

The first Blalock-Taussig shunt surgery in 1945 changed major congenital heart disease (CHD) from a universally lethal condition into a survivable one 1. In the 50 years that followed, rapid progress in anatomic diagnosis via echocardiography, and dramatic improvement in surgical and catheterization techniques, resulted in survival to adulthood and greatly improved quality of life for many individuals with CHD. Despite this, continuing progress in caring for individuals with the most complex forms of CHD has become incremental. One of the particularly frustrating aspects of managing patients with very complex CHD is that the outcomes are highly variable, even when accounting for surgical skill and anatomic complexity. In this issue of Circulation, Nakhleh et al show that a subset of patients with CHD and Heterotaxy has airway ciliary dysfunction that may account for increased morbidity and mortality independent of the complexity of the CHD2. This finding highlights a critical point that will surely affect patient care in the years to come: improving our understanding of the genetic etiology of CHD is likely to impact response to therapy. Individualized therapy for CHD based on the underlying genetics will hopefully contribute to the next major improvement in outcome.
Heterotaxy is defined as any arrangement of organs across the body's left-right axis that differs from complete situs solitus and complete situs inversus. CHD is one of the hallmarks of Htx, and is characterized by a wide range of highly complex cardiac anatomy3,4, including but not limited to anomalous systemic and pulmonary venous return, complex atrioventricular canal, functionally single ventricle and a range of abnormalities of the outflow tract (Table 1). As such, the cardiac lesions associated with Htx represent a significant surgical challenge; however, the surgical outcome in Htx patients remains significantly worse than that in patients with comparably complex CHD as evaluated by the Risk Adjustment in Congenital Heart Surgery-1 (RACHS) score4. This indicates that a subset of patients with Htx-associated CHD may have abnormalities extending beyond their heart disease that affect their response to extensive cardiac surgery. In order to improve outcome, we will need to identify and treat the non-cardiac factors in Htx patients that contribute to their excessive morbidity and mortality.
Table 1. Congenital heart disease in Htx3,4.
| Venous anomalies | Interrupted IVC | 13%-21% |
| Bilateral SVC | 44% | |
| Anomalous Pulmonary Venous Return | 13%-21% | |
| Atria and Ventricles | Atrioventricular septal defect | 32%-79% |
| Single ventricle | 68% | |
| Atrial Septal Defect | 62% | |
| Ventricular Septal Defect | 71% | |
| Ebstein's Malformation | 7% | |
| Outflow and Great Vessels | VA discordance | 70% |
| DORV | 33%-45% | |
| Pulmonary Stenosis/Atresia | 63%-74% | |
| Coarctation of the Aorta | 5%-16% | |
| Right Aortic Arch | 28% |
Extensive evidence points to a predominantly genetic etiology for human Htx, and there are reports of dominant, X-linked, and recessive pedigrees affected by Htx. Further, the incidence of Htx is increased in populations with a high degree of consanguinity, and Htx is seen in association with aneuploidies and genetic syndromes including Primary Ciliary Dyskinesia (PCD) and Bardet-Biedl Syndrome (BBS)5. Understanding the genetics of human Htx is complicated by high lethality leading to few large pedigrees and extensive phenotypic variability. At this time, mutations have been identified in 15 genes accounting for ∼10% of human Htx. In contrast, the list of candidate genes for Htx is much larger 6,7. If one adds all genes associated with human syndromic and non-syndromic Htx, and then adds genes that have been implicated in left-right development in model organisms, the total list of Htx candidate genes currently stands at more than 125 genes encompassing a broad range of biological functions (Figure 1). This indicates that Htx is actually not a monogenic diagnosis, but a manifestation of a broad range of developmental disorders that affect the development of left-right asymmetry. The clinical manifestations observed in Htx patients are further complicated by prominent effects on the development and function of other organ systems; for example, many of the genes affecting LR development, through their role in cilia, also have critical effect(s) on the lung and kidney.
Figure 1.
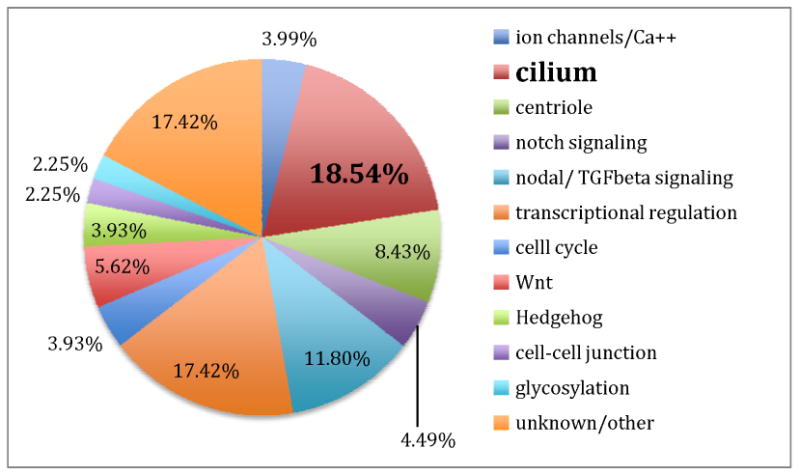
Predicted functional role of 162 genes with a possible role in Heterotaxy.
Almost 20% of the genes known to control LR development have a role in the biogenesis and function of the cilium8. Primary cilia are found on almost all cell types at some time, and they are central to the development of LR asymmetry. The vertebrate LR axis is initiated at the mammalian left-right organizer (LRO) late in gastrulation when motile primary cilia found on LRO cells generate directional flow of extraembryonic fluid, termed “nodal flow”9. The direction of nodal flow is determined by the inherent chirality of the cilium itself. Sensory cilia transduce nodal flow into asymmetric gene expression that is then interpreted by the developing heart to direct asymmetric cardiac morphogenesis10. Cilia, however, have a host of other functions during embryogenesis: they are essential for development and function of the kidney, bones and brain11. They are found on the cells of the developing myocardium12, and after birth, motile cilia in the respiratory tract are required for effective clearance of secretions. Seminal work by Afzelius showed that patients with Primary Ciliary Dyskinesia (PCD) have structural ciliary defects as the cause of their sino-pulmonary disease13. Notably, 6.5% of patients with a primary diagnosis of PCD also have Htx14. Since patients with Htx have a higher incidence of perioperative respiratory complications than non-Htx patients undergoing equally complex cardiac surgery, the question addressed in the article by Nakhleh et al is whether a subset of Htx patients have previously undiagnosed ciliary defects predisposing them to respiratory complications? The authors use a combination of physiological measurements of ciliary function, including direct imaging of cilia obtained from a nasal biopsy, and nasal NO measurement to assess patients for ciliary dysfunction. These techniques are more sensitive than the standard transmission electron microscopy approach more commonly used to diagnose PCD15. Notably, 42% of Htx patients had ciliary dysfunction, compared to none of the healthy controls. Sequencing of 14 PCD genes demonstrated enrichment of novel coding variants in Htx patients with ciliary dysfunction as compared to unaffected controls. Thus during very early embryonic development, a subset of patients with Htx have abnormal left-right and cardiac development due to abnormal ciliary function. Because cilia are also required in the airway throughout life, these patients also have chronic pulmonary disease that is a variant of PCD. The perioperative respiratory complications in at least some Htx patients are thus likely not caused exclusively by their cardiac surgery, but may be the product of intrinsic pulmonary abnormalities coupled with the stress of cardiopulmonary bypass and assisted ventilation.
With the advent of high throughput sequencing, we will be able to define the precise genetic abnormality(s) underlying a significant amount of congenital heart disease. The obvious question this raises is: We can diagnose it, can we treat it? In the case of the Htx patients who have ciliary abnormalities, there are promising approaches to improve their care. PCD without associated heart disease frequently presents with neonatal respiratory distress manifested by unexplained recurrent atelectasis, poor feeding and failure to thrive16. These symptoms resemble those associated with many types of CHD, complicating and delaying the diagnosis of intrinsic pulmonary disease. Even without associated CHD, PCD leads to chronic bronchitis, recurrent pneumonia and eventually bronchiectasis in a subset of patients. The progression of PCD-associated respiratory disease can be significantly slowed by proper therapy, and currently the lifespan of PCD patients without CHD is approaching normal15,17. In the cardiac patient population, the pulmonary manifestations of PCD are very likely exacerbated by CHD itself, and by some of the interventions required in the care of patients with complex CHD. For example, since PCD patients are entirely dependent on cough for airway clearance, intubation and mechanical ventilation that are a necessary evil in CHD surgery are particularly challenging. Careful perioperative management of secretions, and minimizing assisted ventilation may improve immediate perioperative care. It may be beneficial for CHD-PCD patients to have intensive regular chest physiotherapy, even while they appear clinically well. There is some evidence that beta-agonists increase ciliary beat frequency18, and therefore may be beneficial in those CHD-PCD patients with slowed ciliary beat. Finally, in contrast to care of CHD, aggressive antibiotic management of pulmonary symptoms is a cornerstone of PCD care17.
It is highly likely that patients with anatomically similar, but genetically distinct, cardiac lesions will benefit from individualized care tailored in no small part by their genetic diagnosis. An atrioventricular canal defect resulting from trisomy 21 is a different disease than that arising from a defect in cilia and the development of left-right asymmetry. We will need to evaluate whether therapies aimed at specific genetic diagnoses are beneficial to CHD patients. This will require large, multicenter studies combining CHD genomics with clinical research evaluating the benefit of focused clinical management strategies19. The work presented in this issue of Circulation linking ciliary defects and the vexing congenital heart disease seen in heterotaxy is a first step in this direction.
Acknowledgments
The author would like to thank Marie Egan for valuable discussion of the clinical aspects of PCD, and to Mustafa Khokha and Marko Boskovski for critical reading of the manuscript.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Blalock A, Taussig HB. Landmark article may 19, 1945: The surgical treatment of malformations of the heart in which there is pulmonary stenosis or pulmonary atresia. By alfred blalock and helen b. Taussig. JAMA. 1984;251:2123–2138. doi: 10.1001/jama.251.16.2123. [DOI] [PubMed] [Google Scholar]
- 2.Nakhleh N, Francis R, Giese RA, Tian X, Li Y, Zariwala MA, Yagi H, Khalifa O, Kureshi S, Chatterjee B, Sabol SL, Swisher M, Connelly PS, Daniels MP, Srinivasan A, Kuehl K, Kravitz N, Burns K, Sami I, Omran H, Barmada M, Olivier K, Chawla KK, Leigh M, Jonas R, Knowles M, Leatherbury L, Lo CW. High Prevalence of Respiratory Ciliary Dysfunction in Congenital Heart Disease Patients with Heterotaxy. Circulation. 2012;125:XXX–XXX. doi: 10.1161/CIRCULATIONAHA.111.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen MS, Anderson RH, Cohen MI, Atz AM, Fogel M, Gruber PJ, Lopez L, Rome JJ, Weinberg PM. Controversies, genetics, diagnostic assessment, and outcomes relating to the heterotaxy syndrome. Cardiol Young. 2007;17:29–43. doi: 10.1017/S104795110700114X. [DOI] [PubMed] [Google Scholar]
- 4.Swisher M, Jonas R, Tian X, Lee ES, Lo CW, Leatherbury L. Increased postoperative and respiratory complications in patients with congenital heart disease associated with heterotaxy. J Thorac Cardiovasc Surg. 2011;141:637–644. 644 e631–633. doi: 10.1016/j.jtcvs.2010.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutherland MJ, Ware SM. Disorders of left-right asymmetry: Heterotaxy and situs inversus. Am J Med Genet C Semin Med Genet. 2009;151C:307–317. doi: 10.1002/ajmg.c.30228. [DOI] [PubMed] [Google Scholar]
- 6.Fakhro KA, Choi M, Ware SM, Belmont JW, Towbin JA, Lifton RP, Khokha MK, Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bisgrove BW, Morelli SH, Yost HJ. Genetics of human laterality disorders: Insights from vertebrate model systems. Annu Rev Genomics Hum Genet. 2003;4:1–32. doi: 10.1146/annurev.genom.4.070802.110428. [DOI] [PubMed] [Google Scholar]
- 8.Basu B, Brueckner M. Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr Top Dev Biol. 2008;85:151–174. doi: 10.1016/S0070-2153(08)00806-5. [DOI] [PubMed] [Google Scholar]
- 9.Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking kif3b motor protein [published erratum appears in cell 1999;99:117] Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 10.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 11.Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slough J, Cooney L, Brueckner M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev Dyn. 2008;237:2304–2314. doi: 10.1002/dvdy.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, Robinson BV, Minnix SL, Olbrich H, Severin T, Ahrens P, Lange L, Morillas HN, Noone PG, Zariwala MA, Knowles MR. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 15.Lie H, Ferkol T. Primary ciliary dyskinesia: Recent advances in pathogenesis, diagnosis and treatment. Drugs. 2007;67:1883–1892. doi: 10.2165/00003495-200767130-00006. [DOI] [PubMed] [Google Scholar]
- 16.Hossain T, Kappelman MD, Perez-Atayde AR, Young GJ, Huttner KM, Christou H. Primary ciliary dyskinesia as a cause of neonatal respiratory distress: Implications for the neonatologist. J Perinatol. 2003;23:684–687. doi: 10.1038/sj.jp.7210987. [DOI] [PubMed] [Google Scholar]
- 17.Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, Zariwala MA, Knowles MR. Primary ciliary dyskinesia: Diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 18.Wong LB, Miller IF, Yeates DB. Stimulation of ciliary beat frequency by autonomic agonists: In vivo. J Appl Physiol. 1988;65:971–981. doi: 10.1152/jappl.1988.65.2.971. [DOI] [PubMed] [Google Scholar]
- 19.Kaltman JR, Schramm C, Pearson GD. The national heart, lung, and blood institute bench to bassinet program: A new paradigm for translational research. J Am Coll Cardiol. 2010;55:1262–1265. doi: 10.1016/j.jacc.2009.11.055. [DOI] [PubMed] [Google Scholar]