OCT4-mOrange reporter hESCs can respond to differentiation cues and that GFP and mOrange accurately reflects OCT4 expression.
(C) Sequencing results of the non-targeted allele and at the junction of correctly targeted allele in OCT4-eGFP and OCT4-mOrange reporter lines.
We next investigated whether the relatively high targeting efficiency was achieved at the expense of undesirable mutations at the OCT4 locus or any off-target sites. All eight OCT4-eGFP and six OCT4-mOrange lines examined showed the expected sequence at the junction between the endogenous OCT4 sequence and the inserted sequence. This is reassuring, as we made sure that the donor template did not contain the CRISPR target sequence to prevent undesired mutagenesis after reporter gene integration. However, Indel mutations were detected in the non-targeted allele in two of the six OCT4-mOrange reporter lines examined (Figure 3C). These findings underscore the necessity of thorough sequence analysis for eliminating clones with undesired mutations in the non-targeted allele, a point not widely recognized with the CRISPR/Cas-mediated targeting strategy. We also sequenced seven predicted off-target sites based on the 12-bp seed sequence important for target recognition (Jiang et al., 2013; Jinek et al., 2012). Examination of six OCT4-mOrange and eight OCT4-eGFP lines revealed no mutations except that three OCT4-eGFP lines carried mutations at the POU5F1P4 locus, which shares the same 20-nt target sequence with the intended target (Table S1).
Targeting the PDX1 Locus Using a Mini-vector Donor without Drug Selection
We further investigated whether this selection-free approach also applied to genes not expressed in undifferentiated hESCs. We chose to target PDX1, which encodes a transcription factor not expressed in undifferentiated hESCs but in pancreatic progenitors and their differentiated progeny such as pancreatic β cells. The ability to monitor PDX1 expression during in vitro differentiation and to enrich PDX1+ pancreatic progenitor cells or β cells would be valuable for studies of pancreatic development and the use of hESCs for β cell replacement therapies.
We designed two gRNAs (PDX1 cr1 and cr2) to target DNA sequences in proximity to the PDX1 stop codon and used a PDX1-eGFP mini-vector donor for integration of the eGFP reporter into the PDX1 locus (Figures 4A and S4A). Correct targeting should enable expression of eGFP from the endogenous PDX1 locus with minimal impact on PDX1 protein expression or function. Following establishment of clonal lines (Figure 4B), PCR and Southern blot analysis identified three correctly targeted clones without random integration: two clones with eGFP integration in one PDX1 allele and one clone with biallelic integration (Figures 4C, 4D, and S4B). All three PDX1-eGFP reporter lines displayed normal hESC morphology and expressed pluripotency markers OCT4, NANOG, and SOX2 as determined by immunofluorescence staining (Figure S4C). After differentiation into pancreatic progenitors, co-expression of GFP and PDX1 was observed, demonstrating faithful reporter activity (Figure 4E). Thus, we successfully generated multiple faithful knockin reporter lines for PDX1, a gene with a lineage-restricted expression pattern in differentiated hESCs.
Figure 4.
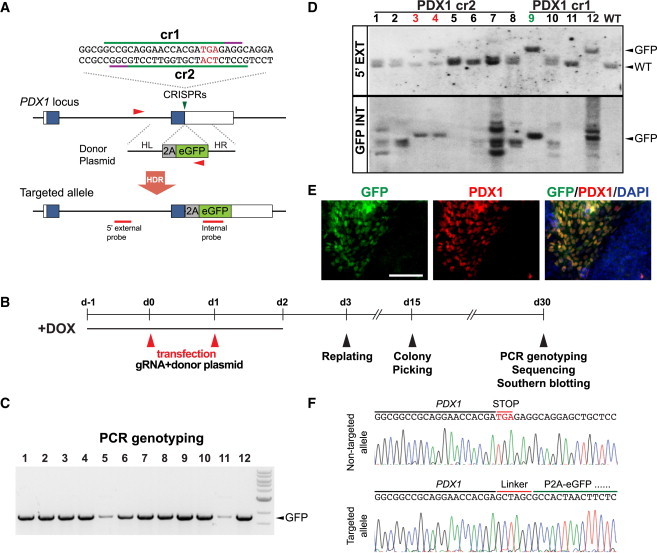
Generation of PDX1-eGFP Reporter Lines without Drug Selection
(A) PDX1 cr1 and cr2 were designed to target sequences in proximity to the PDX1 stop codon. In the presence of the eGFP donor plasmid, HDR resulted in the replacement of the stop codon with 2A-eGFP. PCR primers (F + R) used for genotyping are indicated with red arrowheads.
(B) Timeline for the generation of PDX1-eGFP hESC lines using the iCRISPR platform. DOX, doxycycline.
(C) PCR genotyping of 12 clones, identified from the PCR screen in Figure S4B, that showed the correct PCR product for the targeted allele (GFP: 1,746 bp).
(D) Southern blot analysis using the external probe and internal probe (WT: 3,845 bp; GFP: 4,632 bp) identified three correctly targeted lines. Lines without random integrations and carrying mono-allelic eGFP insertion are labeled in red, and the clone with a bi-allelic eGFP insertion is labeled in green.
(E) Immunofluorescence staining displayed co-expression of GFP and PDX1 in pancreatic progenitors differentiated from PDX1-eGFP hESCs. The scale bar represents 100 μm.
(F) Sanger sequencing of non-targeted and targeted PDX1 alleles showed correct targeting of the PDX1 locus with no undesired mutations.
We further examined the PDX1 reporter lines for potential undesired mutations similar to analysis performed on the OCT4 reporter lines. We found the expected sequences in both the targeted and non-targeted PDX1 allele in all clones (Figure 4F) except for one (no. 3) with a 24-bp deletion in the non-targeted allele. Recent studies suggest that CRISPR/Cas9 tolerates mismatches between CRISPR and target DNA at different positions in a sequence-dependent manner, influenced by the number, position, and distribution of mismatches (Hsu et al., 2013; Tan et al., 2015). Using a prediction program developed by Feng Zhang’s group (http://crispr.mit.edu), we sequenced 20 most likely off-target sites (ten each predicted for PDX1 cr1 and cr2) and detected no mutations (Table S1).
Discussion
Here, we demonstrate the generation of hESC reporter lines without the use of drug selection for both active and silent genes through the use of the iCRISPR system. Selection-free gene targeting eliminates the need for removal of drug-resistance cassette after identification of correctly targeted clones, and mini-vector donors with short homology arms (∼500–1,000 bp each) are convenient to make. Thus, this method significantly reduces the time and effort required for establishing hESC reporter lines. Additionally, conventional gene-targeting strategies typically use the Cre-loxP strategy to remove drug-resistance cassettes, which leaves behind a 34-bp loxP “scar” in the endogenous locus. Although not an issue in most cases, this residual sequence could interfere with the expression of the targeted gene in some situations (Meier et al., 2010). In comparison, our strategy eliminates the need for the selection cassette and thereby minimizes the alteration of the endogenous locus. Although we focused on creating promoter-fusion reporters, the same knockin approach can be readily applied to making protein-fusion reporters for visualizing protein subcellular localization, precisely deleting or replacing specific genomic sequences, and introducing or correcting disease-associated mutations.
Previously, we and others have failed to target the PDX1 locus using traditional targeting approaches (Z.Z. and D.H., data not shown; E. Stanley, personal communication), yet the absolute targeting efficiencies using the selection-free method were comparable between the PDX1 and OCT4 loci. It is known that gene-targeting efficiencies can vary significantly depending on the target locus, though the exact reason is unclear. Traditional gene targeting relies on drug selection; thus, the relative targeting efficiencies after drug selection depend, at least in part, on the expression of the drug-resistance gene from the targeted locus. Because the expression of drug-resistance gene may differ significantly between expressed and silent loci, the relative targeting efficiency after drug selection for a lineage-specific gene may appear much lower compared with a pluripotency gene. For certain loci, the drug-resistance gene may be expressed at such low levels that hinder the identification of a correctly targeted clone using the drug-selection method (Rostovskaya et al., 2012). Because our targeting approach obviates drug selection, it may overcome such bias and facilitate the generation of reporter alleles for genes that were previously difficult to target. One may further use this ability to measure absolute targeting frequencies to compare HDR efficiencies across different genomic contexts.
There have been concerns about potential off-target mutagenesis with the CRISPR/Cas system (Cho et al., 2014; Fu et al., 2013; Hsu et al., 2013; Mali et al., 2013b; Pattanayak et al., 2013). Our analysis so far did not reveal any off-target mutations at sites without perfect complementarity with the CRISPR target sequence. However, we cannot exclude the possibility of off-target mutations elsewhere in the genome, and a thorough analysis may be necessary before the reporter lines are used in future studies. The CRISPR/Cas system is continuously improved with the development of better algorithms for CRISPR design and off-target prediction. It is reassuring that a recent high-coverage whole-genome sequencing study failed to detect significant incidence of off-target mutations in CRISPR-targeted hPSC lines (Veres et al., 2014). On the other hand, we noticed that some correctly targeted clones carried mutations in the non-targeted allele, though the targeting efficiency is sufficiently high that one could simply discard the minority of clones carrying mutations. One may also target intronic regions with low-sequence conservations to further mitigate any concerns associated with Indel mutations in the non-targeted allele.
Our selection-free targeting approach enables rapid generation of knockin reporter lines, though it also requires either using established iCas9 cells or creating new iCas9 lines in a desired hPSC background. The upfront effort for generating iCas9 cells is relatively small due to the efficient TALEN-mediated AAVS1-targeting approach, and it is possible to establish an iCas9 line in about 1 month (González et al., 2014; Zhu et al., 2014). Once an iCas9 line is established, it can be used for making different types of reporters. Our previous study has shown that Cas9 activity is tightly regulated by doxycycline treatment, and established iCas9 lines exhibit no apparent chromosomal aberrations or defects in the maintenance of the pluripotent state (González et al., 2014). A recent study also observed no adverse effects in constitutive Cas9-expressing mice (Platt et al., 2014). An additional benefit of using iCas9 hPSCs for making reporter lines is that the cells can be conveniently used for a variety of downstream genetic studies using gene-editing approaches we already established (González et al., 2014). Thus, we expect this selection-free knockin strategy to further facilitate the use of hESCs for developmental studies, disease modeling, and cell-replacement therapy.
Author Contributions
D.H., Z.Z., N.V., and F.G. conceived the project; Z.Z. performed most experiments related to PDX1 targeting; N.V. performed most experiments related to OCT4 targeting; F.G. and Z.-D.S. performed additional relevant experiments; and D.H., Z.Z., and N.V. wrote the manuscript.
Acknowledgments
We thank Feng Zhang and Rudolf Jaenisch for providing vectors through Addgene and members of the D.H. laboratory for insightful discussions and critical reading of the manuscript. This study was funded in part by NIH (R01DK096239), NYSTEM (C029156), Basil O’Connor Starter Scholar Award from March of Dimes Birth Defects Foundation, Tri-Institutional Stem Cell Initiative, Louis V. Gerstner Jr. Young Investigators Award, and MSKCC Society Special Projects Committee. Z.Z. and F.G. were both supported by the New York State Stem Cell Science (NYSTEM) fellowship from the Center for Stem Cell Biology of the Sloan Kettering Institute. N.V. was supported by the Howard Hughes Medical Institute (HHMI) Medical Research Fellowship.
Published: May 28, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
References
- Byrne S.M., Ortiz L., Mali P., Aach J., Church G.M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res. 2015;43:e21. doi: 10.1093/nar/gku1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.P., Costa M., Grandela C., Holland A.M., Hatzistavrou T., Micallef S.J., Li X., Goulburn A.L., Azzola L., Elefanty A.G., Stanley E.G. A protocol for removal of antibiotic resistance cassettes from human embryonic stem cells genetically modified by homologous recombination or transgenesis. Nat. Protoc. 2008;3:1550–1558. doi: 10.1038/nprot.2008.146. [DOI] [PubMed] [Google Scholar]
- Ding Q., Lee Y.K., Schaefer E.A., Peters D.T., Veres A., Kim K., Kuperwasser N., Motola D.L., Meissner T.B., Hendriks W.T. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q., Regan S.N., Xia Y., Oostrom L.A., Cowan C.A., Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González F., Zhu Z., Shi Z.D., Lelli K., Verma N., Li Q.V., Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Wang H., Kiani S., Lai C.S., Gao Q., Cassady J.P., Cost G.J., Zhang L., Santiago Y., Miller J.C. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z., Zhang Y., Propson N.E., Howden S.E., Chu L.F., Sontheimer E.J., Thomson J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Kim J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier I.D., Bernreuther C., Tilling T., Neidhardt J., Wong Y.W., Schulze C., Streichert T., Schachner M. Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J. 2010;24:1714–1724. doi: 10.1096/fj.09-140749. [DOI] [PubMed] [Google Scholar]
- Merkert S., Wunderlich S., Bednarski C., Beier J., Haase A., Dreyer A.K., Schwanke K., Meyer J., Göhring G., Cathomen T., Martin U. Efficient designer nuclease-based homologous recombination enables direct PCR screening for footprintless targeted human pluripotent stem cells. Stem Cell Reports. 2014;2:107–118. doi: 10.1016/j.stemcr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovskaya M., Fu J., Obst M., Baer I., Weidlich S., Wang H., Smith A.J., Anastassiadis K., Stewart A.F. Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res. 2012;40:e150. doi: 10.1093/nar/gks643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E.P., Li Y., Velasco-Herrera Mdel.C., Yusa K., Bradley A. Off-target assessment of CRISPR-Cas9 guiding RNAs in human iPS and mouse ES cells. Genesis. 2015;53:225–236. doi: 10.1002/dvg.22835. [DOI] [PubMed] [Google Scholar]
- Veres A., Gosis B.S., Ding Q., Collins R., Ragavendran A., Brand H., Erdin S., Cowan C.A., Talkowski M.E., Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Huangfu D. Human pluripotent stem cells: an emerging model in developmental biology. Development. 2013;140:705–717. doi: 10.1242/dev.086165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., González F., Huangfu D. The iCRISPR platform for rapid genome editing in human pluripotent stem cells. Methods Enzymol. 2014;546:215–250. doi: 10.1016/B978-0-12-801185-0.00011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.