

Summary
To ensure accurate genomic segregation, cells evolved the spindle assembly checkpoint (SAC), whose role in adult stem cells remains unknown. Inducible perturbation of a SAC kinase, Mps1, and its downstream effector, Mad2, in skeletal muscle stem cells shows the SAC to be critical for normal muscle growth, repair, and self-renewal of the stem cell pool. SAC-deficient muscle stem cells arrest in G1 phase of the cell cycle with elevated aneuploidy, resisting differentiation even under inductive conditions. p21CIP1 is responsible for these SAC-deficient phenotypes. Despite aneuploidy’s correlation with aging, we find that aged proliferating muscle stem cells display robust SAC activity without elevated aneuploidy. Thus, muscle stem cells have a two-step mechanism to safeguard their genomic integrity. The SAC prevents chromosome missegregation and, if it fails, p21CIP1-dependent G1 arrest limits cellular propagation and tissue integration. These mechanisms ensure that muscle stem cells with compromised genomes do not contribute to tissue homeostasis.
Graphical Abstract
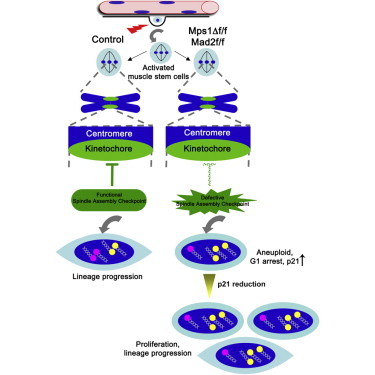
Highlights
-
•
Activation turns on spindle assembly checkpoint (SAC) genes in muscle stem cells
-
•
SAC failure leads to G1 arrest, raises aneuploidy, and blocks differentiation
-
•
G1 arrest is mediated through the p53/p21 pathway
-
•
Activated muscle stem cells from aged mice possess a robust SAC
In this article, Brack and colleagues examine the spindle assembly checkpoint (SAC) in skeletal muscle stem cells. Genetic disruption of the SAC in muscle stem cells promotes aneuploidy, invoking a reversible cell-cycle arrest mediated through p53/p21 to prevent differentiation. Despite the critical role of the SAC for muscle growth and repair, aged muscle stem cells possess robust SAC activity.
Introduction
Stem cells are essential for tissue formation, maintenance, and repair by achieving a timely balance between error-free replicative expansion and differentiation. This balance is frequently jeopardized in cancer and aging, leading to tissue pathology and functional decline (Blanpain et al., 2011; Liu and Rando, 2011; Ricke and van Deursen, 2013). To ensure accurate segregation of chromosomes during mitosis, the spindle assembly checkpoint (SAC) prevents anaphase onset until each chromosome has attached properly to mitotic spindle microtubules via its kinetochore (Foley and Kapoor, 2013; Lara-Gonzalez et al., 2012; Musacchio and Salmon, 2007). Therefore, an active SAC will delay mitosis until all chromosomes have been properly attached and aligned.
Many molecular players participate in a spatiotemporally concerted manner to actuate the SAC. These include MAD2 (mitotic arrest deficient 2), MPS1 (monopolar spindle 1), BUB1 (budding uninhibited by benomyl 1), and BUBR1 (Foley and Kapoor, 2013; Lara-Gonzalez et al., 2012; Musacchio and Salmon, 2007; Suijkerbuijk et al., 2012). In the presence of improperly attached kinetochores, the SAC arrests cells in mitosis by inhibiting the ability of CDC20 to activate APC/C-mediated polyubiquitination and subsequent proteasomal degradation of securin and cyclin B1 (Hwang et al., 1998; Kim et al., 1998).
SAC disruption in different cellular contexts reveals distinct outcomes. Data from immortalized cell lines and single-cell organisms demonstrate that the consequences of SAC failure include premature onset of anaphase, mitotic slippage, chromosome missegregation, and promotion of aneuploidy (Jelluma et al., 2008; Kops et al., 2004). In vivo studies in vertebrates and invertebrates show the consequence of SAC failure to be context dependent. During development, defective SAC activity can be tolerated early during embryogenesis but leads to an eventual loss of viability (Dobles et al., 2000; Fischer et al., 2004). Similarly, a reduction in MAD2 and BUBR1 leads to a SAC defect that can be tolerated but accelerates tumor production (Dai et al., 2004; Michel et al., 2001). In contrast, the SAC is essential during development and adult tissue regeneration in zebrafish (Poss et al., 2002a, 2002b, 2004). Studies on the role of the SAC in stem cells are more limited. Using either germline mutants or developmentally induced Cre drivers to delete SAC genes, both hematopoietic stem cells and epidermal stem cells and their respective committed progeny display differential sensitivity to SAC disruption (Foijer et al., 2013; Ito et al., 2007). Sensitivity to SAC dysfunction in epidermal stem cells was associated with increased aneuploidy and apoptosis (Foijer et al., 2013).
To date, the role of the SAC exclusively in adult mammalian stem cells has not been addressed. Pax7-expressing satellite cells (SCs) possess the function of stem cells and are critical for postnatal growth and repair of adult skeletal muscle (Lepper et al., 2009; Murphy et al., 2011; Sambasivan et al., 2011; Seale et al., 2000). Using inducible Pax7-specific SAC perturbation models, we show that the SAC is essential for normal mammalian SC function, during both early postnatal growth and adult tissue regeneration. Deregulation of the SAC in SC progeny leads to a rapid G1 arrest and missegregation of chromosomes. p21CIP1 is critical for the cellular arrest, and its reduction in SAC-defective progenitors permits the expansion of SCs with faulty genomes. Furthermore, we show that SAC activity and the level of aneuploidy are not altered in cycling satellite cells as a function of physiological aging.
Results
Muscle SCs Have a Functional Spindle Assembly Checkpoint
To investigate whether SCs have a functional spindle assembly checkpoint, we first examined expression of SAC components in quiescent and proliferating SCs. In quiescent SCs isolated by cell-surface markers (lin−, Int-a7+, Vcam+, PI−), Mad2 and Mps1 transcripts were barely detectable. After 3 days in culture in growth media, their levels were significantly upregulated (Figure 1A). To characterize the SAC component MAD2 in SCs residing within their niche, we examined single adult muscle fibers (Zammit et al., 2004). MAD2 protein as detected by immunohistochemistry was not detectable in SCs at 0 hr but was present in activated (KI67+) SCs after 48 hr in culture (Figure 1B). To analyze MAD2 localization during progression through the cell cycle, we costained single fibers for tubulin and MAD2. In interphase, MAD2 localizes to the nuclear envelope and cytoplasm of activated SCs, but not to the nuclei of the postmitotic muscle fiber (Figures 1B and 1C; Figure S1). During prometaphase, MAD2 localizes to improperly attached kinetochores and, by late metaphase, when all the chromosomes are aligned, MAD2 redistributes from the kinetochores to the cytoplasm (presumably due to SAC satisfaction) (Figure 1C; Figure S1). After mitotic exit, MAD2 regains its interphase localization pattern to the nuclear envelope and cytoplasm (Figure 1C; Figure S1).
Figure 1.
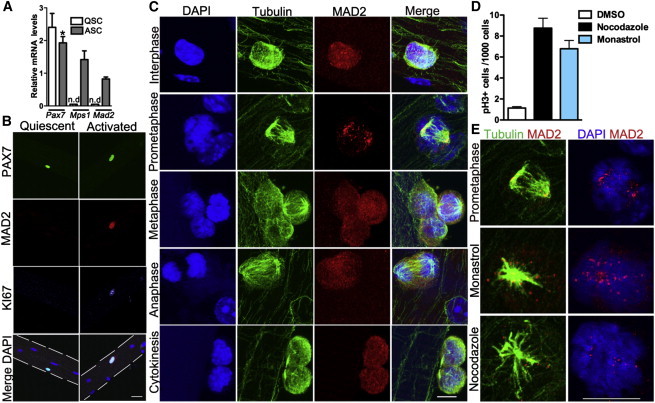
Activated Satellite Cells Have a Functional SAC
(A) Transcript levels of the satellite cell marker Pax7 and SAC genes Mps1 and Mad2 from FACS-purified SCs immediately after isolation (quiescent; QSC) or cultured in high-serum media (activated; ASC) for 3 days. Data were collected from three mice per time point.
(B) Representative MAD2 and KI67 immunostaining pattern in PAX7+ SCs on single muscle fibers either fixed immediately or after 2 days in culture. 4′,6-diamidino-2-phenylindole (DAPI) stains nuclei. n = 3 mice.
(C) MAD2 (red) and tubulin (green) immunostaining at key stages of the cell cycle on single muscle fibers after 2 days in culture. DAPI stains nuclei blue.
(D) Number of cells that arrest in mitosis (pH3+ cells) after exposure to mitotic poisons. Triplicate data from n = 3 mice were pooled, with 800–2,000 cells per condition.
(E) Representative images of MAD2 localization in prometaphase (characterized by the tubulin staining pattern) and in the presence of mitotic poisons.
The scale bars represent 10 μm (B) and 5 μm (C and E). Data are presented as mean ± SEM. ∗p < 0.05. n.d., not detectable. See also Figure S1.
To directly examine whether SCs have a functional SAC, we disrupted the mitotic spindle in proliferating SCs resident in their niche by treating them with either the microtubule-depolymerizing drug nocodazole or the monopolar spindle-inducing drug monastrol. Both treatments arrested SCs in mitosis as detected by phosphohistone 3 (pH3) (Figure 1D) with unattached and/or malattached chromatids (Figure 1E). In both examples, MAD2 localized on the kinetochores, reflecting a functional SAC (Foley and Kapoor, 2013; Lara-Gonzalez et al., 2012; Musacchio and Salmon, 2007) (Figure 1E). These results, for the first time, demonstrate that adult muscle SCs residing in their own niche have a robust SAC.
A Functional SAC Is Necessary for Normal Regenerative Potential of SCs
To examine the role of the SAC in SCs, we disrupted Mps1, a dual-specificity kinase with several cellular functions including regulation of microtubule dynamics, SAC regulation, and centrosomal duplication (Liu and Winey, 2012). The binding of MPS1 to the kinetochore is essential for SAC function (Abrieu et al., 2001; Nijenhuis et al., 2013). Therefore, to target kinetochore function while maintaining kinase activity intact, we used a conditional Mps1 mutant (Mps1Δ f/f) that is unable to localize to the kinetochore (Figure S2) (Foijer et al., 2014; Hached et al., 2011). This was crossed with mice harboring a tamoxifen (Tmx)-inducible Pax7-CreERtm allele and R26R-eYFP allele (hereafter Mps1Δ f/f). Mice containing the mutant loxP allele but lacking the Pax7-CreERtm allele were used as controls.
To determine whether disrupting Mps1 function exclusively in quiescent SCs would impact muscle regeneration, we administered Tmx prior to injury (−14 days) (Figure S3A). Seven days after injury, the fractions of bromodeoxyuridine-positive (BrdU+) cells (Figures S3B and S3C) and muscle fiber size (Figures S3D–S3F) were not different between Mps1Δ f/f and control muscles. This raised the possibility that Mps1 is redundant for SC function in vivo. However, we found incomplete recombination of the Mps1 locus in enhanced yellow fluorescent protein-positive (eYFP+) cells (Figures S3G and S3H). The incongruence between full recombination at the R26R locus (indicated by eYFP) and meager recombination at the Mps1 locus possibly reflects inaccessibility of the CRE recombinase to loxP sites in the Mps1 locus in quiescent SCs.
Due to the elevated expression of SAC genes in proliferating SCs in vitro, we next administered Tmx at early stages of repair (−3 to +3 days after injury) to target Mps1 in proliferating SCs (Figure 2A). Two days after the final Tmx treatment, a high degree of recombination was evident in fluorescence-activated cell sorting (FACS)-purified myogenic cells from regenerating muscle of Mps1Δ f/f mice based on expression of R26R-eYFP (89% of SCs were eYFP+) and disruption of Mps1 at the genomic level (Figures S3G and S3H), suggesting increased accessibility of the Mps1 locus to CRE recombinase as SCs transition from a quiescent to a proliferative state.
Figure 2.
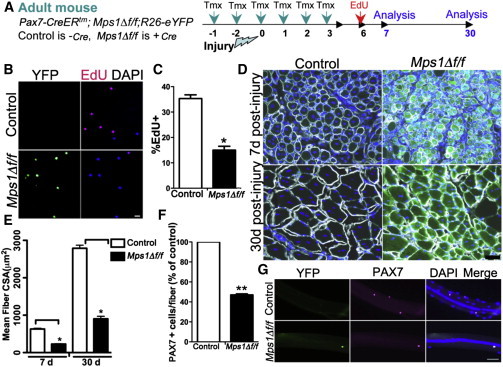
Mps1 Is Required for Normal Muscle Regeneration and Replenishment of the Satellite Cell Pool
(A) Experimental regime showing mouse strains, Tmx strategy, and injury paradigm.
(B and C) Representative images (B) and histogram (C) showing EdU incorporation in control and Mps1Δ f/f myogenic cells collected 7 days after injury by FACS. n = 3 mice per genotype, with 200–300 cells per condition, performed in triplicate.
(D) Transverse sections of regenerating muscle from control and Mps1Δ f/f mice collected 7 and 30 days after injury, stained for laminin, DAPI, and YFP to quantify muscle fiber size. Regenerated muscle fibers are characterized by centrally located nuclei.
(E) Quantification of muscle fiber cross-sectional area (CSA) from regenerating control and Mps1Δ f/f muscle. n = 4 mice per genotype.
(F and G) Number (F) and representative images (G) of PAX7+ SCs/single muscle fibers after 30 days of regeneration in control and Mps1Δ f/f mice. n = 3 mice, with 30–40 myofibers per condition.
The scale bars represent 20 μm (B) and 50 μm (D and G). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, Student’s t test. See also Figures S2–S4.
Analysis of regenerating muscle, 7 days after injury, shows that proliferation of SC-derived progeny, based on 5-ethynyl-2′-deoxyuridine (EdU) incorporation, was significantly reduced in Mps1Δ f/f compared to control muscles (Figures 2A and 2B). In addition, muscle fiber size was smaller, suggesting a delay in muscle repair (Figures 2D and 2E). Thirty days after injury, muscle fiber size in Mps1Δ f/f mice remained smaller than both control regenerated muscle and uninjured contralateral muscle, suggesting a permanent defect in muscle repair in the absence of a functional SAC (Figures 2D and 2E). In addition, we observed 40% fewer sublaminar SCs of regenerated Mps1Δ f/f compared to control muscles (Figures 2F and 2G). In normal muscle, SCs repopulate the niche after muscle injury, restoring the stem cell pool back to homeostatic levels (Sacco et al., 2008; Shea et al., 2010). Therefore, the present results show that the SAC is essential for replenishment and homeostasis of the SC pool after injury.
SAC Is Essential for Normal SC Expansion and Differentiation during Postnatal Muscle Growth
We next examined the requirement for the SAC in SCs during postnatal myogenesis. To this end, we quantified the number of PAX7+ cells, their progeny, and muscle fiber size during postnatal maturation. Tmx was administered from postnatal day 1 (P1) to P4 followed by a 3-day chase (Figure 3A). At P7, we observed a high degree of recombination at both the endogenous Mps1 locus and R26R locus, as determined by eYFP expression (Figures S3G and S3H). Immunotypic analysis at P7 revealed that Mps1Δ f/f muscle had 35%–60% fewer myogenic cells compared to control muscle, as assessed by the number of PAX7+, MYOD+, and myogenin+ cells (Figures 3B and 3C). In support, administration of EdU from P6 to P7 revealed a 50% decline in the number of proliferating (EdU+) myogenic cells (Figure 3D). Furthermore, a 50% decline in muscle cross-sectional area and a commensurate decline in individual muscle fiber cross-sectional area were observed in Mps1Δ f/f compared to control muscle (Figures 3E–3G). Together, these results demonstrate that a defective SAC limits expansion and lineage progression of the SC pool that severely limits muscle growth during postnatal maturation.
Figure 3.
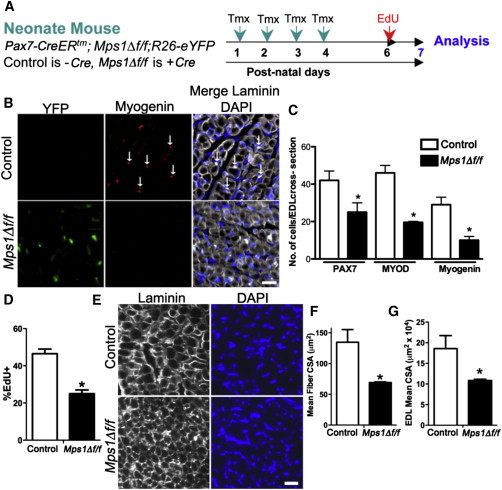
Mps1 Function Is Required for Proper Postnatal Muscle Growth
(A) Experimental regime showing mouse strains and experimental time course.
(B) Representative images of transverse muscle sections from control and Mps1Δ f/f postnatal mice stained with anti-myogenin (red) (arrows), anti-GFP (green), and DAPI (blue).
(C) Quantification of myogenic cells in transverse sections as assessed by staining for PAX7, MYOD, and myogenin. For Mps1Δ f/f animals, only eYFP+ cells were counted. n = 3 mice, with 8–10 muscle sections counted per tissue.
(D) EdU incorporation of myogenic cells collected by FACS fromP7 control and Mps1Δ f/f mice. Only eYFP+ cells were counted in Mps1Δ f/f mice. n = 3 mice per group, with 200–300 cells counted per condition.
(E) Transverse muscle in postnatal control and Mps1Δ f/f mice stained with anti-laminin and DAPI.
(F and G) Quantification of mean extensor digitorum longus (EDL) muscle fiber CSA (F) and whole EDL muscle CSA (G). n = 3 mice.
The scale bars represent 20 μm. Data are presented as mean ± SEM. ∗p < 0.05, Student’s t test. See also Figures S2–S4.
MAD2 Depletion Phenocopies MPS1 Truncation
To assess SAC signaling during myogenesis, we conditionally disrupted Mad2 (Foijer et al., 2013), a downstream effector of Mps1 (Hewitt et al., 2010; Tighe et al., 2008). In agreement with our Mps1 loss-of-function studies, we observed significantly smaller muscle fibers in both regenerating adult muscle (Figure S4A) and postnatal muscle (Figure S4B) in Mad2f/f compared to control mice. In conclusion, the disruption of Mps1 and Mad2 severely compromises mammalian muscle stem cell function and underscores the importance of the SAC pathway during muscle growth and repair.
SAC Defect Induces a Rapid G1 Cell-Cycle Arrest and Aneuploidy
The ability of stem cells to successfully repair tissue is critically dependent on the number and differentiation potential of their progeny. To determine the acute cellular consequence of SAC dysfunction, we disrupted Mps1 and Mad2 function in SC-derived progenitors (hereafter referred to as myoblasts) in vitro using Adeno-GFP-CRE (Ad-CRE) virus. Adeno-GFP was used as a control. We observe that Ad-CRE-treated Mps1Δ f/f and Mad2f/f myoblasts fail to expand and proliferate in high-serum conditions, as determined by quantification of cell numbers (Figure 4A; Figure S6A). In addition, a 24-hr BrdU pulse administered 18 hr after virus treatment revealed a dramatic reduction in BrdU uptake (Figure 4B; Figures S5A and S6B), consistent with a reduction in S phase entry occurring within the following cell cycle.
Figure 4.
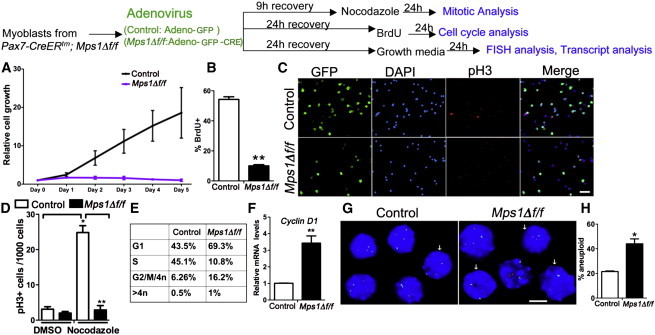
Disruption of Mps1 Function Induces a Rapid G1 Arrest in Myogenic Progenitors
Top: experimental regime showing myoblast treatment and phenotypic assays. Satellite cell-derived myoblasts from Mps1Δ f/f adult mice treated with either Adeno-GFP (control) or Adeno-GFP-CRE (Mps1Δ f/f) virus and recovered in high-serum media.
(A) Cell growth relative to initial plating density (n = 100–200 cells/well on day 0). n = 3 mice per condition, with 100–200 cells plated in triplicate wells.
(B) Quantification of BrdU incorporation in control and Mps1Δ f/f myoblasts after a 24-hr pulse; n = 3 mice per condition, with 200–500 cells counted in triplicate wells.
(C) Representative images of control and Mps1Δ f/f myoblasts treated with nocodazole showing GFP (to detect viral infection) and cells in mitosis (pH3+). DAPI stains nuclei.
(D) Quantification of progenitors stalled in mitosis (pH3+) from control and Mps1Δ f/f myoblasts after nocodazole treatment. Between 1,000 and 2,000 cells were counted per condition. The experiment was done in biological triplicate.
(E) FACS analysis of the cell-cycle distribution of control and Mps1Δ f/f myoblasts.
(F) Relative transcript levels of CyclinD1 between control and Mps1Δ f/f myoblasts Triplicate data are from n = 3 mice.
(G and H) Aneuploidy index scored from interphase FISH analysis of chromosomes 15 (green) and chromosome 19 (red). Arrows indicate aneuploid cells; n = 100–150 cells per condition. The experiment was done in biological triplicate.
The scale bars represent 50 μm (C) and 10 μm (G). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, Student’s t test. See also Figures S5 and S6.
Next, to evaluate SAC competence, Ad-CRE and Ad-GFP virus-treated Mps1Δ f/f myoblasts were subjected to the mitotic poison nocodazole before their entry into mitosis. In this assay, nocodazole normally elicits a mitotic arrest response; cells that bypass the arrest are SAC defective. Compared to controls, Mps1Δ f/f myoblasts failed to arrest in mitosis, as evidenced by a reduction in the fraction of pH3+ cells (Figures 4C and 4D). Therefore, myoblasts lacking Mps1 and Mad2 function cannot mount an appropriate SAC response. Finally, cell-cycle analysis shows that Mps1Δ f/f myoblasts in high-serum conditions are stalled primarily in G1, with far fewer cells entering S phase (Figure 4E; Figure S5B). In support, cyclin D1 transcript levels are enriched (Figure 4F). Thus, SAC disruption leads to mitotic slippage and a rapid arrest in G1 phase of the cell cycle.
SAC dysfunction has been associated with chromosome instability and aneuploidy (Foijer et al., 2014). To investigate whether SAC defects lead to chromosome instability in myoblasts, we assayed chromosome number using interphase fluorescence in situ hybridization (FISH) against chromosomes 15 and 19. We find an almost 2-fold increase in the number of aberrantly segregated chromosomes 24 hr following virus treatment in Mps1Δ f/f and Mad2f/f myoblasts compared to controls (Figures 4G and 4H). These data are consistent with a direct role of the SAC in prevention of aneuploidy in SC progeny.
Differentiation Is Aborted after SAC Disruption
Cell-fate decisions are controlled in G1 phase of the cell cycle (Pauklin and Vallier, 2013). Therefore, we examined the cell fate of myoblasts shortly after SAC-dependent cell-cycle arrest. Analysis of apoptosis by Annexin V staining 48 hr after virus treatment revealed a relatively higher necrotic and apoptotic fraction in Mps1Δ f/f myoblasts. However, almost 90% of Mps1Δ f/f myoblasts are neither apoptotic nor necrotic (Figure 5A). In addition, senescence-associated β-galactosidase staining revealed negligible and comparable senescence between control and Mps1Δ f/f myoblasts (Figures 5B and 5C). Thus, SAC disruption in myoblasts does not substantially increase cell death or senescence.
Figure 5.
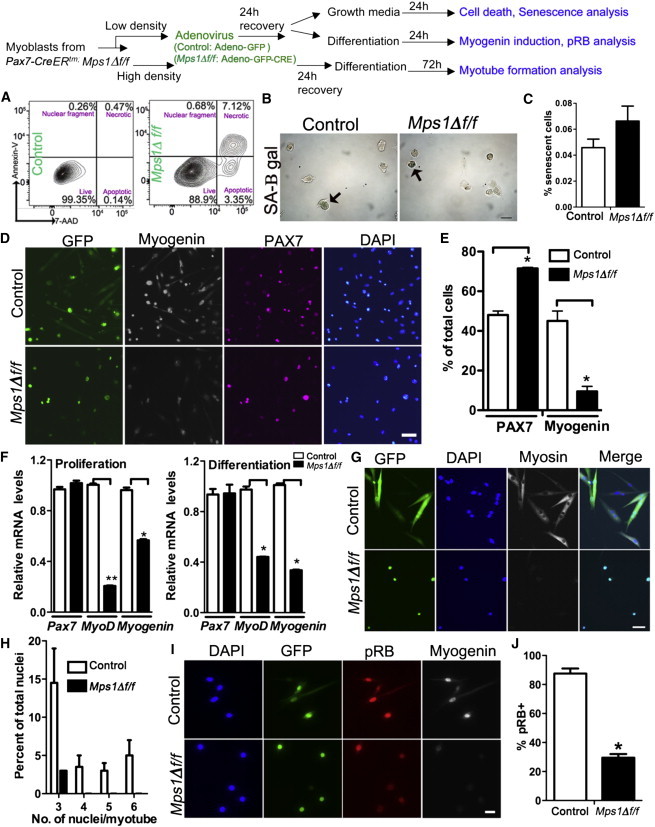
Differentiation Is Aborted in Myoblasts Lacking Kinetochore Binding of Mps1
Top: experimental regime showing myoblast treatment and phenotypic assays. Satellite cell-derived myoblasts from Mps1Δ f/f adult mice treated with either Adeno-GFP (control) or Adeno-GFP-CRE (Mps1Δ f/f) virus and subjected to cell-fate assays in either high- or low-serum culture conditions after recovery.
(A) FACS plots showing Annexin-V and 7AAD staining to quantify necrotic and apoptotic cells in control and Mps1Δ f/f myoblasts 24 hr after virus treatment. Experiments were performed in triplicate; n = 3 mice.
(B and C) Representative images (B) and quantification (C) of senescence-associated β-galactosidase (SA-B gal) assays. Arrows highlight senescent cells. Experiments were counted in triplicate; n = 3 mice.
(D and E) Representative images (D) and quantification (E) of low-density cultures from control and Mps1Δ f/f myoblasts switched to low-serum conditions. Cultures were stained with anti-GFP (green), anti-myogenin (white), anti-PAX7 (violet), and DAPI (blue). Experiments were counted in triplicate; n = 3 mice.
(F) Relative transcript levels of myogenic-fate genes in high-serum (proliferation) and low-serum (differentiation) conditions. Triplicate data from n = 3 mice.
(G and H) Images (G) and quantification (H) of control and Mps1Δ f/f myoblasts after switching to low-serum high-density conditions to test myogenic differentiation (anti-myosin heavy chain; green) and fusion potential. n = 3 trials for each experiment from three mice.
(I and J) Immunofluorescence of pRB under differentiation conditions. n = 3 trials for each experiment from three mice.
The scale bars represent 10 μm (B and I), 50 μm (D), and 20 μm (G). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, Student’s t test. See also Figure S6.
Next, to determine whether G1-arrested Mps1Δ f/f and Mad2f/f myoblasts are capable of entering the myogenic differentiation program, we subjected them to serum withdrawal, an inductive environment for differentiation. In these conditions, control myoblasts lose PAX7 expression and turn on myogenin; Mps1Δ f/f and Mad2f/f myoblasts retain PAX7 and fail to upregulate myogenin (Figures 5D and 5E; Figure S6C). In addition, Mps1Δ f/f myoblasts exhibit 40%–80% lower levels of MyoD and myogenin transcripts under both proliferation and differentiation conditions (Figure 5F). Thus, Mps1Δ f/f myoblasts are incapable of executing a differentiation program. To examine this defect further, we cultured control and Mps1Δ f/f myoblasts in low serum at high cell density to induce myoblast fusion. Control myoblasts effectively form multinucleated myotubes. In contrast, only very rare Mps1Δ f/f myoblasts are able to fuse and form multinucleated myotubes (Figures 5G and 5H). The tumor suppressor pRB is essential for effective differentiation of myoblasts (Hosoyama et al., 2011; Huh et al., 2004; Zacksenhaus et al., 1996; Gu et al., 1993; Puri et al., 2001). Under differentiation conditions, we find that pRB levels are significantly lower in Mps1Δ f/f compared to control myoblasts (Figure 5J). Therefore, an acute loss of SAC function in committed progenitors renders them incompetent for differentiation.
Elevated p21 Levels in SAC-Deficient Myoblasts Induce a Reversible G1 Arrest
Cell-cycle inhibitors balance cell replication and differentiation in normal healthy cells and induce cell-cycle arrest under conditions of DNA damage and chromosomal abnormalities (Halevy et al., 1995; LaBaer et al., 1997). We find elevated transcript levels of the cellular stress component p21Cip1 (hereafter called p21) in adult Ad-CRE-treated Mps1Δ f/f myoblasts and FACS-sorted SCs from Tmx-treated Mps1Δ f/f neonatal mice compared to controls (Figures 6A–6D). At the transcript level, other cell-cycle inhibitors such as p53, p16INK4a, and p19ARF were not responsive to SAC-induced arrest (Figure S7). To investigate whether elevated p21 participates in the SAC-induced cell-cycle arrest and differentiation block, we reduced p21 levels in SAC-deficient cells using small hairpin RNA (shRNA) lentiviral constructs against p21 (p21KD) or scrambled shRNA (scr) (Figure 6E). In high-serum conditions, as expected, the majority of Mps1Δ f/f cells treated with scr do not proliferate, as indicated by the absence of EdU incorporation. In contrast, p21KD-Mps1Δ f/f cells are now able to resume the cell cycle, as determined by EdU incorporation. In addition, when switched to low-serum conditions, myogenin is upregulated, consistent with a reversal of the differentiation block (Figures 6F and 6G). The major cellular component regulating p21 during cellular stress is the tumor suppressor p53 (el-Deiry et al., 1993; Dimitrova et al., 2014; Harper et al., 1993; Laptenko et al., 2011; Reczek et al., 2003; Saramäki et al., 2006). Therefore, we investigated whether reducing p53 levels by lentiviral shRNA would also unlock the cell-cycle arrest in Mps1Δ f/f cells. We find that EdU incorporation and differentiation in p53KD-Mps1Δ f/f cells was significantly increased compared to shRNA-control-Mps1Δf/f cells. In control cells, proliferation and differentiation were not affected after p53KD treatment (Figures 6H–6J). Therefore, p21 upregulation and p53 activity are critical for cell-cycle arrest and aborted differentiation as an acute response to SAC dysfunction.
Figure 6.
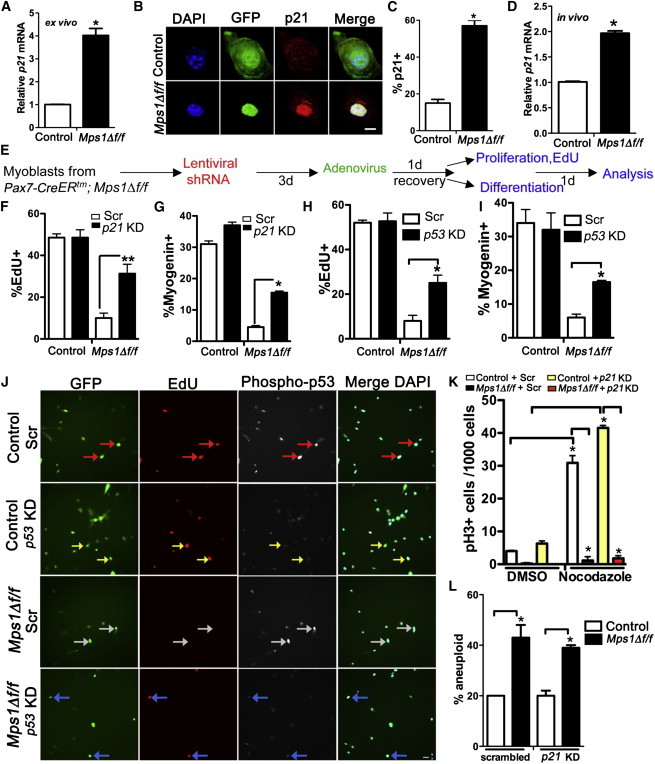
Mps1Δ f/f Myoblasts Undergo a p21-Mediated G1 Arrest
(A–C) Satellite cell-derived myoblasts from Mps1Δ f/f adult mice treated with either Adeno-GFP (control) or Adeno-GFP-CRE (Mps1Δ f/f) virus and recovered in high-serum media.
(A) Relative transcript levels of p21CIP1 (p21). Triplicate data are from n = 3 mice.
(B and C) Representative images (B) and quantification (C) of p21+ cells from control and Mps1Δ f/f cultures. The experiment was performed in biological triplicate, with 200–300 cells counted per condition.
(D) Relative transcript levels of p21 from FACS-sorted myogenic cells from Mps1Δ f/f and control P7 pups as per Figure 3A. Triplicate data are from n = 3 mice. Only eYFP+ myogenic cells were isolated from Mps1Δ f/f pups.
(E) Experimental regime to knock down p21 and p53 in Mps1Δ f/f myoblasts.
(F and H) Cell proliferation (% EdU+ cells).
(G and I) Terminal myogenic differentiation (% myogenin+ cells); 200–300 cells were counted per condition. Experiments were performed in biological triplicate.
(J) Representative images of control and Mps1Δ f/f cultures treated with Scr-shRNA or p53KD and pulsed with EdU to mark cycling cells. Arrows indicate red (p53+, EdU+), white (p53+, EdU−), and yellow and blue (p53−, EdU+).
(K) Mitotic arrest (pH3+) after nocodazole treatment; 1,000–2,000 cells were counted per condition. Experiments were performed in biological triplicate.
(L) Aneuploidy index scored from interphase FISH analysis of chromosomes 15 (green) and chromosome 19 (red). n = 100–150 cells per condition. The experiment was performed in biological triplicate.
The scale bars represent 5 μm (B) and 20 μm (J). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, Student’s t test. See also Figures S6 and S7.
Finally, we examined whether the reentry into the cell cycle after p21KD rescued the mitotic slippage. In sharp contrast to the cell-cycle rescue, reduction of p21 did not ameliorate the SAC defect (mitotic slippage) in the presence of nocodazole (Figure 6K; Figure S6D) or the high levels of aneuploidy (Figure 6L; Figure S6E). Therefore, restoring p21 levels allows SAC-deficient myoblasts to overcome the G1 arrest and proceed through the cell cycle; however, they proliferate with a defective SAC and elevated levels of aneuploidy.
Aged SCs Have a Robust SAC
Aneuploidy confers a proliferative disadvantage on cells and increases across several tissues during aging (Baker et al., 2013; Thompson and Compton, 2008; Torres et al., 2010). Aged SCs demonstrate impaired proliferation in vivo and in vitro (Conboy et al., 2003, 2005; Brack et al., 2007). We hypothesized that SAC defects underlie the age-dependent decline in SC proliferative capacity. To this end, we compared levels of the essential SAC component MAD2 in adult and aged SCs. We find that levels of MAD2 protein do not differ between adult and aged SCs in both interphase and mitosis (Figures 7A and 7B). Moreover, mitotic poisons elicit a similar response in activated SCs from both aged and adult mice, with cells arresting in mitosis with comparable mitotic indices (Figure 7C) and MAD2 localizing to the kinetochores (Figure 7D). Thus, SAC activity is not compromised in SCs as a function of age. In addition, the aneuploidy index, assessed by interphase FISH, in adult and aged SCs is equivalent, whether examined 2 days postisolation to inspect activated SCs or 5 days postisolation to inspect committed progenitor cells (Figure 7E). In conclusion, the impaired proliferative capacity exhibited by aged muscle stem cells is not due to an impaired SAC or abnormal chromosome segregation during the cell cycle.
Figure 7.
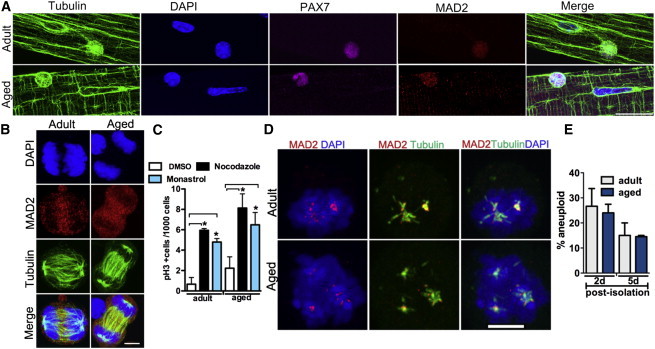
Aging Does Not Impact SAC Function and Aneuploidy in Satellite Cells
(A and B) Representative images of MAD2 protein on activated SCs during interphase (A) and mitosis (B).
(C) Mitotic poison assay using nocodazole and monastrol treatment on adult and aged activated SCs; n = 3 mice, with 800–2,000 cells counted per condition.
(D) Representative images of MAD2 localization in mitotic cells in the presence of nocodazole.
(E) Aneuploidy index on activated satellite cells (2 days postisolation) and progenitor cells (5 days postisolation) measured by FISH on chromosomes 15 and 19. n = 3 mice, with 200 cells counted per condition.
The scale bars represent 20 μm (A) and 5 μm (B and D). Data are presented as mean ± SEM. ∗p < 0.05.
Discussion
The long-lived nature of stem cells imperils them to accumulate genomic damage or errors, which may lead to tissue degeneration or oncogenesis, depending on the cellular response to the insult. The SAC is an essential component of mitosis to ensure fidelity of chromosome segregation during replication. Using targeted approaches to target SAC function specifically in skeletal muscle stem cells, we show that the SAC is essential for muscle stem cell function both during early postnatal growth and adult tissue regeneration. Muscle stem cells lacking a functional SAC arrest in G1 phase of the cell cycle with missegregated chromosomes (aneuploidy) and are incapable of differentiation. The cell-cycle inhibitor p21CIP1 acts as a gatekeeper to protect muscle stem cells from propagating abnormal genomes at the expense of tissue repair.
Until now, only a handful of studies have investigated the role of the SAC in mammalian stem cells. When Mad2 is deleted in the murine epidermal lineage, interfollicular epidermal cells sustain growth; in contrast, hair follicular bulge stem cells fail to grow (Foijer et al., 2013). Haploinsufficiency of Mad2 caused a reduction in the absolute numbers and cycling status of immature but not mature hematopoietic progenitor cells in both the spleen and bone marrow (Ito et al., 2007). Thus, in vivo, depending on the cellular context, the requirement of the SAC is variable.
Satellite cells are a heterogeneous pool containing subsets with stem cell properties and others functioning as progenitors (Chakkalakal et al., 2012, 2014; Kuang et al., 2007; Rocheteau et al., 2012). Based on the present results, we find no evidence for differential requirements of the SAC within satellite cells as they either self-renew or progress along their lineage. However, our results do show that SAC perturbation in quiescent versus activated muscle stem cells results in different phenotypes in terms of cell proliferation and tissue regeneration. We believe this is due to the differential expression patterns of SAC genes in quiescent compared with activated stem cells. Whereas Mad2 and Mps1 are robustly expressed by activated stem cells, they are undetectable in quiescent cells. A common mechanism by which cells repress gene expression is by DNA methylation, and we surmise that the Mad2 and Mps1 gene locus in quiescent cells might be densely methylated. Indeed, it is known that CRE recombinase is unable to access loxP sites inside a methylated locus (Long and Rossi, 2009), explaining the poor recombination efficiency of the Mps1 locus that we observe in quiescent stem cells targeted for recombination when compared to activated stem cells or progenitors. Therefore, the efficient deletion of Mps1 and Mad2 during postnatal growth and adult repair in contrast to adult uninjured muscle suggests that Mps1 and Mad2 loci undergo dynamic changes in methylation status as satellite cells transition between proliferation and quiescence. Although the mechanisms for SAC gene demethylation remain unknown, the findings implicate epigenetic regulation in the stem cell response to injury, specifically acting as a safeguard for genome integrity. Because SAC defects lead to chromosomal instability and tumor formation (Foijer et al., 2014), it is conceivable that a quiescent stem cell that cannot upregulate SAC genes, due to epigenetic repression, may harbor some of the properties that drive a stem or progenitor cell into a tumor-initiating cell.
Genotoxic stress in various forms normally triggers cell-cycle checkpoints that induce cellular arrest (Murray, 1992). In melanocyte stem cells and hematopoietic stem cells, genotoxic stress in the form of low-dose radiation causes premature differentiation (Inomata et al., 2009; Wang et al., 2012). Immortalized myogenic cells (C2C12) exposed to DNA-damaging agents undergo reversible inhibition of differentiation through an ABL-MYOD-regulated checkpoint (Innocenzi et al., 2011; Simonatto et al., 2011, 2013). Aneuploidy is a genotoxic stress and is detrimental to cell survival; cancer cells that tolerate aneuploidy can propagate themselves (Foijer et al., 2014; Thompson and Compton, 2010). In the present study, we demonstrate that activated SCs subjected to genotoxic stress by aneuploidy do not undergo acute cell death, senescence, or terminal differentiation. Instead, they reside in a stable but reversible G1-arrested state. Therefore, distinct genotoxic stressors may create relatively diverse phenotypes in damaged tissues.
Although studied extensively for its effect on cell proliferation, little is known about the impact of aneuploidy on differentiation. Our data suggest aneuploidy to be detrimental to differentiation to limit tissue contribution of faulty genomes. Although diminished, integration of SCs lacking a functional SAC (based on R26R-eYFP expression and genomic recombination at Mps1 and Mad2 loci) into growing and injured regenerating tissue suggests that some SAC-deficient activated SCs are indeed capable of tissue contribution. It will be important to assess the long-term consequences of aneuploidy on the function of a stable postmitotic tissue, such as skeletal muscle. p21 is a cell-cycle inhibitor that can initiate terminal differentiation and cause cell-cycle arrest in response to genotoxic stress, with forced p21 overexpression inducing differentiation even in high serum (Halevy et al., 1995; Skapek et al., 1995). In the present work, we have uncoupled a p21-mediated cell-cycle arrest in response to aneuploidy-induced genotoxic stress and for differentiation. We propose that p21 induction reflects either competence for differentiation or a protective response to cellular stress rather than predicting cell fate.
The differentiation block of myogenic cells undergoing genotoxic stress should be kept in mind while designing therapies for rhabdomyosarcoma (RMS), a soft-tissue sarcoma whose cell of origin is a myogenic precursor (Rubin et al., 2011). RMS is notorious for its inability to terminally differentiate, and this is often correlated with poor prognosis (Saab et al., 2011). We speculate that treating RMS with genome-damaging drugs could potentially exacerbate its inability to differentiate. This information might be valuable in designing myodifferentiation therapies for RMS, especially if the RMS is being treated with DNA-damaging agents such as cisplatin or if the RMS in question might be aneuploid, such as embryonic RMS from mosaic variegated aneuploidy patients, who have mutations in the SAC gene Bub1B.
Genome integrity is compromised across several different tissues during aging via structural (DNA damage, telomere shortening) and numerical (aneuploidy) aberrations of DNA (Behrens et al., 2014; Blanpain et al., 2011; Mandal et al., 2011). In fact, aneuploidization during aging has been observed across many somatic tissues (Baker et al., 2013); moreover, overexpression of the SAC gene BUBR1 can ameliorate many pathologic phenotypes in an accelerated aging model. Despite the dramatic decline in stem cell function during aging (Chakkalakal et al., 2012; Cosgrove et al., 2014; Bernet et al., 2014; Sousa-Victor et al., 2014), satellite cells do not undergo age-induced aneuploidization in noninjured muscle (Baker et al., 2013). Importantly, adult SCs undergo limited divisions throughout life, with estimates ranging from four to eight divisions based on a TetO-H2B-GFP reporter (Chakkalakal et al., 2012, 2014), which suggests that aneuploidization does not increase as a function of chronological aging in stem cells. Thus, the protective effects of BUBR1 overexpression against aging might be independent of its role in the SAC. We now show that when aged SCs are induced to proliferate, they are able to mount an equally robust SAC and have comparable levels of aneuploidy relative to proliferating adult SCs. Despite the robust SAC activity in aged SCs, we cannot rule out malfunction in other mitotic processes as an underlying cause for their limited function.
In multicellular organisms, tissue-specific stem cell proliferation is steered toward self-renewal or differentiation for tissue development or regeneration. Thus, fidelity of chromosome segregation is essential during stem cell proliferation so that organs are not built on cells with compromised genomic integrity. Although quiescent SCs in mammals do not express SAC proteins, they are expressed and used diligently by cycling SCs. To ensure genomic integrity during tissue turnover, adult muscle stem cells utilize a p53- and p21-mediated pathway to block the proliferation of cells with a dysfunctional SAC. Thus, the SAC, along with p53 and p21, helps stem cells to maintain a balance between meeting the demands of building the tissue and preserving genomic integrity favoring genome preservation over tissue homeostasis.
Experimental Procedures
Animals
Conditional alleles for Mad2 and Mps1 were generated and provided by Peter Sorger (Foijer et al., 2014; Hached et al., 2011). They were crossed with Pax7-CreERtm mice (Nishijo et al., 2009) on a C57BL/6 background to carry out tissue-specific deletions. C57BL/6 mice were procured from Charles River Laboratories or from the National Institute on Aging. Mice were used at 4–6 months of age (adult) or 22–24 months (aged). Animals were housed and handled in accordance with the guidelines of the Massachusetts General Hospital Subcommittee on Research Animal Care.
Muscle Injury and Proliferation Assessment
Muscle injury and analysis of regenerating muscle fibers were performed as previously described (Shea et al., 2010). EdU (Carbosynth) in PBS (2.5 mg/ml) was intraperitoneally injected into adults (two doses of 70 μl/day) and subcutaneously injected into pups (two doses of 20 μl/day). EdU detection was carried out according to the manufacturer’s instructions (Click-iT EdU detection; Invitrogen). One dose of 150 μl of 1.5 mg/ml BrdU (Sigma) was intraperitoneally injected into adults along with 2.5 mg/ml BrdU in 5% sucrose water for drinking.
Statistical Analysis
Experiments were performed in triplicate unless otherwise stated. Data are presented as means ± SEM. Statistical significance was assessed using Student’s t test, where results were considered significant at ∗p < 0.05.
Additional methods are discussed in the Supplemental Experimental Procedures.
Author Contributions
Experiments were carried out by S.K., R.A.-K., and C.S. Experimental design, analysis, and preparation of the manuscript were carried out by S.K. and A.S.B.
Acknowledgments
The authors would like to thank Peter Sorger (Harvard University) for mice, Hallie Nelson for technical assistance, members of the A.S.B. laboratory for reading the manuscript, and Laura Pricket-Rice, Kat Foltz-Donahue, and Meredith Weglartz of the MGH FACS Core Facility and Shumei Wang, Anita Hawkins, and Abha Aggarwal of the CytoGenomics Core Facility at Brigham and Women’s Hospital for their contribution. This work was supported by grants from MGH start-up funds and NIH grants (R01 AR060868, R01 AR061002) to A.S.B.
Published: May 7, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
References
- Abrieu A., Magnaghi-Jaulin L., Kahana J.A., Peter M., Castro A., Vigneron S., Lorca T., Cleveland D.W., Labbé J.C. Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 2001;106:83–93. doi: 10.1016/s0092-8674(01)00410-x. [DOI] [PubMed] [Google Scholar]
- Baker D.J., Dawlaty M.M., Wijshake T., Jeganathan K.B., Malureanu L., van Ree J.H., Crespo-Diaz R., Reyes S., Seaburg L., Shapiro V. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol. 2013;15:96–102. doi: 10.1038/ncb2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A., van Deursen J.M., Rudolph K.L., Schumacher B. Impact of genomic damage and ageing on stem cell function. Nat. Cell Biol. 2014;16:201–207. doi: 10.1038/ncb2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernet J.D., Doles J.D., Hall J.K., Tanaka K.K., Carter T.A., Olwin B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C., Mohrin M., Sotiropoulou P.A., Passegué E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell. 2011;8:16–29. doi: 10.1016/j.stem.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Brack A.S., Conboy M.J., Roy S., Lee M., Kuo C.J., Keller C., Rando T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Chakkalakal J.V., Jones K.M., Basson M.A., Brack A.S. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal J.V., Christensen J., Xiang W., Tierney M.T., Boscolo F.S., Sacco A., Brack A.S. Early forming label-retaining muscle stem cells require p27kip1 for maintenance of the primitive state. Development. 2014;141:1649–1659. doi: 10.1242/dev.100842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy I.M., Conboy M.J., Smythe G.M., Rando T.A. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- Conboy I.M., Conboy M.J., Wagers A.J., Girma E.R., Weissman I.L., Rando T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Cosgrove B.D., Gilbert P.M., Porpiglia E., Mourkioti F., Lee S.P., Corbel S.Y., Llewellyn M.E., Delp S.L., Blau H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Wang Q., Liu T., Swamy M., Fang Y., Xie S., Mahmood R., Yang Y.M., Xu M., Rao C.V. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–445. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- Dimitrova N., Zamudio J.R., Jong R.M., Soukup D., Resnick R., Sarma K., Ward A.J., Raj A., Lee J.T., Sharp P.A., Jacks T. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol. Cell. 2014;54:777–790. doi: 10.1016/j.molcel.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobles M., Liberal V., Scott M.L., Benezra R., Sorger P.K. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- el-Deiry W.S., Tokino T., Velculescu V.E., Levy D.B., Parsons R., Trent J.M., Lin D., Mercer W.E., Kinzler K.W., Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Fischer M.G., Heeger S., Häcker U., Lehner C.F. The mitotic arrest in response to hypoxia and of polar bodies during early embryogenesis requires Drosophila Mps1. Curr. Biol. 2004;14:2019–2024. doi: 10.1016/j.cub.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Foijer F., DiTommaso T., Donati G., Hautaviita K., Xie S.Z., Heath E., Smyth I., Watt F.M., Sorger P.K., Bradley A. Spindle checkpoint deficiency is tolerated by murine epidermal cells but not hair follicle stem cells. Proc. Natl. Acad. Sci. USA. 2013;110:2928–2933. doi: 10.1073/pnas.1217388110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foijer F., Xie S.Z., Simon J.E., Bakker P.L., Conte N., Davis S.H., Kregel E., Jonkers J., Bradley A., Sorger P.K. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc. Natl. Acad. Sci. USA. 2014;111:13427–13432. doi: 10.1073/pnas.1400892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley E.A., Kapoor T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 2013;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Schneider J.W., Condorelli G., Kaushal S., Mahdavi V., Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Hached K., Xie S.Z., Buffin E., Cladière D., Rachez C., Sacras M., Sorger P.K., Wassmann K. Mps1 at kinetochores is essential for female mouse meiosis I. Development. 2011;138:2261–2271. doi: 10.1242/dev.061317. [DOI] [PubMed] [Google Scholar]
- Halevy O., Novitch B.G., Spicer D.B., Skapek S.X., Rhee J., Hannon G.J., Beach D., Lassar A.B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Harper J.W., Adami G.R., Wei N., Keyomarsi K., Elledge S.J. The p21 Cdk-interacting protein Cipl is a potent inhibitor of Gl cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hewitt L., Tighe A., Santaguida S., White A.M., Jones C.D., Musacchio A., Green S., Taylor S.S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 2010;190:25–34. doi: 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoyama T., Nishijo K., Prajapati S.I., Li G., Keller C. Rb1 gene inactivation expands satellite cell and postnatal myoblast pools. J. Biol. Chem. 2011;286:19556–19564. doi: 10.1074/jbc.M111.229542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh M.S., Parker M.H., Scimè A., Parks R., Rudnicki M.A. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J. Cell Biol. 2004;166:865–876. doi: 10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang L.H., Lau L.F., Smith D.L., Mistrot C.A., Hardwick K.G., Hwang E.S., Amon A., Murray A.W. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- Innocenzi A., Latella L., Messina G., Simonatto M., Marullo F., Berghella L., Poizat C., Shu C.-W., Wang J.Y.J., Puri P.L., Cossu G. An evolutionarily acquired genotoxic response discriminates MyoD from Myf5, and differentially regulates hypaxial and epaxial myogenesis. EMBO Rep. 2011;12:164–171. doi: 10.1038/embor.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inomata K., Aoto T., Binh N.T., Okamoto N., Tanimura S., Wakayama T., Iseki S., Hara E., Masunaga T., Shimizu H., Nishimura E.K. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 2009;137:1088–1099. doi: 10.1016/j.cell.2009.03.037. [DOI] [PubMed] [Google Scholar]
- Ito S., Mantel C.R., Han M.-K., Basu S., Fukuda S., Cooper S., Broxmeyer H.E. Mad2 is required for optimal hematopoiesis: Mad2 associates with c-Kit in MO7e cells. Blood. 2007;109:1923–1930. doi: 10.1182/blood-2006-06-030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelluma N., Brenkman A.B., McLeod I., Yates J.R., III, Cleveland D.W., Medema R.H., Kops G.J.P.L. Chromosomal instability by inefficient Mps1 auto-activation due to a weakened mitotic checkpoint and lagging chromosomes. PLoS ONE. 2008;3:e2415. doi: 10.1371/journal.pone.0002415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Lin D.P., Matsumoto S., Kitazono A., Matsumoto T. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- Kops G.J.P.L., Foltz D.R., Cleveland D.W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S., Kuroda K., Le Grand F., Rudnicki M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBaer J., Garrett M.D., Stevenson L.F., Slingerland J.M., Sandhu C., Chou H.S., Fattaey A., Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Laptenko O., Beckerman R., Freulich E., Prives C. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc. Natl. Acad. Sci. USA. 2011;108:10385–10390. doi: 10.1073/pnas.1105680108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P., Westhorpe F.G., Taylor S.S. The spindle assembly checkpoint. Curr. Biol. 2012;22:R966–R980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Lepper C., Conway S.J., Fan C.-M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–631. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Rando T.A. Manifestations and mechanisms of stem cell aging. J. Cell Biol. 2011;193:257–266. doi: 10.1083/jcb.201010131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Winey M. The MPS1 family of protein kinases. Annu. Rev. Biochem. 2012;81:561–585. doi: 10.1146/annurev-biochem-061611-090435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M.A., Rossi F.M. Silencing inhibits Cre-mediated recombination of the Z/AP and Z/EG reporters in adult cells. PLoS ONE. 2009;4:e5435. doi: 10.1371/journal.pone.0005435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P.K., Blanpain C., Rossi D.J. DNA damage response in adult stem cells: pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011;12:198–202. doi: 10.1038/nrm3060. [DOI] [PubMed] [Google Scholar]
- Michel L.S., Liberal V., Chatterjee A., Kirchwegger R., Pasche B., Gerald W., Dobles M., Sorger P.K., Murty V.V.V.S., Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–359. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- Murphy M.M., Lawson J.A., Mathew S.J., Hutcheson D.A., Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A.W. Creative blocks: cell-cycle checkpoints and feedback controls. Nature. 1992;359:599–604. doi: 10.1038/359599a0. [DOI] [PubMed] [Google Scholar]
- Musacchio A., Salmon E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Nijenhuis W., von Castelmur E., Littler D., De Marco V., Tromer E., Vleugel M., van Osch M.H.J., Snel B., Perrakis A., Kops G.J.P.L. A TPR domain-containing N-terminal module of MPS1 is required for its kinetochore localization by Aurora B. J. Cell Biol. 2013;201:217–231. doi: 10.1083/jcb.201210033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijo K., Hosoyama T., Bjornson C.R.R., Schaffer B.S., Prajapati S.I., Bahadur A.N., Hansen M.S., Blandford M.C., McCleish A.T., Rubin B.P. Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J. 2009;23:2681–2690. doi: 10.1096/fj.08-128116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauklin S., Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2013;155:135–147. doi: 10.1016/j.cell.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss K.D., Wilson L.G., Keating M.T. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- Poss K.D., Nechiporuk A., Hillam A.M., Johnson S.L., Keating M.T. Mps1 defines a proximal blastemal proliferative compartment essential for zebrafish fin regeneration. Development. 2002;129:5141–5149. doi: 10.1242/dev.129.22.5141. [DOI] [PubMed] [Google Scholar]
- Poss K.D., Nechiporuk A., Stringer K.F., Lee C., Keating M.T. Germ cell aneuploidy in zebrafish with mutations in the mitotic checkpoint gene mps1. Genes Dev. 2004;18:1527–1532. doi: 10.1101/gad.1182604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P.L., Iezzi S., Stiegler P., Chen T.T., Schiltz R.L., Muscat G.E., Giordano A., Kedes L., Wang J.Y., Sartorelli V. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell. 2001;8:885–897. doi: 10.1016/s1097-2765(01)00373-2. [DOI] [PubMed] [Google Scholar]
- Reczek E.E., Flores E.R., Tsay A.S., Attardi L.D., Jacks T. Multiple response elements and differential p53 binding control Perp expression during apoptosis. Mol. Cancer Res. 2003;1:1048–1057. [PubMed] [Google Scholar]
- Ricke R.M., van Deursen J.M. Aneuploidy in health, disease, and aging. J. Cell Biol. 2013;201:11–21. doi: 10.1083/jcb.201301061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheteau P., Gayraud-Morel B., Siegl-Cachedenier I., Blasco M.A., Tajbakhsh S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148:112–125. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- Rubin B.P., Nishijo K., Chen H.I.H., Yi X., Schuetze D.P., Pal R., Prajapati S.I., Abraham J., Arenkiel B.R., Chen Q.R. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell. 2011;19:177–191. doi: 10.1016/j.ccr.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab R., Spunt S.L., Skapek S.X. Myogenesis and rhabdomyosarcoma: the Jekyll and Hyde of skeletal muscle. Curr. Top. Dev. Biol. 2011;94:197–234. doi: 10.1016/B978-0-12-380916-2.00007-3. [DOI] [PubMed] [Google Scholar]
- Sacco A., Doyonnas R., Kraft P., Vitorovic S., Blau H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R., Yao R., Kissenpfennig A., Van Wittenberghe L., Paldi A., Gayraud-Morel B., Guenou H., Malissen B., Tajbakhsh S., Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- Saramäki A., Banwell C.M., Campbell M.J., Carlberg C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006;34:543–554. doi: 10.1093/nar/gkj460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P., Sabourin L.A., Girgis-Gabardo A., Mansouri A., Gruss P., Rudnicki M.A. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Shea K.L., Xiang W., LaPorta V.S., Licht J.D., Keller C., Basson M.A., Brack A.S. Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell. 2010;6:117–129. doi: 10.1016/j.stem.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonatto M., Giordani L., Marullo F., Minetti G.C., Puri P.L., Latella L. Coordination of cell cycle, DNA repair and muscle gene expression in myoblasts exposed to genotoxic stress. Cell Cycle. 2011;10:2355–2363. doi: 10.4161/cc.10.14.15948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonatto M., Marullo F., Chiacchiera F., Musaró A., Wang J.Y., Latella L., Puri P.L. DNA damage-activated ABL-MyoD signaling contributes to DNA repair in skeletal myoblasts. Cell Death Differ. 2013;20:1664–1674. doi: 10.1038/cdd.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek S.X., Rhee J., Spicer D.B., Lassar A.B. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P., Gutarra S., García-Prat L., Rodriguez-Ubreva J., Ortet L., Ruiz-Bonilla V., Jardí M., Ballestar E., González S., Serrano A.L. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Suijkerbuijk S.J.E., van Dam T.J.P., Karagöz G.E., von Castelmur E., Hubner N.C., Duarte A.M.S., Vleugel M., Perrakis A., Rüdiger S.G.D., Snel B., Kops G.J.P.L. The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev. Cell. 2012;22:1321–1329. doi: 10.1016/j.devcel.2012.03.009. [DOI] [PubMed] [Google Scholar]
- Thompson S.L., Compton D.A. Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 2008;180:665–672. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S.L., Compton D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A., Staples O., Taylor S. Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J. Cell Biol. 2008;181:893–901. doi: 10.1083/jcb.200712028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres E.M., Dephoure N., Panneerselvam A., Tucker C.M., Whittaker C.A., Gygi S.P., Dunham M.J., Amon A. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Sun Q., Morita Y., Jiang H., Gross A., Lechel A., Hildner K., Guachalla L.M., Gompf A., Hartmann D. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148:1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E., Jiang Z., Chung D., Marth J.D., Phillips R.A., Gallie B.L. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zammit P.S., Golding J.P., Nagata Y., Hudon V., Partridge T.A., Beauchamp J.R. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J. Cell Biol. 2004;166:347–357. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.