

Abstract
After treatment with ultraviolet radiation (UV), human fibroblasts that express the HPV type 16 E6 oncoprotein display defects in repair of cyclobutane pyrimidine dimers, hypersensitivity to inactivation of clonogenic survival and an inability to sustain DNA replication. To determine whether these effects are specific to depletion of p53 or inactivation of its function, fibroblast lines were constructed with ectopic expression of a dominant-negative p53 allele (p53-H179Q) to inactivate function or a short-hairpin RNA (p53-RNAi) to deplete expression of p53. Only the expression of HPV16E6 sensitized fibroblasts to UV or the chemical carcinogen, benzo[a]pyrene diolepoxide I (BPDE). Carcinogen-treated cells expressing p53-H179Q or p53-RNAi were resistant to inactivation of colony formation and did not suffer replication arrest. CHK1 is a key checkpoint kinase in the response to carcinogen-induced DNA damage. Control and p53-RNAi-expressing fibroblasts displayed phosphorylation of Ser345 on CHK1 45–120 min after carcinogen treatment with a return to near baseline phosphorylation by 6 h after treatment. HPV16E6-expressing fibroblasts displayed enhanced and sustained phosphorylation of CHK1. This was associated with enhanced phosphorylation of Thr68 on CHK2 and Ser139 on H2AX, both markers of severe replication stress and DNA double strand breaks. Incubation with the phosphatase inhibitor okadaic acid produced more phosphorylation of CHK1 in UV-treated HPV16E6-expressing cells than in p53-H179Q-expressing cells suggesting that HPV16E6 may interfere with the recovery of coupled DNA replication at replication forks that are stalled at [6-4]pyrimidine-pyrimidone photoproducts and BPDE-DNA adducts. The results indicate that HPV16E6 targets a protein or proteins other than p53 to deregulate the activity of CHK1 in carcinogen-damaged cells.
Keywords: HPV16E6, checkpoint, kinase, carcinogen, replication, p53
Introduction
As “guardian” of the genome, the tumor suppressor gene product p53 regulates many elements of DNA damage response.1 The p53 protein homotetramer transactivates p21Waf1 as the major effector of the G1 checkpoint response to DNA double-strand breaks.2 p53 transactivates two genes that are required for nucleotide excision repair, XPC3 and p48/XPE.4 Cells with defects in p53 function display reduced global repair of UV-induced cyclobutane pyrimidine dimers (CPDs)5 and reduced apoptosis after treatment with UV.6 p53 also is known to interact with several DNA helicases including XPB, XPD and WRN.7,8 Thus, p53 is central to many DNA metabolic transactions that serve to stabilize the genome.
Oncogenic viruses encode gene products that interfere with p53 function or deplete p53 expression. HPV16E6 acts as an E3 ligase targeting p53, and other proteins, for ubiquitin-mediated proteolysis.9 The selective ability of E6 gene products from oncogenic strains of HPV, but not from non-oncogenic strains, to deplete p53 suggested that viral carcinogenesis was in part a consequence of inactivation of p53 tumor suppressor function.10
Because of the facility with which HPV16E6 depletes and inactivates p53 in human cells, this oncogene has been commonly used as a tool to study the effects of p53 inactivation. Diploid human fibroblasts expressing HPV16E6 displayed severe phenotypes associated with depletion of p53 including ablation of p53-dependent G1 checkpoint function,11 attenuation of nucleotide excision repair,12 and progressive chromosomal destabilization.13 These phenotypes seen in HPV16E6-expressing human cells have been reproduced using other means to inactivate p53 including expression of dominant-negative p53 alleles and RNAi-mediated depletion of p53 protein expression,13–15 demonstrating that the phenotypes are likely derived from the effects of HPV16E6 on p53. However, HPV16E6 is known to affect other cellular proteins that are less well recognized than p53,16 suggesting that the oncogene also may influence cellular response to environmental carcinogens through p53-independent mechanisms.
To explore the requirement for p53 in human cell responses to environmental carcinogens, we depleted or inactivated p53 in telomerase-expressing diploid human fibroblasts by expression of HPV16E6, a dominant-negative p53 allele (p53-H179Q), or a short hairpin RNAi (p53-RNAi). Depletion of p53 with HPV16E6 was found to sensitize fibroblasts to inactivation of clonogenic survival by UV and benzo[a]pyrene diolepoxide I (BPDE), while inactivation of p53 with the p53-H179Q or depletion of p53 with p53-RNAi made cells resistant to these carcinogens. The severe sensitization provoked by expression of HPV16E6 was associated with severe replication arrest after carcinogen challenge. Analysis of intra-S checkpoint responses to UV and BPDE revealed that HPV16E6-expressing cells displayed enhanced and sustained phosphorylation of ser345 on CHK1, that was associated with enhanced expression of phospho-CHK2 and phospho-H2AX, as markers of replication stress and/or DNA double-strand breaks. The results demonstrate that HPV16E6 targets at least one other component of the machinery of DNA damage response to deregulate CHK1 and block the recovery of DNA replication after environmental DNA damage.
Results
Effect of inactivation of p53 on fibroblast sensitivity to environmental carcinogens
Previous studies have demonstrated that expression of HPV16E6 in human fibroblast strains sensitizes cells to inactivation of colony formation by UV.12,21 To determine whether this effect was due to inactivation of p53 function, telomerase-expressing human skin fibroblast lines were transduced with various genetic constructs to inactivate p53 function directly. As shown in Figure 1A, expression of HPV16E6 and p53-RNAi to knock down expression of p53 reduced p53 abundance to a similar degree. Reduced expression of p53 was associated with reduced expression of p21Waf1, a target of p53 transactivation and effector of G1 checkpoint function.11 Expression of p53-H179Q raised the total p53 level but also severely attenuated expression of p21Waf1. UV induces and activates p53,22 but high doses are usually required and the time course is slow relative to that seen after induction of DNA double strand breaks with IR.21,23 Treatment of F1-LXIN fibroblasts with 6 J/m2 UV produced a modest increment in p53 protein, a clear signal of phospho-15 p53 but little induction of p21Waf1 at 6 h. No induction of p21Waf1 was observed in p53-defective cells after treatment with UV. Quantitative analysis of ionizing radiation-induced G1 arrest by flow cytometry indicated that all three p53-inactivating alleles severely attenuated G1 checkpoint function.13 While the G1 to S transition was inhibited by 93% in the isogenic control line that was transduced with the empty vector LXIN and then treated with 1.5 Gy IR, this inhibition was 3%, 21% and 11% in the HPV16E6-, p53-RNAi- and p53-H179Q-expressing lines, respectively. The panel of isogenic human skin fibroblast lines provided the opportunity to determine whether HPV16E6 expression conferred hypersensitivity to UV and BPDE independently of its effect on p53 function.
Figure 1.

Effect of p53 on clonogenic survival after treatment with UV or BPDE. Fibroblasts were infected with an empty retroviral vector LXIN (◆), or with retroviral vectors expressing HPV16E6 (■), p53-RNAi (○), or p53-H179Q (*). (A) Western immunoblot analysis. Cells were either sham-treated or irradiated with 6 J/m2 UV followed by 6 hours incubation. Equivalent amounts of total protein were analyzed by western immunoblot for expression of p53, phospho-ser15-p53, p21Waf1 and β-actin (as loading control). (B and C) Logarithmically growing cells were plated at a density of 1,000 cells per 100 mm dish and incubated 8 hours followed by UV irradiation or 15 minutes BPDE treatment. Colonies were allowed to grow 14 days before quantification. The means with standard deviations of triplicate experiments for each cell line are shown.
The control fibroblast line with intact p53 function, F1-LXIN, displayed normal sensitivity to inactivation of colony formation after treatment with UV and BPDE (Fig. 1B) equivalent to that reported previously for the parental F1 fibroblast strain24 and similar to that of other normal human skin fibroblast strains.21 The HPV16E6-expressing line, F1-HPV16E6, displayed hypersensitivity to UV equivalent to that reported previously for other HPV16E6-expressing normal human fibroblast lines.12,21 The F1-HPV16E6 line also displayed hypersensitivity to BPDE (Fig. 1C). In contrast, the F1-p53RNAi and F1-p53H179Q lines displayed resistance to inactivation of colony formation by both UV and BPDE (Fig. 1B and C). These results indicated that expression of HPV16E6 sensitized human fibroblasts to carcinogen-induced toxicity through a mechanism that was independent of p53. Inactivation of p53 function by protein depletion (p53-RNAi) or overexpression of a mutant allele (p53-H179Q) rendered fibroblasts resistant to carcinogen-induced cytotoxicity.
Effect of inactivation of p53 on DNA replication and intra-S checkpoint signaling in carcinogen-damaged cells
Hypersensitivity of HPV16E6-expressing fibroblasts to UV cytotoxicity was associated with development of severe inhibition of DNA replication 6–8 h after irradiation.21 As shown in Figure 2, treatment with 6 J/m2 caused little change in the profile of incorporation of BrdU as a measure of replicative DNA synthesis in F1-LXIN control fibroblasts 6–8 h after irradiation. Although DNA replication was transiently inhibited by UV,21,25 F1-LXIN cells displayed near control levels of incorporation of BrdU by 6–8 h after treatment with UV. The HPV16E6-expressing fibroblasts differed from the F1-LXIN control line in their response to UV in two ways. First, the fraction of BrdU-labeled cells in the first quarter of S (i.e., with 2–2.5 N DNA content) was increased by UV treatment, and second, incorporation of BrdU by cells in the second, third and fourth quarters of S (i.e., with 2.5–4 N DNA content) was severely depressed. Inactivation of p53 function with p53-RNAi or p53-H179Q also caused a modest UV-induced accumulation of BrdU-labeled cells in the first quarter of S, but DNA synthesis by UV-damaged cells in the second half of S proceeded at near control rates, as seen in F1-LXIN.
Figure 2.

Effects of carcinogens on DNA replication throughout S. Logarithmically growing cells were either irradiated with 6 J/m2 UV or treated with 100 nM BPDE for 15 minutes followed by 6 hours incubation. Cells were labeled with BrdU for the final 2 h of this incubation, then processed for flow cytometric analysis of BrdU incorporation and DNA content. Rectangles enclose BrdU-labeled cells in the first quarter of S-phase with 2–2.5 N DNA content. The percentages of S-phase cells in this compartment are shown above the rectangles.
Cellular responses to BPDE were identical to those after treatment with UV. HPV16E6-expressing cells that were treated with BPDE displayed accumulation of BrdU-labeled cells in the first quarter of S, and severe inhibition of BrdU incorporation in the second, third and fourth quarters of S. Inactivation of p53 with p53-RNAi and p53-H179Q did not sensitize cells to treatment with BPDE. These results demonstrate that hypersensitivity to carcinogen toxicity in HPV16E6-expressing cells was associated with severe inhibition of DNA replication in the second through fourth quarters of S.
To determine the kinetics of inhibition of DNA synthesis, cells were incubated with [3H]thymidine at various times after carcinogen-treatment (Fig. 3A and B). The cell lines displayed the same degree of inhibition of DNA synthesis at 45 min and 2 h after treatment with UV. While F1-LXIN control fibroblasts and fibroblasts expressing p53-RNAi to deplete p53 recovered DNA synthesis rates between 2 and 6 h after irradiation, the HPV16E6-expressing line displayed persistent and severe inhibition of DNA synthesis up to 6 h after treatment. The degree of inhibition of DNA synthesis 6 h after 6 J/m2 UV in F1-LXIN and F1-HPV16E6 fibroblasts (35% and 75% inhibition, respectively) approximated the relative inactivation of colony formation by UV, as was reported previously with another set of HPV16E6-expressing fibroblasts.21 Similarly, at 6 h after treatment with BPDE, the HPV16E6-expressing fibroblasts displayed more severe inhibition of DNA replication than the F1-LXIN and F1-p53RNAi fibroblasts.
Figure 3.
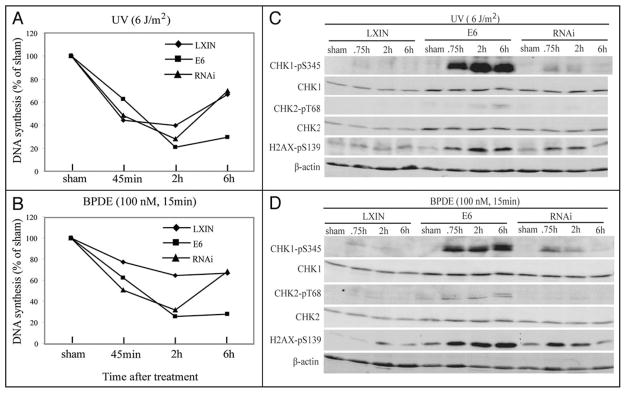
Inhibition of DNA synthesis and activation of DNA damage checkpoints in carcinogen-treated human fibroblasts. (A and B) At various times after treatment with 6 J/m2 of UV (A) or 100 nM BPDE for 15 minutes (B), incorporation of [3H]thymidine was quantified and expressed relative to the sham-treated control. (C and D) Fibroblasts were treated with 6 J/m2 of UV (A) or 100 nM BPDE for 15 minutes (B). At different times after treatment with carcinogen (45 min, 2 or 6 h) or sham-treatment (2 h), equivalent amounts of total protein were analyzed by western immunoblot for phospho-Ser345-CHK1, CHK1, phospho-Thr68-CHK2, CHK2, γH2AX (phospho-Ser139-H2AX) and β-actin.
UV and BPDE are known to induce a CHK1-dependent intra-S checkpoint response that slows the rate of replicon initiation.18,26 As expression of HPV16E6 was associated with severe, persistent inhibition of DNA replication after carcinogen treatment, DNA damage checkpoint function was monitored in carcinogen-damaged fibroblasts. Treatment of F1-LXIN and F1-p53RNAi fibroblasts with 6 J/m2 UV and 300 nM BPDE induced a transient phosphorylation of ser345 in CHK1 that was maximal 45 min to 2 h after treatment (Fig. 3C and D). CHK1 phosphorylation returned to near basal levels by 6 h after treatment of these lines. The HPV16E6-expressing fibroblasts displayed a modestly increased basal level of expression of phospho-CHK1 in the absence of DNA damage; however, in response to UV and BPDE, CHK1 was rapidly and strongly phosphorylated and phosphorylation did not diminish for up to 6 h after treatment.
The minor histone variant H2AX is phosphorylated in response to various forms of DNA damage including UV.27,28 Phosphorylation of H2AX was induced by carcinogen-treatment in all fibroblast lines (Fig. 3). Again, while levels of phospho-H2AX declined to near the basal state within 6 h of treatment of the F1-LXIN and F1-p53RNAi lines, the levels of phospo-H2AX remained elevated at 6 h in the F1-HPV16E6 fibroblasts. CHK2 is phosphorylated by ATM in response to DNA double strand breaks and by ATR under conditions of severe replication arrest.29 Little phospho-CHK2 was observed in F1-LXIN and F1-p53RNAi after treatment with the carcinogens. In contrast HPV16E6-expressing cells developed increased expression of phospho-CHK2 that was of greatest intensity at 6 h after carcinogen treatment. These results show that HPV16E6-expressing cells displayed significantly increased checkpoint signaling in response to carcinogen challenge. The persistent activation of checkpoint kinases CHK1 and CHK2 was associated with sustained replication arrest in HPV16E6-expressing cells.
To test the role of CHK1 in the inhibition of DNA replication in UV-damaged cells, F1-LXIN and F1-HPV16E6 cells were electroporated with siRNA to deplete expression of CHK1 (Fig. 4). Depletion of CHK1 in F1-LXIN was associated with complete reversal of the UV-induced inhibition of DNA replication seen at 6 h after treatment (Fig. 4B). Depletion of CHK1 in F1-HPV16E6 produced only a partial reversal of the inhibition of DNA replication by UV. Incomplete reversal of the inhibition of DNA replication by UV in F1-HPV16E6 cells was associated with incomplete depletion of CHK1 (Fig. 4A). Even though the levels of total protein were reduced by >80%, HPV16E6-expressing cells still expressed levels of phospho-CHK1 equivalent to that seen in F1-LXIN cells that were electroporated with a scrambled control siRNA and then treated with UV. These results demonstrate that the inhibition of DNA replication that persists 6 h after treatment of normal human fibroblasts with a moderately cytotoxic dose of UV (6 J/m2) is due to signaling through the intra-S checkpoint effector kinase CHK1. Enhanced, persistent activation of CHK1 in HPV16E6-expressing fibroblasts appears to account for the severe and persistent inhibition of DNA replication after carcinogen treatments.
Figure 4.

Reversal of UV-induced inhibition of DNA synthesis by knockdown of CHK1. F1-LXIN control fibroblasts and fibroblasts expressing HPV16E6 were electroporated with siRNA against CHK1 or a scrambled control siRNA. Twenty four hours later cells were sham-treated or treated with 6 J/m2 of UV irradiation, then harvested 6 h after treatment. Equivalent amounts of total protein were analyzed by western immunoblot for phospho-Ser345-CHK1, CHK1 and β-actin (A). DNA synthesis in cell cultures was determined by quantification of [3H]thymidine incorporation during the final 15 min of post-treatment incubation (B). Results show the mean (+standard deviation) levels of DNA synthesis in treated cells relative to sham-treated controls in three independent experiments.
Formation and repair of UV-induced DNA damage
Previous studies have demonstrated a requirement for p53 for global excision repair of UV- and BPDE-induced DNA damage.5,15,30 A radioimmunoassay was used to determine the formation and repair of UV-induced CPDs and [6-4]PPs in the human fibroblast lines. All fibroblast lines repaired [6-4]PPs with high efficiency. When sampled 6 h after treatment with 12 J/m2, the levels of [6-4] PPs were reduced by >90% from the levels measured immediately after irradiation, regardless of p53 function. This result indicates that NER of [6-4]PPs does not require p53. UV-induced CPD’s were repaired much more slowly than [6-4]PP’s and by 6 h after treatment the levels of CPD’s were reduced by 30% or less. These results were congruent with a recent report showing rapid repair of [6-4]PP’s in HPV16E6-expressing fibroblasts and slow repair of CPD’s.31 Thus, expression of HPV16E6 did not inactivate NER of [6-4]PP’s.
Regulation of CHK1 in hydroxyurea-treated F1-HPV16E6 cells
The deregulation of CHK1 phosphorylation in carcinogen-damaged F1-HPV16E6 fibroblasts could reflect a generalized defect in response to replication stress. To test this possibility fibroblasts were treated with 2 mM hydroxyurea (HU) for 6 h to deplete DNA precursor pools and induce a condition of severe replication stress. After treatment with HU, cells were incubated an additional 6 h in fresh medium to permit recovery of DNA replication. Cells were incubated with BrdU for 2 h immediately before cell harvest at the end of the treatment with HU or after the 6 h recovery phase. Treatment with HU inhibited DNA replication severely as evidenced by the lack of incorporation of BrdU in cells with ~2.5–3.5 N DNA content (Fig. 5A). After wash-out of HU and incubation in fresh medium, a fraction of F1-LXIN cells resumed DNA synthesis and incorporated BrdU to the same level seen in cells that were not incubated with HU. A large bolus of cells with 4 N DNA content was observed to incorporate BrdU at control rates. These cells likely were collected at the beginning of S phase during incubation with HU, and upon removal of HU synchronously transited S phase and nearly doubled their DNA content during the subsequent 6 h incubation. Another fraction of about 6% of cells with ~2.5–3.5 N DNA content displayed markedly reduced incorporation of BrdU suggestive of impaired recovery of DNA replication. This population of cells appeared to have suffered greater damage during incubation with HU so that recovery of DNA replication was impaired. Thus, in the control F1-LXIN cells, treatment with HU was associated with severe inhibition of DNA replication, and upon removal of the drug, a minor subfraction of cells that were arrested in S phase displayed impaired recovery of DNA replication.
Figure 5.

HPV16E6-expressing cells display normal dephosphorylation of CHK1 and recovery of DNA synthesis after HU-induced replication arrest. Proliferative fibroblasts were incubated with 2 mM HU for 6 h and then after removal of HU incubated in normal growth medium for an additional 6 h. (A) BrdU was added to culture medium 2 h before cell harvest for flow cytometric analysis of DNA synthesis. Rectangles enclose cells with ~2.5–3.5 N DNA content and reduced incorporation of BrdU. The percentages of cells in this compartment are shown above the rectangles. (B) Cell protein was analyzed for expression of phospho-ser345-CHK1, phospho-thr68-CHK2 and β-actin.
Inactivation of p53 function by expression of HPV16E6 or overexpression of the mutant allele p53-H179Q did not significantly alter the pattern of cellular response to HU. DNA replication was severely inhibited by HU and a majority of cells recovered control rates of DNA replication. The subfraction of cells with impaired recovery of DNA replication was similar to that seen in F1-LXIN cells with active p53. Western immunoblot confirmed that inhibition of DNA replication with HU activated CHK1 by phosphorylation, and upon washout of HU, phospho-ser345-CHK1 returned to basal levels within 6 h in all three cell lines (Fig. 5B). F1-HPV16E6 cells with hypersensitivity to UV and BPDE did not appear to be hypersensitive to HU, and recovered DNA replication after reversal of HU-induced replication stress. Thus, F1-HPV16E6 cells did not display a generalized defect in CHK1 regulation in response to replicative stress.
Analysis of CHK1 phosphatase expression
The enhanced expression of phospho-CHK1 in carcinogen-treated HPV16E6 cells resembles a condition that is induced in human cells by knockdown of the protein phosphatase, PPM1D/Wip1.32 PPM1D appears to have a comparatively broad range of substrates (including p38 MAP kinase, p53 and CHK1) that are phosphorylated in response to DNA damage. Western immunoblot analysis indicated that F1-HPV16E6 cells expressed levels of PPM1D equivalent to those seen in F1-LXIN and F1-p53RNAi (results not shown). While PPMID expression in F1-LXIN was induced 6 h after treatment with 6 J/m2, as expected, it was not induced in F1-HPV16E6 and F1-p53RNAi cells, also as expected. The enhanced and persistent activation of CHK1 in carcinogen-treated HPV16E6-expressing fibroblasts could not be accounted for by reduced expression of PPM1D as the F1-p53-RNAi cells with equivalent expression of PPM1D did not display hyperactivity of CHK1 after UV treatment.
PP2A was recently shown also to regulate CHK1 phosphorylation in human cells.33 Western immunoblot indicated equal levels of expression of the large 70 kDa subunit that contains PP2A catalytic activity in F1-LXIN and F1-HPV16E6 cells (results not shown). To examine the possibility that expression of the catalytic subunit did not predict phosphatase activity, cell extracts were incubated under conditions previously reported to detect dephosphorylation of CHK1 by PP2A.33 Extracts of HPV16E6-expressing cells were as effective at dephosphorylating CHK1 in vitro as extracts of F1-LXIN control cells or cells expressing p53-H179Q (results not shown). Dephosphorylation of CHK1 in cell extracts was inhibited fully by 100 nM okadaic acid (OA) an inhibitor of PPP family phosphatases.
As the enhanced expression of phospho-CHK1 in carcinogen-damaged F1-HPV16E6 fibroblasts did not appear to be due to reduced dephosphorylation, an experiment was done to test for enhanced phosphorylation. CHK1 phosphorylation was analyzed in cells that were incubated with OA to inhibit phosphatase activities after treatment with UV or HU. As shown in Figure 6, treatment with HU caused phosphorylation of CHK1 that was fully reversed 6 h after withdrawal of HU. Incubation with OA after withdrawal of HU prevented dephosphorylation of CHK1 in both HPV16E6- and p53-H179Q-expressing fibroblasts. CHK1 phosphorylation 6 hr after UV was reversed in F1-p53-H179Q cells but not in F1-HPV16E6 cells. Post-irradiation incubation with OA blocked the dephosphorylation of CHK1 in both cell lines. However, the signal of phospho-CHK1 was much greater in UV- and OA-treated F1-HPV16E6 cells than in UV- and OA-treated F1-p53-H179Q cells. Incubation with OA alone also induced expression of phospho-CHK1 consistent with a previous report showing that PP2A regulates the activity of CHK1.33 These results suggest that the increased expression of phospho-CHK1 in UV- and BPDE-treated HPV16E6-expressing cells was due to enhanced phosphorylation by a CHK1 protein kinase.
Figure 6.
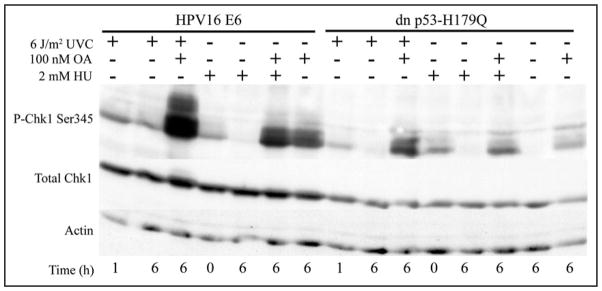
HPV16E6-expressing cells display enhanced phosphorylation of CHK1 after UV irradiation. Proliferative fibroblasts were treated with 6 J/m2 UV and then incubated for 1 or 6 h without OA or for 6 h with 100 nM OA to inhibit PPP family phosphatases. Control cultures were incubated for 6 h with the DMSO solvent for OA or OA alone. Parallel cultures were incubated with HU for 6 h then immediately harvested (0 h) or after withdrawal of HU, incubated for an additional 6 h with or without OA. Equal amounts of cell protein were separated by SDS-PAGE, and CHK1, phospho-ser345-CHK1 and actin were monitored by immunoblot analysis.
Expression of HPV16E6 sensitizes p53-defective fibroblasts
The results described above indicated that depletion of p53 by expression of HPV16E6 sensitized fibroblasts to carcinogenic stress while inactivation of p53 function by expression of a dominant-negative allele or short-hairpin RNA did not. To further confirm that the effect of E6 was independent of p53, the F1-p53-RNAi cells were transduced with E6 and UV sensitivity was determined. Expression of E6 in F1-p53-RNAi cells induced all of the phenotypes that were induced in the F1-LXIN line, including sensitization to UV-induced inactivation of colony formation, replicative arrest, and CHK1 phosphorylation (Fig. 7). Expression of E6 also reduced the low level of p53 remaining in the F1-p53-RNAi line (Fig. 1), confirming that the oncoprotein was functional in these cells.
Figure 7.

HPV16E6 sensitizes p53-defective fibroblasts to UV. The F1-p53-RNAi line was infected with retrovirus carrying HPV16E6 or the LXIN empty vector then selected for growth in G418. E6- and LXIN-transduced cells were assayed for UV-induced inhibition of colony formation as in Figure 1, BrdU incorporation as in Figure 2, and CHK1 phosphorylation as in Figure 3 (cells were harvested 6 h after 6 J/m2 UVC). Colony formation in UV-treated E6-expressing cells was expressed relative to that measured in UV-treated empty-vector controls (mean + sd, n = 3).
Discussion
We report here a new feature of cellular dysfunction associated with expression of HPV16E6, deregulation of CHK1 after treatment with the carcinogens UV and BPDE. Although the two carcinogens produce very different forms of DNA damage, cellular responses to the DNA damage were quite similar supporting a hypothesis that cell cycle checkpoint responses are stereotypic (Table 1). The hypersensitivity of HPV16E6-expressing cells to inactivation of colony formation and inhibition of DNA replication was not a consequence of inactivation of p53 function, as expression of a dominant-negative p53 allele or depletion of p53 did not sensitize cells to carcinogen challenge. Indeed, inactivation of p53 function by these two alternative means was associated with cellular resistance to inactivation of colony formation by carcinogen challenge. Expression of HPV16E6 also sensitized p53-depleted cells to UV-induced cell killing, replicative arrest and activation of CHK1. In response to UV-induced DNA damage, p53 induces pro-apoptotic genes that kill cells and inactivate colony formation.6 Consequently, cells in which p53 was inactivated by HPV16E6 displayed severe hypersensitivity to UV and BPDE in comparison to isogenic lines in which p53 was inactivated by other means.
Table 1.
Comparison of effects on DNA damage responses in NHF1-TERT fibroblast lines with expression of HPV16E6, p53-H179Q, p53-RNAi or p53-RNAi + E6
| Lines | IR G1 chkpt |
UV CFE |
UV DNA syn |
UV CHK1 act’n |
BPDE CFE |
BPDE DNA syn |
BPDE CHK1 act’n |
HU DNA syn |
HU CHK1 inact’n |
|---|---|---|---|---|---|---|---|---|---|
| TERT | eff | wt | wt | wt | wt | wt | wt | wt | wt |
| E6 | def | sens | sens | sens | sens | sens | sens | wt | wt |
| p53-H179Q | def | res | wt | wt | res | wt | wt | wt | wt |
| p53-RNAi | def | res | wt | wt | res | wt | wt | nd | nd |
| p53-RNAi + E6 | def | sens | sens | sens | nd | nd | nd | nd | nd |
G1 checkpoint (chkpt) response to IR was quantified as the reduction in the fraction of cells in the first half of S phase 6–8 h after 1.5 Gy. UV- and BPDE-induced inactivation of colony formation efficiency (CFE) was measured by treating single cells with the carcinogens then counting colonies two weeks later. UV- and BPDE-induced inhibition of DNA synthesis (syn) was measured by evaluation of BrdU incorporation 6 h after treatment with 6 J/m2 UV or 100 nM BPDE. UV- and BPDE-induced activation (act’n) of CHK1 was determined by western immunoblot analysis of phospho-ser345 in CHK1 6 h after treatment with 6 J/m2 UV or 100 nM BPDE. Recovery of DNA synthesis was determined as BrdU incorporation 6 h after removal of 2 mM HU. CHK1 inactivation (inact’n) was measured as loss of phospho-ser345 in CHK1 6 h after removal of 2 mM HU. Eff = effective response with >90% G1 arrest; def = defective response with <22% G1 arrest; wt = wildtype response equivalent to that seen in diploid human fibroblast lines; sens = sensitive response with greater inactivation of colony formation, greater inhibition of DNA synthesis, and greater than wildtype activation of CHK1 after carcinogen challenge; res = resistant response with less than wildtype inactivation of colony formation after carcinogen challenge; nd = not determined.
The p53-independent, HPV16E6-dependent sensitization of human fibroblasts to UV and BPDE was associated with development of a state of severe replication arrest after carcinogen challenge. By 6 h after treatment with carcinogen, HPV16E6-expressing cells had accumulated at the beginning of S phase, and cells in the second, third and fourth quarters of S displayed little or no incorporation of BrdU as a measure of DNA synthesis. Under the same conditions, the F1-LXIN control cells and the lines with dominant-negative p53-H179Q or depletion of p53 displayed very little alteration in the profiles of BrdU incorporation. DNA damage responses including rapid [6,4]PP NER and CHK1-dependent intra-S checkpoint signaling were largely able to ameliorate the inhibitory effects on DNA replication of UV-induced DNA damage, even in cells without p53 function. The ~35% inhibition of DNA replication that persisted in F1-LXIN cells 6 h after 6 J/m2 UV was due to active CHK1-dependent checkpoint signaling as knockdown of CHK1 fully reversed the inhibition. We interpret the accumulation of carcinogen-treated, HPV16E6-expressing cells at the beginning of S to reflect progression of damaged G1 cells into S phase, with further progression through S being retarded. Cells that were in S phase at the time of irradiation proceeded to replicate part of their DNA but progressively arrested replication, culminating in cells with 3–4 N DNA content being capable of little or no incorporation of BrdU. This severe inhibition of DNA replication (replication collapse) was associated with a strong and persistent activation of CHK1. While substantial depletion of CHK1 expression partially reversed the inhibition of DNA replication in UV-treated F1-HPV16E6 cells, significant levels of activated CHK1 were detected and could account for the incomplete reversal of the inhibition of DNA replication. We reported previously that overexpression of CHK1 severely inhibited DNA replication in HeLa cells.18 Similar levels of expression of a kinase-inactive CHK1 allele enhanced DNA synthesis in un-damaged control cells and fully reversed the UV-induced intra-S checkpoint response of inhibition of replicon initiation. Thus, CHK1 exerts significant control over the rate of DNA replication in human cells.
The phenotype of HPV16E6-expressing cells resembled that of mammalian cells with targeted inactivation of PPM1D, a p53-inducible protein phosphatase with substrates that include p53 and CHK1.32 Inactivation of PPM1D enhances and sustains CHK1 phosphorylation in response to DNA damage. Expression of HPV16E6 did not produce a generalized defect in regulation of CHK1 phosphorylation, however, as F1-HPV16E6 cells recovered DNA replication and CHK1 phosphorylation was diminished after reversal of HU-induced replication stress. Western immunoblot also demonstrated expression of PPM1D in HPV16E6-expressing cells, although it was not induced by UV treatment.
The difference in the persistence of CHK1 phosphorylation in HPV16E6-expressing cells after treatment with HU or the carcinogens, UV and BPDE, may be related to the structure of arrested or stalled replication forks. Incubation with 2 mM HU depleted DNA precursors and produced a severe inhibition of DNA polymerase on both leading and lagging template strands. Upon withdrawal of HU and replenishment of DNA precursors, replication resumed to near control levels in most of the arrested cells. Treatment with UV and BPDE produces widely spaced template lesions that block DNA polymerization. While lesions on the lagging template strand produce gaps in nascent DNA of less than the size of Okazaki fragments, lesions on the leading template strand may produce large stretches of single-stranded DNA as coordination between leading and lagging strand synthesis is uncoupled. Evidence for uncoupling of leading and lagging strand synthesis at sites of irreparable damage has been obtained in yeast34 and human cells,35 and during in vitro replication by human cell extracts of a plasmid molecules with a single site-specific lesion.36 The extended region of single-stranded template DNA that is generated after uncoupling of leading and lagging strand synthesis should be coated with RPA, accumulate ATR/ATRIP/TopBP1,37 and through mediators such as Claspin,38 Timeless and Tipin39 lead to phosphorylation and activation of CHK1.40
The recovery of coupled DNA replication after arrest of DNA growing points at carcinogen-induced DNA damage appears to be required to extinguish the CHK1 signal and allow resumption of replicon initiation. Bypass of the blocking lesion by an error-free or error-prone translesion DNA polymerase should restore coupled replication. Rapid bypass of CPD’s is accomplished by pol eta, and pol kappa can bypass BPDE-DNA adducts. Mutational inactivation of pol eta in xeroderma pigmentosum variant cells and inactivation of pol kappa in murine cells caused more persistent activation of CHK1 and inhibition of replicon initiation after carcinogen challenge.25,41,42 The mechanisms of bypass of [6-4]PPs in human cells have not yet been established although evidence suggests the participation of Rev1 and Rev3, subunits of pol zeta.43,44 In yeast strains, cooperation between pol iota, pol delta and pol zeta has been implicated for bypass of [6-4]PPs.45 A second mechanism for bypass of DNA lesions involves a process of template switching46 with denaturation of daughter DNA strands, renaturation of template strands and pairing of daughter strands to produce a “chicken foot”-like intermediate. The blocked terminus is switched from the damaged template strand to an undamaged daughter template and DNA polymerase extends the terminus. After completion of synthesis on the daughter DNA template, the process of denaturation and renaturation is reversed to regenerate a replication fork with the daughter strand terminus beyond the site of template damage. In yeast this process appears to require Rad5 and MMS2 as well as PCNA and pol delta.47 The bypass of carcinogen-induced DNA damage during DNA replication should restore coupled DNA replication and remove the substrate that causes phosphorylation and activation of CHK1.
The demonstration that HPV16E6-expressing cells recovered from HU-induced replication stress, but not from inhibition of DNA replication by UV and BPDE, suggests that HPV16E6 targets some component of the machinery of DNA repair that restores coupled DNA replication on damaged templates. The failure to restore coupled DNA replication after treatment with UV or BPDE causes sustained phosphorylation of CHK1 by ATR exceeding the capacity of PPM1D and PP2A to dephosphorylate CHK1 and extinguish the intra-S checkpoint signal to inhibit replicon initiation. The substantial reduction in CHK1 phosphorylation seen in NHF1-hTERT-LXIN and NHF1-hTERT-p53RNAi fibroblasts between 45 min and 6 h after treatment with UV (Fig. 3) was associated with >90% repair of [6-4]PPs suggesting that replication arrest at [6-4]PPs is largely responsible for uncoupling of replication forks, activation of CHK1 and intra-S checkpoint signaling. Only a small fraction of CPD’s were repaired by 6 h after treatment with UV (<30%), and the persisting CPD’s appear to be rapidly bypassed by pol eta. The attenuation of CHK1 phosphorylation with time in BPDE-treated NHF1-hTERT-LXIN and NHF1-hTERT-p53RNAi cells also implies fast repair of BPDE-DNA lesions that block DNA replication. Failure to restore coupled DNA replication may lead to replication fork collapse and/or formation of DNA double-strand breaks.34 The enhanced and sustained phosphorylation of H2AX and CHK2 in carcinogen-damaged HPV16E6-expressing fibroblasts is consistent with such a model.
Smoking is a risk factor for cervical cancer that interacts synergistically with infection of HPV16.48 Benzo[a]pyrene is a carcinogen in cigarette smoke that can be delivered throughout the body when it enters the blood. The results described here provide a plausible mechanism whereby expression of HPV16E6 in infected cervix enhances the genotoxicity of chemical carcinogens in cigarette smoke.
Materials and Methods
Cell lines and culture
The normal human fibroblast strain, NHF1, was derived from neonatal foreskin. An immortal derivative was obtained by ectopic expression of human telomerase (NHF1-hTERT).13 NHF1-hTERT cells were infected with a replication-defective retrovirus containing the neomycin resistance gene (LXIN) with and without the HPV16E6 oncogene and denoted as F1-hTERT-LXIN and F1-hTERT-HPV16E6.13 p53 was also inactivated in NHF1-hTERT by infection with retro-viruses encoding a dominant-negative p53 allele (p53-H179Q, provided by Dr. Howard Liber, Colorado State University)13 or a short interfering RNA to knockdown expression of p53 (p53-RNAi). The retroviral short interfering RNA vector to knockdown p53 was purchased from Oligoengine. Vesicular stomatitis virus glycoprotein G-pseudotyped, replication-defective retroviruses were produced as previously described following transient transfection of viral vector and helper plasmids into HEK 293T cells.13 As F1-hTERT was initially selected in 200 μg/ml puromycin, stable expression of the p53 RNAi was done by growth in 400 μg/ml puromycin.
The fibroblast lines were cultured in DMEM supplemented with 2 mM L-glutamine and 10% fetal bovine serum. All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2, and were tested and shown to be free of mycoplasma contamination using a commercial kit (Gen-Probe).
Genotoxin treatment
After a single rinse with phosphate-buffered saline (PBS), culture dishes were irradiated with UV (254 nm) at a fluence rate of 0.5 J/m2/s, then reserved medium was replaced and cells incubated at 37°C for the indicated periods of time. BPDE (from the NCI carcinogen repository) was dissolved in anhydrous dimethylsulfoxide and added to serum-free medium at 1/1,000 dilution to yield the specified concentrations. Cultures were incubated in serum-free medium containing BPDE for 15 minutes, rinsed with warm PBS, reserved medium was replaced, and cells were returned to the incubator. Prior to each BPDE treatment, the reactivity of the carcinogen was determined by the method of MacLeod and Lew17 to be >90%. For IR irradiation, cultures were maintained in their culture medium and exposed to a 137Cs source at a dose rate of 0.86 Gy/min. For hydroxyurea (HU) treatment, cultures were incubated with medium containing 2 mM HU for six hours. After this treatment, cultures were rinsed once with PBS, then incubated in fresh medium for an additional six hours. Sham-treated cultures were handled exactly the same way as described above, except they were not exposed to genotoxin.
Clonogenic survival assays
Logarithmically growing cells were plated in 10-cm dishes at a density of 1,000 cells/dish, incubated for 8 hours, then irradiated with various fluences of UV or treated with BPDE at various concentrations. Medium was changed every 3 days. After 2 weeks, colonies on the dishes were fixed and stained with a solution containing 40% methanol and 0.05% crystal violet. Colonies of >50 cells were counted. Assays were repeated three times, each time with three dishes for each dose.
Flow cytometric analysis of BrdU incorporation
After various experimental treatments cells were incubated with 10 μM 5-bromo-2′-deoxy-uridine (BrdU) for 2 hours and analyzed by flow cytometry as previously described13 to determine incorporation of BrdU in S phase cells.
DNA synthesis assay
Logarithmically growing cells were plated into 60-mm dishes at a density of 2.5 × 105 cells per dish and grown at 37°C for 30–40 h in medium containing 10 nCi/ml [14C]-thymidine to uniformly label DNA. Radioactive medium was replaced with fresh medium to chase [14C]-labeled precursors into DNA for at least 3 h. Cells were irradiated with 6 J/m2 of UV, or treated with 100 nM of BPDE, incubated for 45 min, 2 h or 6 h, and then labeled for 15 min with 25 μCi/ml [3H]-thymidine. Sham-treated cells were incubated for 2 h then labeled 15 min with [3H]-thymidine. For HU treatment, cells were incubated in medium with or without 2 mM of HU for 6 h, then incubated 15 min with [3H]-thymidine immediately before or following another 6 h incubation in fresh medium. The effect of genotoxin treatment on DNA synthesis was measured from [3H]/[14C] ratios as previously described.18
Immunoblotting and antibodies
After experimental treatments, cells were harvested by trypsinization, washed once in PBS and dissolved in lysis buffer [10 mM sodium phosphate buffer (pH 7.2), 1 mM EDTA, 1 mM EGTA, 150 mM NaCl and 1% NP-40, supplemented with 10 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 10 mM β-glycerophosphate, 10 mM sodium orthovanadate and 10 μg/ml leupeptin and aprotinin]. Protein concentrations were determined using the Bio-Rad DC protein assay (Bio-Rad laboratories). An equivalent amount of protein (100 μg) was mixed with 2× Laemmli sample buffer [125 mM Tris-HCl (pH 6.8), 4% sodium dodecyl sulfate (SDS), 20% glycerol containing 5% β-mercaptoethanol], boiled for 5 min, and separated by electrophoresis through 8% or 12% polyacrylamide gels (SDS-PAGE). Proteins were transferred to nitrocellulose membranes and probed with antibodies against p53 (Neomarkers), phospho-Ser15 p53 (Cell Signaling), p21Waf1 (Neomarkers), CHK1 (Santa Cruz Biotechnology), phospho-Ser345 CHK1 (Cell Signaling), CHK2 (Upstate Biotechnology Institute), phospho-Thr68 CHK2 (Cell Signaling), phospho-Ser139 H2AX (Upstate Biotechnology Institute) or β-actin (Santa Cruz Biotechnology) as previously described.18,19
Electroporation
Fibroblasts were electroporated with CHK1 siRNA (Dharmacon) or scrambled control (SC) siRNA using an Amaxa Nucleofector™ apparatus (Amaxa GmbH, Koeln, Germany) according to the manufacturer’s protocol. DNA synthesis and expression of CHK1 were measured 6 h after irradiation with 6 J/m2 of UV as described above.
Radioimmunoassay (RIA) of CPDs and [6-4] photoproducts
Briefly, antisera were raised against DNA dissolved in 10% acetone and irradiated with UVB light under conditions that have been shown to produce CPDs exclusively or against DNA irradiated with a high dose of UVC to produce also (6-4) pyrimidine-pyrimidone photoproducts ([6-4]PPs). For the RIA 2–5 μg of heat-denatured sample DNA was incubated with 5–10 pg of poly(dA):poly(dT) (labeled to >5 × 108 cpm/μg by nick translation with [32P]-dTTP) in a total volume of 1 ml of 10 mM Tris, pH 7.8, 150 mM NaCl, 1 mM EDTA and 0.15% gelatin (Sigma). Antiserum was added at a dilution that yields 30–60% binding to labeled ligand and after incubation overnight at 4°C the immune complex was precipitated with goat anti-rabbit immunoglobulin (Calbiochem) and carrier serum from non-immunized rabbits (UTMDACC, Science Park/Veterinary Division, Bastrop, TX). After centrifugation, the pellet was dissolved in NCS tissue solubilizer (Amersham), mixed with ScintiSafe (Fisher) containing 0.1% glacial acetic acid, and [32P] quantified by liquid scintillation spectrometry. Under these conditions, antibody binding to an unlabeled competitor inhibits antibody binding to the radiolabeled ligand. These details, as well as those concerning the specificities of the RIAs and standards used for quantification, are described elsewhere.20
Acknowledgments
This work was supported in part by PHS grants CA55065, CA81343, ES11391, ES11012 and P30-ES10126. We thank Cyrus Vaziri for critical reading of this manuscript prior to submission.
References
- 1.Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 2.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–7. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 3.Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA. 2002;99:12985–90. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA. 1999;96:424–8. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ford JM, Hanawalt PC. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem. 1997;272:28073–80. doi: 10.1074/jbc.272.44.28073. [DOI] [PubMed] [Google Scholar]
- 6.Itoh T, O’Shea C, Linn S. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: mutual regulatory interactions between p48(DDB2) and p53. Mol Cell Biol. 2003;23:7540–53. doi: 10.1128/MCB.23.21.7540-7553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang XW, Yeh H, Schaeffer L, Roy R, Moncollin V, Egly JM, et al. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat Genet. 1995;10:188–95. doi: 10.1038/ng0695-188. [DOI] [PubMed] [Google Scholar]
- 8.Spillare EA, Robles AI, Wang XW, Shen JC, Yu CE, Schellenberg GD, et al. p53-mediated apoptosis is attenuated in Werner syndrome cells. Genes Dev. 1999;13:1355–60. doi: 10.1101/gad.13.11.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S, Talis AL, Howley PM. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J Biol Chem. 1999;274:18785–92. doi: 10.1074/jbc.274.26.18785. [DOI] [PubMed] [Google Scholar]
- 10.zur Hausen H. Papillomavirus infections—a major cause of human cancers. Biochimica et Biophysica Acta. 1996;1288:55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 11.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–23. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 12.Ford JM, Baron EL, Hanawalt PC. Human fibroblasts expressing the human papilloma-virus E6 gene are deficient in global genomic nucleotide excision repair and sensitive to ultraviolet irradiation. Cancer Res. 1998;58:599–603. [PubMed] [Google Scholar]
- 13.Simpson DA, Livanos E, Heffernan TP, Kaufmann WK. Telomerase expression is sufficient for chromosomal integrity in cells lacking p53 dependent G1 checkpoint function. J Carcinog. 2005;4:18. doi: 10.1186/1477-3163-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu PK, Kraus E, Wu TA, Strong LC, Tainsky MA. Analysis of genomic instability in Li-Fraumeni fibroblasts with germline p53 mutations. Oncogene. 1996;12:2267–78. [PMC free article] [PubMed] [Google Scholar]
- 15.Ford JM, Hanawalt PC. Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc Natl Acad Sci USA. 1995;92:8876–80. doi: 10.1073/pnas.92.19.8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massimi P, Gammoh N, Thomas M, Banks L. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation. Oncogene. 2004;23:8033–9. doi: 10.1038/sj.onc.1207977. [DOI] [PubMed] [Google Scholar]
- 17.MacLeod MC, Lew L. A rapid, spectrophotometric assay for the integrity of diol epoxides. Carcinogenesis. 1988;9:2133–5. doi: 10.1093/carcin/9.11.2133. [DOI] [PubMed] [Google Scholar]
- 18.Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, et al. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol. 2002;22:8552–61. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heffernan TP, Unsal-Kacmaz K, Heinloth AN, Simpson DA, Paules RS, Sancar A, et al. Cdc7-Dbf4 and the human S checkpoint response to UVC. J Biol Chem. 2007;282:9458–68. doi: 10.1074/jbc.M611292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell D. Quantification of DNA photoproducts in mammalian cell DNA using radioimmunoassay. In: Henderson D, editor. Methods in Molecular Biology: DNA Repair Protocols. 2. Totowa: The Humana Press; 2005. pp. 239–49. [Google Scholar]
- 21.Cistulli CA, Kaufmann WK. p53-dependent signaling sustains DNA replication and enhances clonogenic survival in 254 nm ultraviolet-irradiated human fibroblasts. Cancer Research. 1998;58:1993–2002. [PubMed] [Google Scholar]
- 22.Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–94. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu X, Lane DP. Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell. 1993;75:765–78. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 24.Boyer JC, Kaufmann WK, Cordeiro-Stone M. Role of postreplication repair in transformation of human fibroblasts to anchorage independence. Cancer Res. 1991;51:2960–4. [PubMed] [Google Scholar]
- 25.Kaufmann WK, Cleaver JE. Mechanisms of inhibition of DNA replication by ultraviolet light in normal human and xeroderma pigmentosum fibroblasts. J Mol Biol. 1981;149:171–87. doi: 10.1016/0022-2836(81)90297-7. [DOI] [PubMed] [Google Scholar]
- 26.Guo N, Faller DV, Vaziri C. Carcinogen-induced S-phase arrest is Chk1 mediated and caffeine sensitive. Cell Growth Differ. 2002;13:77–86. [PubMed] [Google Scholar]
- 27.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 28.Ward IM, Minn K, Chen J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem. 2004;279:9677–80. doi: 10.1074/jbc.C300554200. [DOI] [PubMed] [Google Scholar]
- 29.Brown AL, Lee CH, Schwarz JK, Mitiku N, Piwnica-Worms H, Chung JH. A human Cds1-related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc Natl Acad Sci USA. 1999;96:3745–50. doi: 10.1073/pnas.96.7.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lloyd DR, Hanawalt PC. p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res. 2000;60:517–21. [PubMed] [Google Scholar]
- 31.Ferguson BE, Oh DH. Proficient global nucleotide excision repair in human keratinocytes but not in fibroblasts deficient in p53. Cancer Res. 2005;65:8723–9. doi: 10.1158/0008-5472.CAN-05-1457. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–74. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leung-Pineda V, Ryan CE, Piwnica-Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol. 2006;26:7529–38. doi: 10.1128/MCB.00447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Meneghini R, Cordeiro-Stone M, Schumacher RI. Size and frequency of gaps in newly synthesized DNA of xeroderma pigmentosum human cells irradiated with ultraviolet light. Biophys J. 1981;33:81–92. doi: 10.1016/S0006-3495(81)84873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cordeiro-Stone M, Makhov AM, Zaritskaya LS, Griffith JD. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J Mol Biol. 1999;289:1207–18. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 37.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–55. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 38.Chini CC, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem. 2003;278:30057–62. doi: 10.1074/jbc.M301136200. [DOI] [PubMed] [Google Scholar]
- 39.Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, et al. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007;27:3131–42. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 41.Bi X, Slater DM, Ohmori H, Vaziri C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J Biol Chem. 2005;280:22343–55. doi: 10.1074/jbc.M501562200. [DOI] [PubMed] [Google Scholar]
- 42.Bomgarden RD, Lupardus PJ, Soni DV, Yee MC, Ford JM, Cimprich KA. Opposing effects of the UV lesion repair protein XPA and UV bypass polymerase eta on ATR checkpoint signaling. EMBO J. 2006;25:2605–14. doi: 10.1038/sj.emboj.7601123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Zhang H, McManus TP, McCormick JJ, Lawrence CW, Maher VM. hREV3 is essential for error-prone translesion synthesis past UV or benzo[a]pyrene diol epoxide-induced DNA lesions in human fibroblasts. Mutat Res. 2002;510:71–80. doi: 10.1016/s0027-5107(02)00253-1. [DOI] [PubMed] [Google Scholar]
- 44.Gibbs PE, Wang XD, Li Z, McManus TP, McGregor WG, Lawrence CW, et al. The function of the human homolog of Saccharomyces cerevisiae REV1 is required for muta-genesis induced by UV light. Proc Natl Acad Sci USA. 2000;97:4186–91. doi: 10.1073/pnas.97.8.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibbs PE, McDonald J, Woodgate R, Lawrence CW. The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6–4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005;169:575–82. doi: 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol. 1976;101:417–25. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- 47.Torres-Ramos CA, Prakash S, Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol. 2002;22:2419–26. doi: 10.1128/MCB.22.7.2419-2426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gunnell AS, Tran TN, Torrang A, Dickman PW, Sparen P, Palmgren J, et al. Synergy between cigarette smoking and human papillomavirus type 16 in cervical cancer in situ development. Cancer Epidemiol Biomarkers Prev. 2006;15:2141–7. doi: 10.1158/1055-9965.EPI-06-0399. [DOI] [PubMed] [Google Scholar]