

Summary
Traditionally, lipid rafts have been defined by their insolubility in ice-cold Triton X-100 and low-buoyant density. These low-density membrane microdomains have been referred to as detergent-resistant membranes, Triton-insoluble membranes, and Triton-insoluble floating fraction. They are enriched in cholesterol, often sphingomyelin and various gangliosides (GM1, GM2, and GM3). The ability of the B-subunit of cholera toxin to bind GM1 has been exploited to visualize membrane rafts by confocal microscopy in patching and capping experiments. Biochemically, membrane rafts are isolated by solubolization in ice-cold Triton X-100 and separation of the low-buoyant density fractions from soluble material on sucrose density gradients. We describe the isolation of Jurkat cell-specific membrane rafts using 2% Triton X-100. This procedure yielded a consistent raft product that was enriched in cholesterol, gangliosides sphingo-myelin and membrane raft protein markers including lck and lat 1. Moreover, rafts were visualized using Alexa Fluor 647 cholera toxin capped with anti-cholera toxin antibody. Co-localization of the C subunit of cytolethal distending toxin to rafts was determined using patching techniques.
Keywords: Membrane microdomains, detergent resistance, Jurkat cells, Triton X-100
1. Introduction
The physical relationship between hydrophilic aqueous medium and hydro-phobic fat-like molecules has intrigued scientists since the 1770s, when Benjamin Franklin observed that any oily substance clearly covered half the surface area when compared with an equal volume of aqueous solution, implying a bilayer structure. As early as 1925, Gorter and Grendel (1) proposed the now-classic deduction that membrane lipids are arranged in a bilayer configuration in which parallel sheets of phospholipids have polar or charged headgroups oriented toward the aqueous environment and acyl chains interacting within the hydrophobic membrane core. In 1972, Singer and Nicolson provided a model that took into consideration the dynamic nature of lipid–protein interactions, providing a matrix in which proteins have a degree of motion that, in turn, can have a dramatic impact on activity. Thus, the fluid mosaic model (2) became the framework and benchmark for our current understanding of membrane bilayers and their physiological function. The assumed homogeneous nature of membrane bilayers proposed in this model was called into question in the 1970s, when it was observed that membranes contain a unique composition of lipid and protein components that are specific to cell type and subcellular localization. A heterogeneous distribution of lipids and proteins is even observed within spatially separated regions of the same membrane of Golgi (3) or apical and basolateral plasma membranes of polarized cells (4). Within the past decade, a unifying theme describing the organization of lipid and membrane proteins has focused on localized regions within the membrane known collectively as membrane microdomains. The last 5 yr have seen an emergence of interest in a specific type of microdomain, known colloquially as a membrane raft. More precisely, these regions are globally defined as cholesterol-rich domains in the liquid-ordered phase. These microdomains are proposed to be involved in a wide variety of cellular processes including, protein sorting (5), signal transduction (6), calcium homeostasis (7), transcytosis (8), potocytosis (9), alternative routes of endocytosis (10), internalization of toxins, bacteria, and viruses (11–13), HIV-1 assembly and release (14), and cholesterol transport (15,16).
The association of cytolethal distending toxin (cdt) with Jurkat cells will be used to illustrate the methods used to analyze membrane raft functionality using biochemical and microscopic techniques.
2. Materials
2.1. Isolation of Triton X-100 Resistant Membrane Microdomains
0.5 to 2% Triton X-100 in MOPS buffer.
0.5 to 2% octyglucopyranoside (wt/v) in MOPS buffer.
MOPS buffer: 10 mM MOPS, pH 7.2, 60 mM KCl, 30 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT; see Note 1), 5 μM aprotinin, and 1 μM leupeptin.
0.5, 0.6, 0.65, 0.7, 0.75, 0.80, and 2.4 M sucrose in MOPS buffer.
Beckman Optima LE 80K Ultracentrifuge, including SW-41 rotor, SW-41 titanium buckets, and ultra-clear tubes.
Wheaton glass-homogenizer; 7.5-mL or 15-mL volume.
T-cell leukemia cell line Jurkat (E6-1; T1B152, lot no. 2113016, ATCC) or other cells of interest (cell count approx 5 × 108).
2.2. Visualization of Membrane Rafts in Jurkat Cells and Localization of Proteins to Raft Microdomains
Hank's balanced salt solution (HBSS): Gibco 10× HBSS, dilute with H2O to make 1× HBSS.
Cholera toxin B subunit-Alexa Fluor 647. Reconstituted with 100 μL of phosphate-buffered saline (PBS). Stock concentration of 1 mg/mL. Use 1 μL of stock per 1 mL of cell suspension/appropriate tube.
Anticholera toxin, subunit B, Vibrio cholera, (goat). Reconstituted with 100 μL of dH2O, 10 μL of reconstituted antitoxin + 240 μL of HBSS = 1:25 dilution. Use 100 μL of the antitoxin dilution per appropriate tube.
CGM: RPMI-1640 Glutamax, 10% fetal bovine serum, 2% Pen/Strep, 1 mM sodium pyruvate.
ABChis (see Note 2): B:021004, 163 μg/mL: 37 μL of stock + 263 μL of CGM = 20 μg/mL.
Buffer: PBS/1% bovine serum album (ice-cold).
Goat immunoglobulin (Ig; Southern Biotech) diluted to 0.5 mg/mL in PBS (stock). Make 1:50 dilution of the stock in PBS.
Anti-ABChis purified Ig monoclonal antibody: anti-ABChis, 17A1.15, use 10 μL of a 100 μg/mL stock to 1 μg/mL.
Goat anti-mouse Ig biotin. Dilution: 25 μL of stock solution at 500 μg/mL + 225 μL of buffer = 50 μg/mL. Use 20 μL of 50 μg/mL dilution/appropriate tube = 1 μg/appropriate tube.
Alexa Fluor 488-streptavidin reconstitued with 1 mL of PBS, 030904 TLM. Dilute 4 μL of stock + 996 μL of buffer to equal 1:250 dilution. (Centrifuge for 5 min at 10,000g before use.)
Radiance 2000 laser confocal microscope with argon, green He/Ne, Red diode and Blue diode lasers.
3. Methods
The Methods described below outline the biochemical isolation of rafts and the visualization and localization of proteins to membrane rafts.
3.1. Detergents Used in the Isolation of Membrane Rafts
The isolation of membrane microdomains or rafts relies on the relative insolubility of the less-fluid cholesterol-rich liquid-ordered membrane regions in Triton X-100. Recently, the repertoire of detergents used to isolate low-buoyant density membrane microdomains and signaling complexes has been expanded to include Brij 98, NP-40, CHAPS, and Lubrol. These detergents differ in their critical micelle concentration (CMC) and thus are postulated to solubi-lize mixed “raft-like domains” and tetraspanin protein complexes (17). Lastly, although not reviewed in this chapter, detergent-independent modes of raft and/ or caveolae isolation have been developed. These include sodium carbonate lysis, sonication and sucrose gradient centrifugation (see Note 3 [18]), and the isolation of plasma membrane-specific rafts using Percoll gradient-purified membranes, which are sonicated and rafts isolated by floatation in continuous Opti-prep gradients (19).
3.2. Isolation of Signaling Complexes in Detergent-Resistant Membranes
The isolation of detergent-resistant membranes (DRMs) from bacterial toxin-treated cells, HIV-infected cells, and cells stimulated with a variety of ligands have provided valuable information on the mode of action of these agonists. Stimulation of cells with growth factors and the isolation rafts has allowed investigators to determine in which compartment a specific signaling event has occurred. Using this approach, tyrosine kinases appear to activate mitogen-activated protein kinase (MAPK) and other signal transduction pathways from within rafts; phosphatidylinositol turnover occurs in lipid rafts in response to growth factor stimulation; and cholera toxin's mode of action requires the association of the B unit with gangliosides enriched in membrane rafts. Because membrane rafts are stabilized through the interaction of cholesterol with other lipid components, as a complementary approach to studying raft function is the depletion of cell membrane cholesterol, membrane raft integrity can be disrupted with the addition of β-methylcyclodextrin (Sigma), an agent that sequesters and removes membrane cholesterol (20). Using this cholesterol depletion approach, the role of lipid rafts in specific signaling events can be studied directly in intact cells (for review, see ref. 28). Although there are countless examples of the role of membrane rafts in a variety of biological processes, the basic techniques used to isolate rafts are largely similar to those described here.
3.3. Isolation of Jurkat Cell Membrane Rafts on Toxin Association
The T-cell leukemia cell line Jurkat (E6-1) was maintained in RPMI-1640 supplemented as described (21). Cells were harvested in mid-log growth phase, and for membrane raft preparations, the cells were grown at 2.0 × 106 cells/mL in T-75 flasks. The cells were exposed to medium or toxin for 2 h, isolated, and washed in MOPS buffer (22). To distinguish between Triton X-100-resistant membranes and simply a partial detergent-dependent nonraft-specific solubilzation, control cells were homogenized in 2% octyl glucopyranoside: parallel to the Triton X-100-treated samples. Membrane rafts were isolated from the Jurkat cells as described in the following steps.
Resuspend isolated cells in a final volume of 1.1 mL of MOPS buffer; if the cells appear to aggregate, resulting in a nonhomogenous suspension, add an additional 1 mL of MOPS buffer (see Note 4).
Transfer 1 mL of cells to a Wheaton glass homogenizer (on ice), add 0.77 mL of ice-cold 2% (v/v) Triton X-100 in MOPS buffer (see Note 5). For control cells, add 0.77 mL of 2% octyl glucopyranoside.
Homogenize five strokes on ice, taking care to keep bubbles and foaming to a minimum.
Let sit on ice for 15 min.
Add 1.24 mL of 2.4 M sucrose in MOPS buffer (see Note 6), vortexing immediately.
Transfer samples to clear SW-41 centrifuge tubes.
Sequentially layer 1 mL of each of the following: 0.8, 0.7, 0.65, 0.6, and 0.5 M sucrose solutions onto sample to create a sucrose step gradient (see Note 7).
Top samples off with requisite volume of MOPS buffer so that the tubes are filled approx 0.10 cm from the top.
Place samples in SW-41 buckets.
Spin at 400,000g for 20 h at 4°C.
3.4. Fractionation of Sucrose Density Gradient
Detergent-insoluble membrane fractions are isolated as low-buoyant density fractions, as shown in Fig. 1. Jurkat cells both with or without toxin exhibit a characteristic low-buoyant density band. These bands are collected either directly with a Pasteur pipet or the sucrose density gradient fractionated as described (23). The low-buoyant density bands, DRMs, are analyzed for cholesterol (24), phospholipid (25), and total protein (Bio-Rad). To further confirm that these membranes are rafts, the level of GM1 is analyzed by immunoblotting using anticholera toxin antibody. In addition, or as an alternative to analysis of the gangliosides, total lipid extracts may be prepared as described (26) and analyzed by thin-layer chromatography (TLC) for total sph-ingolipid content (27). The protein markers used to confirm that a DRM is a raft are described immediately below in Subheading 3.5.
Fig. 1.
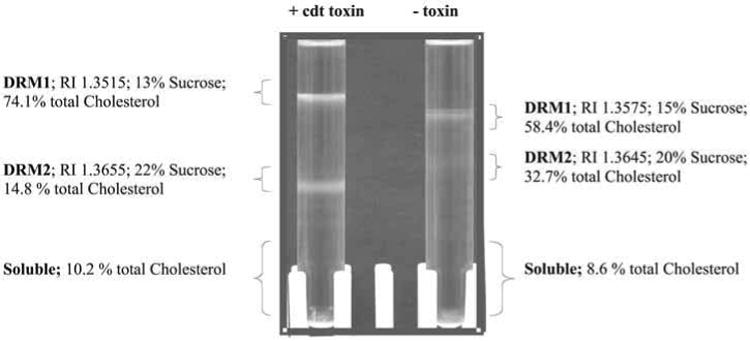
Cdt toxin treatment alters the buoyant density of Jurkat cell membrane rafts. In order to determine whether the toxin, or any of its subunits, localize to lipid microdomains, detergent-resistant membranes (DRMs) from both untreated Jurkat cells and cells exposed to CdtABC were isolated. After a 2-h incubation, Jurkat cells were disrupted, homogenized in ice-cold Triton X-100, and separated on a sucrose gradient. Two distinct low-buoyant density bands, designated DRM1 and DRM2, were obtained and the position the sucrose density gradient determined as a measure of refractive index as indicated. The cholesterol content composition of these bands was analyzed (24) and is presented as percentage of the total membrane cholesterol.
3.5. Membrane Raft Protein Markers
In addition to higher levels of cholesterol, GM1, phospholipids with saturated fatty acyl side chains, and sphingomyelin, lipid rafts can be characterized based on the presence of specific lipid raft marker proteins. Although these proteins vary from cell to cell, often flotilin-1 and 2, LAT, Thy, and as a rule of thumb, most GPI-anchored proteins and src family kinases are membrane raft-associated. A comprehensive list of such proteins can be found in refs. 28, 28a, and 29. In addition, caveolin 1 and 2 (see Note 8) are often are associated with high cholesterol caveolae or membrane caves, a subset of the membrane microdomain family. Conversely, the transferrin receptor and geranylated proteins are routinely nonraft markers. As shown in Fig. 2, the DRMs isolated from Jurkat cells (with or without cdt toxin) were enriched in the raft marker, Lck and deficient in the nonraft marker, CD71. Collectively in these studies, both DRM1 and 2 were identified as membrane rafts based on position in the sucrose density gradient, the increased levels of GM1, high percentage of cholesterol, increased sphingomyelin, and the presence of raft-specific protein markers (shown is Lck).
Fig. 2.
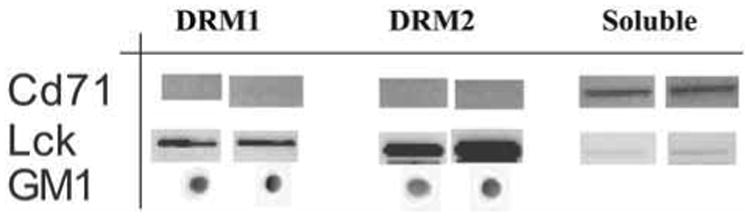
DRM1 and DRM2 are enriched in membrane raft markers: GM1 and Lck. As shown, both DRM1 and DRM2 were enriched in GM1 (dot blot) and in total cholesterol. Moreover, the raft-associated protein Lck was enriched in these fractions. In contrast, the transferrin receptor (CD71), a nonraft-associated protein, was found in the soluble fraction.
3.6. Visualization and Localization of Proteins to Membrane Rafts
3.6.1. Membrane Raft Capping and Patching Techniques
The visualization of membrane rafts in cells is limited by the resolution of the techniques used. Rafts have been visualized in model membranes, giant unilammellar vesicles (GUVs) composed of distinct raftophilic lipids using fluorescent membrane probes designed to detect lipid ordering (30). Visualization of rafts in intact cells is somewhat more difficult for a number of reasons. GPI-anchored proteins and GM1 markers appear uniformly distributed since their concentration may be only three- to fourfold higher in rafts than in the remaining membrane or membrane rafts may be transient complexes formed in response to agonists or antigens (31). Thus, to overcome these problems, membrane rafts are visualized buy exploiting the fact that GPI-anchored proteins and GM1 molecules will cluster in response to antibodies. In the method described below, GM1 is crosslinked by the cholera toxin B subunit (this process is referred to as capping), and the capped cholera toxin is subsequently treated with anti-cholera toxin antibody resulting in the clustering of GM1 in a process referred to as patching (32). Moreover, we describe the co-localization of the C-subunit of cdt toxin to these membrane clusters using biotin– streptavadin labeling techniques.
Harvest Jurkat cells, wash one time with HBSS, centrifuge at 800g for 8 min, discard supernatant, resuspend cell pellet in HBSS, and count.
Prepare a tube with 6 mL of cells in HBSS at 2 × 106 cells/mL.
Add 6 μL of Alexa Fluor 647 cholera toxin (stock 1 mg/mL) to the 6 mL of cells (final 1 μg/mL).
Incubate the cells on ice for 30 min.
Add 1 mL of the cholera toxin-treated cell suspension to each of five tubes and label them 1 through 5.
Wash cells with 2 mL of HBSS, centrifuge at 800g for 8 min, and discard supernatant.
Repeat step 6.
Add 100 μL of HBSS to tube 1 (no capping control).
Add 100 μL of 1:25 dilution of anticholera toxin to all remaining tubes (positive capping).
Incubate for 30 min on ice; incubate at 37°C for 40 min.
Wash cells with 2 mL of HBSS, centrifuge at 800g for 8 min, and discard supernatant.
Repeat step 11.
Resupend each tube with 500 μL of CGM.
Add 500 μL of CGM to the “cells only” tubes.
Add 400 μL of CGM + 100 μL of ABChis at 20 μg/mL to the “cells + ABChis” tubes. (ABChis, B:021004, stock at 163 μg/mL.)
Incubate all tubes at 37°C for 2 h.
Wash cells with 2 mL of buffer, centrifuge at 1000g for 8 min, and discard supernatant.
Repeat step 17.
Add 10 μL of goat Ig in buffer at 10 μg/mL to tubes 1 through 5.
Incubate on ice for 10 min.
Add 10 μL of either buffer to tubes 1 and 2, and 10 μL of Anti-ABChis purified Ig 17A1.15 at 100 μg/mL to tubes 3 through 5 (final, 2 μg/tube).
Incubate on ice for 30 min.
Wash all tubes with 2 mL of buffer, centrifuge at 1000g, for 8 min, and discard supernatant.
Repeat step 23.
Add 20 μL of 50 (g/mL dilution of goat anti-mouse Ig biotin to all tubes (1 μg/ tube).
Incubate on ice for 30 min.
Wash all tubes with 2 mL of buffer, centrifuge at 1000g for 8 min, and discard supernatant.
Repeat step 27.
Add 50 μL of a 1:250 dilution of Alexa Fluor 488-SA to all tubes.
Incubate on ice for 30 min.
Wash cells with 2 mL of buffer, centrifuge at 800g for 8 min, and discard supernatant.
Repeat step 31.
Resuspend cells in 500 μL of 2% formaldehyde.
As shown in Fig. 3, the CdtC subunit localizes to membrane lipid rafts. Utilizing confocal fluorescence microscopy we demonstrate co-localization of the C subunit with the cholera toxin B subunit (CtB) bound to GM1. To control for nonspecific staining, isotype matched control IgG was used instead of the anti-Cdt monoclonal antibody. As shown below, virtually all fluorescence associated with either CdtC co-localizes with GM1 (i.e., CtB fluorescence).
Fig. 3.
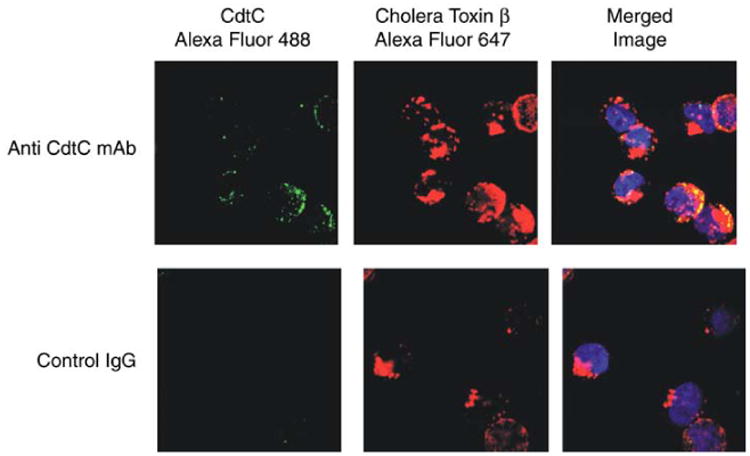
Visualization and co-localization of cdt to membrane rafts. To demonstrate that the Cdt subunits localize to membrane lipid rafts, confocal fluorescence microscopy to demonstrate co-localization of the C subunit with the cholera toxin B subunit (CtB) bound to GM1 was used. Jurkat cells were first exposed to CtB-Alexa Fluor 647 for 20 min; the cells were then treated with anti-CtB antisera to induce patch formation. Cells were then exposed to CdtABC for 2 h, washed, and sequentially stained with monoclonal antibody (MAb) to CdtC, goat anti-mouse Ig conjugated to biotin, and streptavidin-Alexa Fluor 488. To control for nonspecific staining, isotype-matched control IgG was used instead of the anti-Cdt MAb. As shown, virtually all fluorescence associated with either CdtC colocalized with GM1 (i.e., CtB fluorescence).
4. Notes
Buffers containing DTT should be prepared fresh daily. Routinely, MOPS buffer is prepared in the absence of DTT. DTT is added to the desired volume prior to the start of each experiment.
The ABChis is the active holotoxin of the cytolethal distending toxin. In individualized experiments, this may be a ligand for a receptor or any bacterial toxin of interest.
The sodium carbonate lysis method relies on a pH of 11.0 and is often is used to remove excess peripheral proteins from the membranes. This method is described in detail in Chapter 10.
It is important that the cells be a homogenous suspension. Thus, the volume of buffer used in the resuspension may be increased; however, with this increase there must be an increase in the amount of 2% ice-cold Triton X-100 added. For example, for 1 mL of cell suspension, we add 0.77 mL of Triton; for 2 mL of a cell suspension, 1.44 mL of Triton X-100 is required, etc. (see Note 6 for sucrose amounts).
Different concentrations of Triton X-100 have been used by a variety of investigators to isolate DRMs; the relative solubility of the components of interest determines the protein composition of the membrane raft. The final concentration of Triton X-100 in the sample is expected to be less than 1% and limited solubility is seen with decreasing amounts of Triton X-100 (see ref. 21).
If the volume of cells was increased (as per Note 4) to maintain an appropriate sucrose concentration (i.e., >0.8 M), then the amount of 2.4 M sucrose must be increased. For example, to a preparation containing 1 mL of cells and 0.77 mL of Triton X-100, add 1.25 mL of 2.4 M sucrose; to a preparation containing 2 mL of cells and 1.44 mL of Triton X-100, add 2.5 mL of 2.4 M sucrose.
As an alternative to sucrose step gradients, some investigators prefer a continuous gradient from 5 to 30% sucrose.
A number of cells, Jurkats included, do not contain caveolin. In addition, membrane caves, i.e., membrane microdomains enriched in caveolae, are isolated using a nondetergent-based Opti-prep gradient (19) or sodium carbonate lysis procedures (18). The isolation of caveolin-enriched microdomains is described in Chapter 10.
Acknowledgments
The author would like to thank Cheryl Gretzula and Lisa Pankoski for their critical reading of the manuscript and Terry McKay for her expert technical assistance. Supported by NIH: DE06014 and EY10420.
References
- 1.Gorter E, Grendel F. On biomolecular layers of lipids on the chromatocytes of blood. J Exp Med. 1925;41:439–443. doi: 10.1084/jem.41.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 3.Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Boulan E, Nelson WJ. Morphogenesis of the polarized epithelial cell phenotype. Science. 1989;245:718–725. doi: 10.1126/science.2672330. [DOI] [PubMed] [Google Scholar]
- 5.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 6.Zajchowski LD, Robbins M. Lipid rafts and little caves: compartmentalized signalling in membrane microdomains. Eur J Biochem. 2002;269:737–752. doi: 10.1046/j.0014-2956.2001.02715.x. [DOI] [PubMed] [Google Scholar]
- 7.Isshiki M, Anderson RGW. Calcium signal transduction from cav-eolae. Cell Calcium. 1999;26:201–208. doi: 10.1054/ceca.1999.0073. [DOI] [PubMed] [Google Scholar]
- 8.Simionescu N. Cellular aspects of transcapillary exchange. Physiol Rev. 1983;63:1536–1560. doi: 10.1152/physrev.1983.63.4.1536. [DOI] [PubMed] [Google Scholar]
- 9.Anderson RGW, Kamen BA, Rothberg KG, Lacey SW. Potocytosis: sequestration and transport of small molecules by caveolae. Science. 1992;255:410–411. doi: 10.1126/science.1310359. [DOI] [PubMed] [Google Scholar]
- 10.Smart EJ, Graf GA, McNiven MA, et al. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. 1999;19:7289–7304. doi: 10.1128/mcb.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fivaz M, Abrami L, van der Goot FG. Landing on lipid rafts. Trends Cell Biol. 1999;9:212–213. doi: 10.1016/s0962-8924(99)01567-6. [DOI] [PubMed] [Google Scholar]
- 13.Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–788. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- 14.Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci USA. 2001;98:13,925–13,930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oram JF, Yokoyama S. Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J Lipid Res. 1996;37:2473–2491. [PubMed] [Google Scholar]
- 16.Smart EJ, Ying YS, Donzell WC, Anderson RGW. A role for caveolin in transport of cholesterol from endoplasmic reticulum to plasma membrane membrane. J Biol Chem. 1996;271:29,427–29,435. doi: 10.1074/jbc.271.46.29427. [DOI] [PubMed] [Google Scholar]
- 17.Claas C, Stipp CS, Hemler ME. Evaluation of prototype trans-membrane 4 superfamily protein complexes and their relation to lipid rafts. J Biol Chem. 2001;276:7974–7984. doi: 10.1074/jbc.M008650200. [DOI] [PubMed] [Google Scholar]
- 18.Song KS, Li S, Okamoto T, Quiliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 19.Smart EJ, Ying YS, Mineo C, Anderson RGW. A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc Natl Acad Sci USA. 1995;92:10,104–10,108. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2274. [PubMed] [Google Scholar]
- 21.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Shenker BJ, Besack D, Mc Kay T, Pankoski L, Zekavat A, Demuth D. actinobacillus actinomycetemcomitans cytolethal Distending Toxin (Cdt): Evidence evidence that the holotoxin is composed of three subunits: CdtA, CdtB, and CdtC. J Immunol. 2004;172:410–417. doi: 10.4049/jimmunol.172.1.410. [DOI] [PubMed] [Google Scholar]
- 23.Boesze-Battaglia K, Besack D, Pankoski L, Mc Kay T, Jordan-Sciutto K, Shenker BJ. Association of actinobacillus actinomycetemcomitans cytolethal distending toxin (cdt) with membrane rafts, submitted 2004 [Google Scholar]
- 24.Boesze-Battaglia K, Hennessey T, Albert AD. Cholesterol heterogeneity in bovine retinal rod outer segment disk membranes. J Biol Chem. 1989;264:8151–8155. [PMC free article] [PubMed] [Google Scholar]
- 25.Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem. 1974;20:470–475. [PubMed] [Google Scholar]
- 26.Bartlett GR. Phosphorous assay in column chromatography. J Biol Chem. 1959;234:466–473. [PubMed] [Google Scholar]
- 27.Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipides from animal tissue. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 28.Boesze-Battaglia K, Dispoto J, Kahoe MA. Association of a pho-toreceptor-specific tetraspanin protein, ROM-1, with triton X-100-resistant membrane rafts from rod outer segment disk membranes. J Biol Chem. 2002;277:41,843–41,849. doi: 10.1074/jbc.M207111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.van der Goot FG, Harder T. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin Immunol. 2001;13:89–97. doi: 10.1006/smim.2000.0300. [DOI] [PubMed] [Google Scholar]
- 30.Dietrich C, Volovyk ZN, Levi M, Thompson NL, Jacobson K. Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc Natl Acad Sci USA. 2001;98:10,642–10,647. doi: 10.1073/pnas.191168698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem. 2000;275:17,221–17,224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell JS, Kanca O, Mc Intyre BW. Lipid microdomain clustering induces a resdistribution of antigen recognition and adhesion molecules on human T lymphocytes. J Immunol. 2002;168:2738–2744. doi: 10.4049/jimmunol.168.6.2737. [DOI] [PubMed] [Google Scholar]