

Missense mutations in membrane proteins cause dominant diseases by unknown mechanisms. Pathogenic mutations in adenine nucleotide translocase 1 affect protein folding and the assembly of multiple protein complexes. The misfolding of one single protein is therefore sufficient to induce proteostatic stress on the membrane.
Abstract
Approximately one-third of proteins in the cell reside in the membrane. Mutations in membrane proteins can induce conformational changes and expose nonnative polar domains/residues to the lipid environment. The molecular effect of the resulting membrane stress is poorly defined. Adenine nucleotide translocase 1 (Ant1) is a mitochondrial inner membrane protein involved in ATP/ADP exchange. Missense mutations in the Ant1 isoform cause autosomal dominant progressive external ophthalmoplegia (adPEO), cardiomyopathy, and myopathy. The mechanism of the Ant1-induced pathologies is highly debated. Here we show that equivalent mutations in the yeast Aac2 protein cause protein misfolding. Misfolded Aac2 drastically affects the assembly and stability of multiple protein complexes in the membrane, which ultimately inhibits cell growth. Despite causing similar proteostatic damages, the adPEO- but not the cardiomyopathy/myopathy-type Aac2 proteins form large aggregates. The data suggest that the Ant1-induced diseases belong to protein misfolding disorders. Protein homeostasis is subtly maintained on the mitochondrial inner membrane and can be derailed by the misfolding of one single protein with or without aggregate formation. This finding could have broad implications for understanding other dominant diseases (e.g., retinitis pigmentosa) caused by missense mutations in membrane proteins.
INTRODUCTION
Adenine nucleotide translocase (Ant) belongs to a large family of proteins known as mitochondrial carriers, which transport nucleotides, cofactors, signaling molecules, and metabolic intermediates across the mitochondrial inner membrane (Palmieri, 2004, 2014). Ant primarily catalyzes the export of ATP out of mitochondria by exchanging with cytosolic ADP (Klingenberg, 2008). Under low–membrane potential conditions, Ant reverses its transport directionality, allowing the import of cytosolic ATP into mitochondria against matrix ADP. In addition to ATP/ADP exchange, Ant also mediates proton leak across the membrane (Brand et al., 2005). This activity mildly uncouples the membrane, which prevents hyperpolarization and excessive production of superoxide.
Ant is one of the most conserved proteins through evolution and is present in multiple isoforms in different species. In humans, Ant1 is predominantly expressed in skeletal muscle and heart (Stepien et al., 1992). Yeast cells express three isoforms of Ant, with Aac2 being the major ADP/ATP carrier under respiring conditions (Lawson and Douglas, 1988; Lawson et al., 1990). Ant contains three repeats of a sequence module of ∼100 amino acids, with each predicted to form two transmembrane α-helices (Saraste and Walker, 1982; Walker and Runswick, 1993). The compact organization was confirmed by atomic structures established for the bovine Ant1 and yeast Aac2 and Aac3 isoforms in the presence of the competitive inhibitor carboxyatractyloside (Pebay-Peyroula et al., 2003; Ruprecht et al., 2014). These structures revealed that the six transmembrane helices in an Ant monomer form a central channel that is sufficiently large to accommodate the bulky ADP and ATP. This led to the alternating-access transport model, predicting that an Ant monomer alternates between the cytosolic and matrix conformations, which allows the alternate access and then translocation of cytosolic ADP and matrix ATP during the nucleotide exchange cycle (Kunji and Crichton, 2010).
Several missense mutations in Ant1 (A90D, L98P, D104G, A114P, and V289M) cause autosomal dominant progressive external ophthalmoplegia (adPEO), characterized by late or adult-onset muscle weakness and exercise intolerance (Kaukonen et al., 2000; Napoli et al., 2001; Komaki et al., 2002; Siciliano et al., 2003; Deschauer et al., 2005). Another missense mutation, A123D, leads to hypertrophic cardiomyopathy and myopathy in homozygous patients (Palmieri et al., 2005). These diseases are commonly manifested by multiple mitochondrial DNA (mtDNA) deletions. Several models have been proposed to explain the Ant1-induced pathogenesis. Fontanesi et al. (2004) found that the adPEO-type mutants preferentially import ATP over ADP in in vitro reconstituted proteoliposomes. This novel property was proposed to promote a futile ATP/ATP homoexchange and elevated mitochondrial ATP and dATP levels, which consequently affect the accuracy of mtDNA replication and stability. In mouse C2C12 myotube cells, Kawamata et al. (2011) found that mitochondria of myotubes expressing the ANT1A114P allele are switched to the reversal ATPcytosol4−/ADPmatrix3− exchange mode at a higher membrane potential. This may also lead to increased ATP level and possibly, nucleotide imbalance in the mitochondrial matrix.
More recently, Ravaud et al. (2012) showed that the missense pathogenic variants of Ant1 have altered nucleotide-binding affinity and translocation kinetics when expressed on the plasma membrane of Escherichia coli. This observation raises the possibility that loss of nucleotide transport function may be responsible for the pathogenesis, possibly by a haploinsufficient mechanism. However, this model is inconsistent with the findings that both humans and mice heterozygous for a complete loss-of-function ant1 allele are healthy (Graham et al., 1997; Palmieri et al., 2005; Echaniz-Laguna et al., 2012) and that homozygous ant1-null alleles do not cause ophthalmologia or ocular motility defect (Echaniz-Laguna et al., 2012; Strauss et al., 2013). The latter findings tend to suggest that the pathogenesis induced by the missense mutations involves a novel mechanism that is different from the loss-of-transport-function mode.
Our previous studies showed that the adPEO-type Ant mutations in a yeast model severely affect cell viability in a dominant manner and that mtDNA instability in the mutant cells arises independent of adenine nucleotide transport (Chen, 2002; Liu and Chen, 2013; Wang et al., 2008a, b). We proposed that the mutant proteins may interfere with general mitochondrial biogenesis in addition to their effect on oxidative phosphorylation and mtDNA stability and that mtDNA instability likely arises as a secondary effect of mitochondrial damage. By using the filamentous fungus Podospora anserine as a model system, El-Khoury and Sainsard-Chanet (2009) also showed that expression of the adPEO-type Ant variants leads to severe mitochondrial damage, as manifested by low mitochondrial membrane potential. The mechanism by which the mutant Ant causes mitochondrial damage is unknown. In the present study, we provide evidence suggesting that pathogenic missense mutations cause the misfolding of Ant, which subsequently induces proteostatic stress on the membrane and loss of cell viability.
RESULTS
Mutant Aac2 proteins affect protein quality control on the mitochondrial inner membrane
We previously showed that the A106D, M114P, A128P, and A137D alleles of AAC2, equivalent to the pathogenic A90D, L98P, A114P, and A123D mutations in human ANT1, are synthetically lethal, with the disruption of YME1 even in cells expressing an endogenous wild-type copy of AAC2 (Wang et al., 2008a). YME1 encodes the i-AAA protease involved in the quality control of proteins on the mitochondrial inner membrane (Thorsness et al., 1993; Leonhard et al., 1999). This observation suggested that expression of the mutant Aac2 may dominantly affect protein homeostasis on the membrane, which cannot be tolerated in cells lacking Yme1. In support of this idea, we found that cells expressing a mutant allele of AAC2 are hypersensitive to diverse conditions that affect proteostasis and the biochemical properties and functionality of the membrane. First, meiotic spores cosegregating AAC2A128P with the disruption of OXA1 failed to form viable colonies on yeast extract/peptone/dextrose (YPD) medium (Figure 1A). OXA1 encodes the mitochondrial inner membrane insertase that mediates the insertion of mtDNA-encoded proteins from the matrix side (Bonnefoy et al., 1994; Herrmann et al., 1997; Stuart, 2002). Loss of Oxa1 directly affects the assembly of respiratory complexes on the inner membrane. Second, AAC2A128P was found to be synthetically lethal with ybr238cΔ (Supplemental Figure S1). Ybr238c is an inner membrane–associated protein that, together with its paralogue Rmd9, controls mRNA processing/stability in mitochondria (Nouet et al., 2007; Williams et al., 2007). Given that the ybr238CΔ single mutant has barely detectable defect in respiratory growth, the synthetic lethality suggests that mitochondria expressing Aac2A128P are sensitive to subtle disturbance to the respiratory complexes on the membrane due to reduced mitochondrial gene expression. Third, the AAC2A128P allele was also synthetically lethal, with the disruption of PSD1 (Figure 1B), encoding the phosphatidylserine decarboxylase on the inner membrane involved in the synthesis of phosphatidylethanolamine (Figure 1C). Compositional alterations to the lipid environment on the membrane are therefore detrimental to mitochondria expressing Aac2A128P. Finally, we found that haploid cells coexpressing the wild-type AAC2 and AAC2A128P are hypersensitive to antimycin on YPD, which inhibits the bc1 complex in the electron transport chain (Supplemental Figure S2). This indicates that the defect in the electron transport chain synergizes with AAC2A128P, which leads to membrane stress and the inhibition of cell growth.
FIGURE 1:

Expression of mutant Aac2 induces proteostatic stress and affects protein quality control on the mitochondrial inner membrane. (A) The AAC2A128P allele is synthetically lethal, with the disruption of yme1∆ and oxa1∆. The diploid strains were sporulated and dissected on YPD medium. All the meiotic segregants express an endogenous copy of the wild-type AAC2. Circled are the nonviable spores deduced to cosegregate AAC2A128P and yme1∆ or oxa1∆. (B) Growth phenotype of a complete tetrad from CS1422 on YPD showing that expression of AAC2A128P leads to severe growth defect in the psd1Δ background. (C) Schematic representation for a role of PSD1 in the synthesis of phosphophatidylethanolamine (PE) from phosphatidylserine (PS) on the mitochondrial inner membrane. PC, phosphophatidylcholine. (D) Representative Western blot showing steady-state levels of Nde1-3HA and the mtDNA-encoded Cox2 in the strains CS1546/2 (WT), CS1551-8C (yme1∆), CS1552-2C (WT + AAC2A128P), CS1772-1B(WT + AAC2A137D), CS1771-1A (WT + AAC2A106D), and CS1770-2A (WT + AAC2M114P) at the indicated time points after the inhibition of cytosolic protein synthesis by cycloheximide at 37°C. (E–I) Stability of Nde1-3HA in the yme1∆ mutant and in strains coexpressing the wild-type and a mutant allele of AAC2. Error bars are SDs of three independent experiments. *p < 0.05 (unpaired Student's t test).
To provide direct evidence for the proteostatic stress hypothesis, we examined whether general protein quality control is affected on the inner membrane in cells coexpressing the wild-type and a mutant variant of Aac2. The hemagglutinin (HA)-tagged NADH dehydrogenase Nde1 is unstable in vivo and is stabilized in cells defective in the Yme1 protease (Figure 1, D and E; Augustin et al., 2005). We found that the Yme1-dependent turnover of Nde1-3HA is marginally delayed by AAC2A128P and AAC2A137D (Figure 1, F and G) but significantly reduced by AAC2A106D and AAC2M114P (Figure 1, H and I). These data suggest that the mutant Aac2 proteins dominantly affect the turnover of misfolded proteins on the inner membrane, including Nde1-3HA.
Aac2 variants with the adPEO-type mutations are prone to aggregation
We hypothesized that the mutant Aac2 proteins may be misfolded, which causes global proteostatic stress on the inner membrane. Misfolded proteins often have increased propensity to form aggregates. To test whether the mutant Aac2 proteins form aggregates, mitochondria were isolated from cells cultured at 30°C. After detergent solubilization, the proteins were analyzed by Blue Native PAGE (BN-PAGE). As shown in Figure 2A, the monomeric wild-type Aac2 migrated as a band of ∼60 kDa, in contrast to an estimated molecular weight of 34.4 kDa for the protein. The increased mass in BN-PAGE is related to the association of detergent-lipid micelles and Coomassie blue G-250, as previously reported (Crichton et al., 2013). The electrophoretic properties of the four mutant Aac2 proteins are indistinguishable from the wild type under these conditions. Of interest, when mitochondria were preincubated at 25°C before BN-PAGE, we found that Aac2A106D, Aac2M114P, and Aac2A128P formed large aggregates of >720 kDa (Figure 2B). The monomeric form of these proteins was accordingly reduced or became undetectable. The increased propensity of aggregation indicates that the mutant Aac2 proteins are misfolded. Parallel analysis of the cold-treated protein samples by SDS–PAGE revealed that the Aac2 aggregates can be resolubilized by SDS (Figure 2C). No obvious aggregates were detected when mitochondria were isolated from cells grown at 25°C and analyzed by BN-PAGE without prior incubation at low temperature (Figure 2D).
FIGURE 2:

Aac2A106D, Aac2M114P, and Aac2A128P, but not Aac2A137D, are prone to aggregation. (A) Analysis of freshly prepared mitochondria isolated from cells grown in YPD at 30°C. Mitochondria were isolated from strains W303-1B (WT), CS341/1 (aac2∆), CS1434/4 (aac2∆, trp1Δ::AAC2A106D), CS1433/3 (aac2∆, trp1Δ::AAC2M114P), CY4193 (aac2∆, trp1Δ::AAC2A128P), and CS1762-8A (aac2∆, lys2Δ::AAC2A137D). A 40- μg amount of DDM (2%)-solubilized proteins was separated by BN-PAGE, followed by immunoblotting using anti-Aac2 antibody. (B) BN-PAGE, followed by immunoblotting of mitochondria after incubation at 25°C for 16 h before being solubilized by 2% DDM. (C) Immunoblotting of a SDS–PAGE gel with mitochondrial proteins after incubation at 25°C for 16 h. The level of the mitochondrial matrix Ilv5 protein was determined as a loading control. (D) BN-PAGE of freshly prepared mitochondria isolated from cells grown in YPD at 25°C. The same strains were used as in A. (E) Time-dependent aggregation of mutant Aac2. Isolated mitochondria were incubated at 25°C for 4, 8, 12, or 16 h and solubilized by 2% DDM. A 40-μg amount of proteins was analyzed by BN-PAGE followed by immunoblotting using an anti-Aac2 antibody.
In contrast to the adPEO-type mutant Aac2 proteins, no apparent aggregates were detected with the cardiomyopathy- and myopathy-type Aac2A137D variant under the same conditions (Figure 2B). The monomeric form of Aac2A137D was noticeably reduced in BN-PAGE. This was not caused by protein degradation, as SDS–PAGE analysis of n-dodecyl-β-d-maltoside (DDM)–extracted mitochondrial proteins showed that the level of Aac2A137D was little changed after the 16-h incubation (Figure 2C). It is possible that Aac2A137D has a poor reactivity with the antibody under the native conditions.
Further, in organello aggregation assays revealed that Aac2A106D, Aac2M114P, and Aac2A128P formed aggregates 8–12 h after the incubation at 25°C (Figure 2E). Extended incubation eventually led to noticeable reduction of signals for the aggregates. This could be due to the formation of excessively large aggregates that become resistant to DDM extraction and separation by BN-PAGE. The time-course study also confirmed that Aac2A137D does not form aggregates during the entire period of incubation at 25°C.
The mutant Aac2 proteins affect the assembly and stability of multiple protein complexes on the inner membrane
The three adPEO-type mutations occur at Ala-106, Met-114, and Ala-128, which are located at the end of α-helices 2 (H2) and 3 (H3) on the intermembrane space side, with their side chains facing the lipid phase in the carboxylatractyloside-inhibited conformation (Figure 3A). The cardiomyopathy- and myopathy-type mutation occurs at Ala-137, which is also located on H3 but is deeply embedded in the membrane with its side chain relatively facing inward. To evaluate the possible effect of Aac2 misfolding on membrane function, we first determined whether the assembly state of the oxidative phosphorylation pathway is dominantly affected in strains coexpressing the wild-type and mutant AAC2 alleles. In yeast mitochondria, the complexes III (coenzyme Q:cytochrome c oxidoreductase) and IV (cytochrome c oxidase) in the electron transport chain form the III2IV2 and III2IV1 supercomplexes to facilitate substrate channeling, whereas complex V (ATP synthase) is present in both monomeric and dimeric forms (Schagger and Pfeiffer, 2000). These supramolecular structures in wild-type mitochondria can be readily resolved by BN-PAGE (Figure 3B). We found that expression of the four mutant Aac2 proteins drastically affected the formation of these structures. III2IV1 was reduced by AAC2M114P and AAC2A128P and became barely detectable in cells expressing AAC2A106D and AAC2A137D. III2IV2 was severely reduced by all the four mutant alleles (Figure 3, B and C). Immunoblotting of the native gels revealed that the dimeric form of the ATP synthase (V2) was severely reduced by AAC2M114P and AAC2A128P and mildly decreased by AAC2A137D. The monomeric form of the ATP synthase (V1) was drastically decreased in cells expressing all the four mutant alleles (Figure 3D). The reduction of V2 and V1 correlated with the appearance of a band with a molecular weight corresponding to an α/β dimer, which likely derives from disassembled V1 and V2. We also observed that the level of free F1-ATPase, which is not associated with the inner membrane, was unaffected by mutant Aac2, indicating that the mutant Aac2 protein specifically affects protein complexes within the inner membrane.
FIGURE 3:

Expression of mutant Aac2 affects the assembly/stability of protein complexes in the oxidative phosphorylation pathway. (A) Crystal structure of yeast Aac2 (Ruprecht et al., 2014). The positions of amino acids equivalent to the mutated residues in human Ant1 are indicated by arrows. H2 and H3, α-helices 2 and 3, respectively. Carboxylatractyloside trapped in the putative substrate translocation channel is highlighted in magenta. IMS, intermembrane space. (B) BN-PAGE showing the assembly state of respiratory supercomplexes and the ATP synthase. A representative gel from two independent experiments is presented. Yeast cells were grown at 25°C in YPD. Mitochondria were isolated from M2915-6A (WT), 6A/UDU (WT + AAC2A106D [A106D]), 6A/UP2U (WT + AAC2M114P [M114P]), 6A/UPU (WT + AAC2A128P [A128P]), and CS1482/1 (WT + AAC2A137D [A137D]) and solubilized by 2% digitonin. A 25-μg amount of proteins was loaded on the BN-PAGE gel. V1 and V2, monomeric and dimeric forms of FoF1-ATP synthase (or complex V), respectively. III2IV2, respiratory supercomplex containing dimeric complex III and dimeric complex IV. III2IV1, respiratory supercomplex containing dimeric complex III and monomeric complex IV. (C) Relative levels of respiratory supercomplexes in strains coexpressing wild-type and mutant Aac2. (D) Western blotting analysis of a native gel, showing the assembly state of complex V using antisera against the α and β subunits.
To assess further the damage to membrane protein complexes, we expanded our analysis to the TIM22 and TIM23 protein translocases. The TIM22 complex is involved in the membrane insertion of mitochondrial carriers, including Aac2, whereas TIM23 promotes the translocation of precursor proteins across the inner membrane to the mitochondrial matrix (Chacinska et al., 2009). We found that AAC2A106D, AAC2M114P, and AAC2A128P reduce the level of assembled TIM22 complex on the native gel (Figure 4A). Multiple bands of lower molecular weight were detected, suggesting that the TIM22 protein translocation channels may be disassembled or delayed in assembly. The disassembled or unassembled TIM22 complex may become unstable, because the levels of the Tim22 protein were accordingly reduced, as revealed by SDS–PAGE (Figure 4C). The level of neither the TIM22 complex nor the Tim22 protein was reduced by AAC2A137D, although fast-migrating bands suggestive of TIM22 disassembly/unassembly were detected in cells expressing this particular allele. Because no bands with a molecular weight higher than the ∼400-kDa TIM22 complex were detected, it does not appear that the misfolded Aac2 proteins are stably trapped inside the twin-pore TIM22 translocation channels.
FIGURE 4:
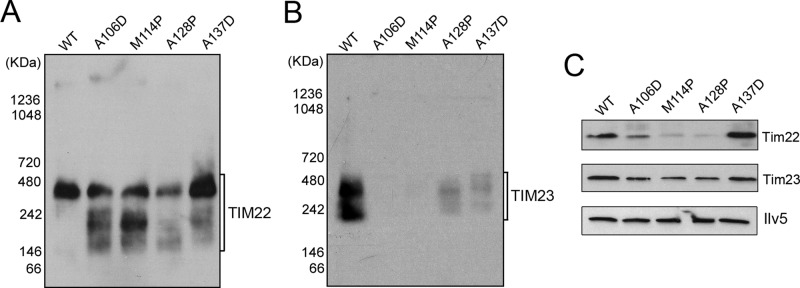
Expression of mutant Aac2 affects the assembly/stability of the TIM22 and TIM23 complexes. (A, B) Immunoblotting of native gels showing the assembly state of the TIM22 and TIM23 protein translocases, respectively. Mitochondria were isolated from M2915-6A (WT), 6A/UDU (WT + AAC2A106D [A106D]), 6A/UP2U (WT + AAC2M114P [M114P]), 6A/UPU (WT + AAC2A128P [A128P]), and CS1482/1 (WT + AAC2A137D [A137D]). (C) Western blots of SDS–PAGE gels showing the steady-state levels of Tim22 and Tim23 in mitochondria isolated from the same strains used in A and B. The level of the mitochondrial matrix protein Ilv5 was used as loading control.
Finally, we determined the effect of the misfolded Aac2 on the TIM23 complex. As shown in Figure 4B, TIM23 was barely detectable in cells coexpressing the wild-type AAC2 and AAC2A106D or AAC2M114P. The level of TIM23 was strongly reduced by AAC2A128P and AAC2A137D. The Tim23 protein level was only slightly reduced by the mutant AAC2 alleles, as shown by SDS–PAGE analysis (Figure 4C). These observations indicate that the assembly of TIM23 is also dominantly deterred by the mutant Aac2 proteins. The overall loss of Tim23 signal in the mutant mitochondria on the BN-PAGE gel may be caused by the aggregation of unassembled Tim23 or the poor solubility of structurally altered TIM23 complex, which may cause poor protein recovery before electrophoretic separation.
The i-AAA protease Yme1 plays a critical role in the quality control of misfolded Aac2
Misfolded membrane proteins are expected to be degraded by specific protein quality control machineries. To provide further support to the model that the mutant Aac2 proteins are misfolded, we first determined the in vivo stability of Aac2A128P compared with the wild type. We observed that both Aac2A128P and its wild-type control remain stable 24 h after cycloheximide chase (Figure 5A). We then determined the stability of Aac2A128P by an in organello assay. Isolated mitochondria were incubated at different temperatures, and the levels of Aac2 were monitored by immunoblotting (Figure 5B). We found that the wild-type Aac2 remained stable 3.5 h after incubation at all three temperatures tested. Aac2A128P was stable at 25°C but became unstable at 30°C. Aac2A128P degradation was further accelerated at 37°C. The temperature-dependent turnover of Aac2A128P correlated with the growth phenotype of the AAC2A128P mutant (Figure 5C). Incubation at 25°C, but not 37°C, inhibited cell growth on complete medium containing glucose or glycerol plus ethanol as carbon sources. It remains unclear as to why AAC2A128P turnover is detected in the in organello but not in vivo assays. We speculate that the in organello conditions may exacerbate the misfolding of the mutant protein, which accelerates its recognition and then turnover by protein quality control systems.
FIGURE 5:
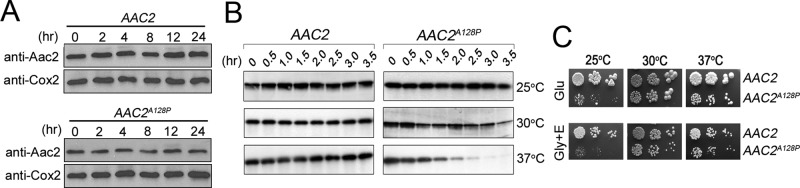
Increased turnover of Aac2A128P. (A) Cycloheximide chase experiment showing the stability of Aac2A128P in vivo. Cells were grown at 37°C in YPD. Cycloheximide (1 mg/ml), was added and cell lysates were extracted from M2915-6A (AAC2) and UPU5-6B (AAC2A128P) at the indicated time points. Western blotting was performed using antisera against Aac2 and Cox2. (B) In organello assay showing increased turnover of Aac2A128P. Mitochondria were isolated from M2915-6A (AAC2) and UPU5-6B (AAC2A128P) and incubated at 25, 30, and 37°C. The levels of Aac2 at different time points were analyzed by Western blotting. (C) Growth phenotype of M2915-6A (AAC2) and UPU5-6B (AAC2A128P) on YPD (Glu) and YPGly plus ethanol (Gly+E) medium.
We next attempted to identify potential protein quality control mechanisms and other cellular pathways that might reduce Aac2A128P-induced mitochondrial damage, by screening for genes that suppress Aac2A128P-induced growth defect (Figure 6A). Expression of AAC2A128P from the galactose-inducible GAL10 promoter strongly inhibited cell growth on galactose medium. We found that overexpression of YME1, but not RCA1 and AFG3, suppressed growth inhibition by Aac2A128P. In contrast to YME1, which encodes the i-AAA protease to preferentially degrade misfolded membrane proteins from the intermembrane space side, YTA12/RCA1 and YTA10/AFG3 encode the two subunits of the hetero-oligomeric m-AAA protease, which degrades misfolded and unassembled proteins on the inner membrane from the matrix side (Arlt et al., 1996). Overexpression of SOD2, CTA1 and CTT1, encoding the mitochondrial superoxide dismutase and catalases that reduce oxidative stress, failed to suppress aac2A128P. These observations suggest that the Yme1 protease likely plays a major role in the quality control of Aac2.
FIGURE 6:

Yme1 protease plays a key role in the quality control of Aac2. (A) Suppression of Aac2A128P-induced cell death by YME1 overexpression. Aac2A128P was expressed from the GAL10 promoter on complete medium containing galactose and raffinose as carbon sources at 30°C. All the genes were cloned into a multicopy vector and introduced into the PG1 strain by transformation. (B) BN-PAGE coupled with immunoblotting showing the aggregation of Aac2A128P in fresh mitochondria prepared from a strain with the yme1Δ background. The strains CS1153/1 (rpl6BΔ, AAC2), CS1227-4A (rpl6BΔ, AAC2, yme1∆), CS1374-2D (rpl6BΔ, AAC2A128P), and CS1699/1 (rpl6BΔ, AAC2A128P, yme1∆) were grown at 30°C in YPD. Mitochondria were isolated and solubilized by 2% DDM. Approximately 40 μg of solubilized proteins was applied to BN-PAGE, followed by immunoblotting using anti-Aac2 antibody. (C) Steady-state levels of Aac2A128P compared with wild-type Aac2 in the presence or absence of Yme1. Mitochondria were prepared as in B, and 30 μg of proteins was analyzed by SDS–PAGE and Western blotting. The matrix Ilv5 protein was used as a loading control. (D) Relative steady-state levels of Aac2 in C. Error bars indicate the SDs of three independent experiments. *p < 0.05 (unpaired Student's t test). (E) A model for the membrane damage caused by misfolded Aac2. See Discussion for details. The red star denotes the pathogenic missense mutations. IM, inner membrane; IMS, intermembrane space.
To further support a role of Yme1 in the surveillance of Aac2 quality, we asked whether loss of Yme1 accelerates Aac2A128P aggregation. The pathogenic Aac2 variants are synthetically lethal with yme1Δ (Wang et al., 2008a). We previously showed that the synthetic lethality is suppressed by the disruption of RPL6B, which partially reduces cytosolic protein synthesis and improves global protein homeostasis (Wang et al., 2008b). We thus constructed viable strains that combine AAC2A128P with yme1Δ in the rpl6BΔ background. Cells were first grown at 30°C, and freshly prepared mitochondria were analyzed by BN-PAGE. As shown in Figure 6B, yme1Δ reduced the level of monomeric form of the wild-type Aac2, but no obvious aggregates were detected. No Aac2A128P aggregates were formed in cells expressing YME1 under these experimental conditions. In contrast, yme1Δ induced the formation of visible Aac2A128P aggregates that migrated as a distinct band of ∼1.0 mDa in the native gel. Aac2A128P aggregation was concomitant to the loss of its monomeric form. Because these conditions did not induce Aac2A128P aggregation in cells expressing YME1 (Figure 2A), the data strongly suggest that Yme1 plays a role in quality control of Aac2 and prevention of Aac2A128P aggregation. SDS–PAGE revealed that the levels of both Aac2A128P and wild-type Aac2 were reduced instead of increased in the absence of Yme1 (Figure 6, C and D). It is possible that yme1Δ-induced inner membrane stress affects the membrane insertion of both wild-type and mutant Aac2. A fraction of the Aac2 proteins may have been degraded by other proteases before maturing on the inner membrane. The remaining mutant Aac2 that is successfully matured on the inner membrane is sufficient to affect cell survival because of severe misfolding in the absence of Yme1.
DISCUSSION
We found that the adPEO-type Aac2A106D, Aac2M114P, and Aac2A128P share similar biochemical properties. Of note, they all have increased propensity to form large aggregates and are capable of inducing proteostatic stress and severe defects in the assembly and structural maintenance of multiple protein complexes on the mitochondrial inner membrane. The data strongly support the view that Ant1-induced adPEO is a protein misfolding disorder. In addition to damages to the oxidative phosphorylation pathway, the mutant Ant1 likely reduces cell viability by affecting the function of multiple membrane protein complexes, including TIM22 and TIM23, which directly participate in mitochondrial biogenesis. The mutant Aac2 proteins therefore gain a toxic function, which is consistent with the dominant nature of the disease.
We found that Aac2A137D, mimicking Ant1A123D, which causes cardiomyopathy and myopathy (Palmieri et al., 2005), does not form visible aggregates. However, cells expressing AAC2A137D share many common phenotypes with those expressing the adPEO-type alleles (Wang et al., 2008a). These include uncoupled respiration, cold sensitivity, ρo-lethality, mtDNA instability, and synthetic lethality with yme1Δ. Furthermore, as shown in the present study, Aac2A137D also interferes with the assembly and stability of multiple protein complexes on the membrane. These findings argue that Aac2A137D may also be misfolded and affect mitochondrial function by the same mechanism as the adPEO-type Aac2 variants. Although Aac2A137D does not aggregate in in organello assay, it may have altered conformation that causes proteostatic stress on the membrane. Definitive proof for Aac2A137D misfolding is lacking. If Aac2A137D were misfolded, an extended speculation would be that protein misfolding, rather than aggregation per se, may be the primary cause of membrane damage (Figure 6E). Because the A137D mutation (or A123D in human Ant1) completely abolishes nucleotide transport activity (Palmieri et al., 2005), the cardiomyopathy and myopathy phenotypes observed in the reported homozygous patient may result from a double hit. The pathogenesis may involve both Ant1 misfolding and defective ATP/ADP exchange. In the future, it would be interesting to see whether ANT1A123D can cause adPEO in a dominant manner in heterozygous individuals if such clinical cases become available.
The quality control mechanism of plasma membrane proteins has been extensively studied. It involves the recognition of misfolded domains by E3 ubiquitin ligases on the endoplasmic reticulum (ER) membranes, followed by ER-associated degradation (ERAD; Hirsch et al., 2009; Houck and Cyr, 2012). The removal of the misfolded proteins by ERAD decreases the delivery of functional proteins on the cell surface, leading to recessive pathologies such as cystic fibrosis. However, in other clinical cases, destabilizing mutations in transmembrane domains cause dominant diseases (e.g., retinitis pigmentosa). These diseases often involve the toxic accumulation of misfolded proteins (Sanders and Myers, 2004; Mendes et al., 2005). Not much is known about the extent to which the delivery of a misfolded protein might affect membrane functionality and cell viability. We provide evidence that the misfolded Aac2 proteins affect the assembly and stability of multiple protein complexes on the mitochondrial inner membrane. Among the adPEO-type mutations, M114P and A128P are expected to introduce a proline kink that would prematurely terminate α-helices 2 and 3 (Figure 3A). A106D introduces a negative charge in the proximity of the membrane surface, which could stretch the helix 2 toward the positively charged intermembrane space. The cardiomyopathy- and myopathy-type A137D mutation introduces a negative charge at a deeper position within the membrane. Possibly, a common consequence of these structural and conformational changes could be the exposure of nonnative, especially polar residues to the membrane environment. This may induce structural perturbations (e.g., helix misalignment) in order to solvate the unfavorable energy potential within the membrane. In this regard, aggregation of Aac2A106D, Aac2M114P, and Aac2A128P may be a compensatory molecular strategy that helps to shield the exposed nonnative patches to defuse the unfavorable energy potential. Although we detected visible aggregates of mutant Aac2 in freshly prepared mitochondria in cells lacking the Yme1 protease, Aac2 aggregates were not observed in fresh mitochondria isolated from YME1 cells, even when cells were grown at 25°C. Aggregates were readily detected in isolated mitochondria after extended incubation at 25°C. It remains unknown what exactly triggers the aggregation of the mutant Aac2 during the in organello incubation. Nonetheless, the data clearly indicate that the mutant proteins are biophysically distinguishable from the wild-type Aac2. Compared with Ala-106, Met-114, and Ala-128, which have side chains facing directly to the lipid phase, the side chain in Ala-137 faces relatively inwardly. It is possible that Aac2A137D adopts a specific conformation that is unfavorable for oligomerization and aggregation.
In considering possible mechanisms of mitochondrial damage (Figure 6E), one can speculate that the misfolded Aac2 alters the surface property of the protein and exposes nonnative residues. This may in turn promote ectopic interactions with other membrane proteins. Interactions with unassembled proteins may prevent their incorporation into protein complexes. Previous studies suggested that Aac2 physically associates with multiple protein complexes in the membrane, including the III-IV supercomplexes and TIM23 (Claypool et al., 2008; Dienhart and Stuart, 2008; Mehnert et al., 2014). The intrinsic affinity to Aac2 may explain the severe defects in the assembly of these complexes in cells expressing a misfolded Aac2. On the other hand, as Aac2 is inserted into the membrane through the TIM22 complex, the interaction with the misfolded Aac2 appears to directly destabilize the TIM22 structure, as manifested by the breakdown of the complex. As alternative mechanisms, it is also possible that the misfolded Aac2 proteins deplete the molecular chaperones and proteases required for the biogenesis and quality control of other protein complexes. The damage to TIM22 and defective membrane protein import may also contribute to the defect in the biogenesis of respiratory complexes and the TIM23 preprotein translocase.
Two AAA proteases play key roles in the quality control of proteins on the mitochondrial inner membrane (Gerdes et al., 2012). The i-AAA protease (or Yme1) has its catalytic domain facing the intermembrane space, whereas the m-AAA protease, composed of the Yta12 (or Rca1) and Yta10 (or Afg3) subunits, has the catalytic domain facing the matrix. These proteases sense the folding state of solvent-exposed domains on either side of the membrane and specifically degrade unfolded membrane proteins (Arlt et al., 1996; Leonhard et al., 1996, 1999, 2000). The suppression of Aac2A128P-induced cell lethality by YME1 overexpression and accelerated accumulation of Aac2A128P aggregates in cells lacking Yme1 suggest that the i-AAA protease plays a key role in the quality control of Aac2A128P. The premature termination of α-helix 2 likely distorts the conformation of the solvent-exposed loop between helices 2 and 3, which is recognized and attacked by Yme1. Whether other mutants are also recognized by Yme1 for degradation has yet to be determined.
In summary, our data strongly suggest that Ant1-induced adPEO is a protein misfolding disease, which distinguishes it from clinical conditions caused by null mutations in ANT1. Of greater importance, our work provides evidence that the misfolding of one single protein could have detrimental effects on the biogenesis and stability of other membrane complexes on the membrane and that proteostatic stress could be a significant source of damage to the oxidative phosphorylation apparatus under pathophysiological conditions. Our finding could have general implications for understanding the physiological consequences of other destabilizing mutations in transmembrane proteins that are associated with an increasing number of dominant diseases, including retinitis pigmentosa (Sanders and Myers, 2004; Mendes et al., 2005; Hirsch et al., 2009; Houck and Cyr, 2012).
MATERIALS AND METHODS
Cell growth conditions and yeast strains
Standard media were used to cultivate yeast cells. The genotypes and sources of Saccharomyces cerevisiae strains used in this study are listed in Supplemental Table S1. To generate the NDE1-3HA allele, the HA epitope was added to the C-terminus of NDE1 by the integration of a PCR product amplifying from the 3HA-KanMX6 cassette. The strain PG1 (MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, trp1::GAL10-AAC2A128P-HIS3) was derived from BY4741.
Determination of protein stability
Yeast cells were grown in YPD to late exponential phase and shifted to 37°C after addition of cycloheximide (1 mg/ml). At indicated time points, three OD600 equivalents of cells were harvested. The cell extract was prepared as previously described (Chen, 2001). Approximately 5 μl of cell extracts was analyzed by SDS–PAGE, followed by Western blot using antisera against Aac2, Cox2, or HA.
Mitochondrial preparation and BN-PAGE analysis
Yeast cells were grown at 25 or 30°C in YPD. Crude mitochondria were isolated as described (Boldogh and Pon, 2007), except that the cell wall was removed by enzymatic digestion at 5 mg of Zymolyase 20T/g of wet cells at 25 or 37°C. Isolated mitochondria were resuspended in SEH buffer (0.6 M sorbitol, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]-KOH, pH 7.4, 2 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, 1 mM phenylmethylsulfonyl fluoride). For BN-PAGE analysis, crude mitochondria were solubilized in a 2% DDM or digitonin solution containing 50 mM NaCl, 2.5 mM MgCl2, 20 mM HEPES-KOH (pH 7.4), 10% (vol/vol) glycerol, and 1 mM EDTA (pH 7.4) on ice for 30 min. Insoluble material was removed by centrifugation of 10,000 × g at 4°C for 15 min. Approximately 30–40 μg of mitochondrial proteins were mixed with 5% G-250 sample additive (Life Technologies, Carlsbad, CA) such that the final concentration of G-250 was 0.5%. The protein samples were loaded to a 3–12% NativePAGE Bis-Tris gel (Life Technologies), which was run at 150 V in dark cathode buffer for 1 h and then 250 V in light cathode buffer for 45 min at 4°C. Staining and destaining of native gels were performed according to the protocols of the manufacturer (Life Technologies). For immunoblotting, the native gel was prewashed in a transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) containing 2% SDS for 30 min before being transferred on a polyvinylidene fluoride membrane at 30 V overnight. The membrane was then incubated in 8% acetic acid for 15 min to fix the proteins, followed by rinse with deionized water. Before immunoblotting, the membrane was rewetted with methanol to remove excessive dye bound to the membranes.
Acknowledgments
This work was supported by grants from National Institute on Aging and a predoctoral fellowship from the American Heart Association to Y.L. We thank Stephan Wilkens for critical reading of the manuscript.
Abbreviations used:
- adPEO
autosomal dominant progressive external ophthalmoplegia
- Ant
adenine nucleotide translocase
- BN-PAGE
blue native polyacrylamide gel electrophoresis
- DDM
n-dodecyl β-d-maltoside
- HA
hemagglutinin
- mtDNA
mitochondrial DNA
- YPD
yeast extract/peptone/dextrose medium.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-01-0030) on April 1, 2015.
REFERENCES
- Arlt H, Tauer R, Feldmann H, Neupert W, Langer T. The YTA10–12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell. 1996;85:875–885. doi: 10.1016/s0092-8674(00)81271-4. [DOI] [PubMed] [Google Scholar]
- Augustin S, Nolden M, Muller S, Hardt O, Arnold I, Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem. 2005;280:2691–2699. doi: 10.1074/jbc.M410609200. [DOI] [PubMed] [Google Scholar]
- Boldogh IR, Pon LA. Purification and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 2007;80:45–64. doi: 10.1016/S0091-679X(06)80002-6. [DOI] [PubMed] [Google Scholar]
- Bonnefoy N, Chalvet F, Hamel P, Slonimski PP, Dujardin G. OXA1, a Saccharomyces cerevisiae nuclear gene whose sequence is conserved from prokaryotes to eukaryotes controls cytochrome oxidase biogenesis. J Mol Biol. 1994;239:201–212. doi: 10.1006/jmbi.1994.1363. [DOI] [PubMed] [Google Scholar]
- Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392:353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ. Activity of the Kluyveromyces lactis Pdr5 multidrug transporter is modulated by the Sit4 protein phosphatase. J Bacteriol. 2001;183:3939–3948. doi: 10.1128/JB.183.13.3939-3948.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ. Induction of an unregulated channel by mutations in adenine nucleotide translocase suggests an explanation for human ophthalmoplegia. Hum Mol Genet. 2002;11:1835–1843. doi: 10.1093/hmg/11.16.1835. [DOI] [PubMed] [Google Scholar]
- Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol. 2008;182:937–950. doi: 10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton PG, Harding M, Ruprecht JJ, Lee Y, Kunji ER. Lipid, detergent, and Coomassie Blue G-250 affect the migration of small membrane proteins in blue native gels: mitochondrial carriers migrate as monomers not dimers. J Biol Chem. 2013;288:22163–22173. doi: 10.1074/jbc.M113.484329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschauer M, Hudson G, Muller T, Taylor RW, Chinnery PF, Zierz S. A novel ANT1 gene mutation with probable germline mosaicism in autosomal dominant progressive external ophthalmoplegia. Neuromuscul Disord. 2005;15:311–315. doi: 10.1016/j.nmd.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Dienhart MK, Stuart RA. The yeast Aac2 protein exists in physical association with the cytochrome bc1-COX supercomplex and the TIM23 machinery. Mol Biol Cell. 2008;19:3934–3943. doi: 10.1091/mbc.E08-04-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Chassagne M, Ceresuela J, Rouvet I, Padet S, Acquaviva C, Nataf S, Vinzio S, Bozon D, Mousson de Camaret B. Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J Med Genet. 2012;49:146–150. doi: 10.1136/jmedgenet-2011-100504. [DOI] [PubMed] [Google Scholar]
- El-Khoury R, Sainsard-Chanet A. Suppression of mitochondrial DNA instability of autosomal dominant forms of progressive external ophthalmoplegia-associated ANT1 mutations in Podospora anserina. Genetics. 2009;183:861–871. doi: 10.1534/genetics.109.107813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi F, Palmieri L, Scarcia P, Lodi T, Donnini C, Limongelli A, Tiranti V, Zeviani M, Ferrero I, Viola AM. Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum Mol Genet. 2004;13:923–934. doi: 10.1093/hmg/ddh108. [DOI] [PubMed] [Google Scholar]
- Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases—towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Neupert W, Stuart RA. Insertion into the mitochondrial inner membrane of a polytopic protein, the nuclear-encoded Oxa1p. EMBO J. 1997;16:2217–2226. doi: 10.1093/emboj/16.9.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458:453–460. doi: 10.1038/nature07962. [DOI] [PubMed] [Google Scholar]
- Houck SA, Cyr DM. Mechanisms for quality control of misfolded transmembrane proteins. Biochim Biophys Acta. 2012;1818:1108–1114. doi: 10.1016/j.bbamem.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- Kawamata H, Tiranti V, Magrane J, Chinopoulos C, Manfredi G. adPEO mutations in ANT1 impair ADP-ATP translocation in muscle mitochondria. Hum Mol Genet. 2011;20:2964–2974. doi: 10.1093/hmg/ddr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Komaki H, Fukazawa T, Houzen H, Yoshida K, Nonaka I, Goto Y. A novel D104G mutation in the adenine nucleotide translocator 1 gene in autosomal dominant progressive external ophthalmoplegia patients with mitochondrial DNA with multiple deletions. Ann Neurol. 2002;51:645–648. doi: 10.1002/ana.10172. [DOI] [PubMed] [Google Scholar]
- Kunji ER, Crichton PG. Mitochondrial carriers function as monomers. Biochim Biophys Acta. 2010;1797:817–831. doi: 10.1016/j.bbabio.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Lawson JE, Douglas MG. Separate genes encode functionally equivalent ADP/ATP carrier proteins in Saccharomyces cerevisiae. Isolation and analysis of AAC2. J Biol Chem. 1988;263:14812–14818. [PubMed] [Google Scholar]
- Lawson JE, Gawaz M, Klingenberg M, Douglas MG. Structure-function studies of adenine nucleotide transport in mitochondria. I. Construction and genetic analysis of yeast mutants encoding the ADP/ATP carrier protein of mitochondria. J Biol Chem. 1990;265:14195–14201. [PubMed] [Google Scholar]
- Leonhard K, Guiard B, Pellecchia G, Tzagoloff A, Neupert W, Langer T. Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol Cell. 2000;5:629–638. doi: 10.1016/s1097-2765(00)80242-7. [DOI] [PubMed] [Google Scholar]
- Leonhard K, Herrmann JM, Stuart RA, Mannhaupt G, Neupert W, Langer T. AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J. 1996;15:4218–4229. [PMC free article] [PubMed] [Google Scholar]
- Leonhard K, Stiegler A, Neupert W, Langer T. Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature. 1999;398:348–351. doi: 10.1038/18704. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen XJ. Adenine nucleotide translocase, mitochondrial stress, and degenerative cell death. Oxid Med Cell Longev. 2013:146860. doi: 10.1155/2013/146860. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert CS, Rampelt H, Gebert M, Oeljeklaus S, Schrempp SG, Kochbeck L, Guiard B, Warscheid B, van der Laan M. The mitochondrial ADP/ATP carrier associates with the inner membrane presequence translocase in a stoichiometric manner. J Biol Chem. 2014;289:27352–27362. doi: 10.1074/jbc.M114.556498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Napoli L, Bordoni A, Zeviani M, Hadjigeorgiou GM, Sciacco M, Tiranti V, Terentiou A, Moggio M, Papadimitriou A, Scarlato G, Comi GP. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology. 2001;57:2295–2298. doi: 10.1212/wnl.57.12.2295. [DOI] [PubMed] [Google Scholar]
- Nouet C, Bourens M, Hlavacek O, Marsy S, Lemaire C, Dujardin G. Rmd9p controls the processing/stability of mitochondrial mRNAs and its overexpression compensates for a partial deficiency of oxa1p in Saccharomyces cerevisiae. Genetics. 2007;175:1105–1115. doi: 10.1534/genetics.106.063883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- Palmieri F. Mitochondrial transporters of the SLC25 family and associated diseases: a review. J Inherit Metab Dis. 2014;37:565–575. doi: 10.1007/s10545-014-9708-5. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet. 2005;14:3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Ravaud S, Bidon-Chanal A, Blesneac I, Machillot P, Juillan-Binard C, Dehez F, Chipot C, Pebay-Peyroula E. Impaired transport of nucleotides in a mitochondrial carrier explains severe human genetic diseases. ACS Chem Biol. 2012;7:1164–1169. doi: 10.1021/cb300012j. [DOI] [PubMed] [Google Scholar]
- Ruprecht JJ, Hellawell AM, Harding M, Crichton PG, McCoy AJ, Kunji ER. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc Natl Acad Sci USA. 2014;111:E426–E434. doi: 10.1073/pnas.1320692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CR, Myers JK. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- Saraste M, Walker JE. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982;144:250–254. doi: 10.1016/0014-5793(82)80648-0. [DOI] [PubMed] [Google Scholar]
- Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano G, Tessa A, Petrini S, Mancuso M, Bruno C, Grieco GS, Malandrini A, DeFlorio L, Martini B, Federico A, et al. Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in the ANT1 gene. Neuromuscul Disord. 2003;13:162–165. doi: 10.1016/s0960-8966(02)00221-3. [DOI] [PubMed] [Google Scholar]
- Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]
- Strauss KA, Dubiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA. 2013;110:3453–3458. doi: 10.1073/pnas.1300690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart R. Insertion of proteins into the inner membrane of mitochondria: the role of the Oxa1 complex. Biochim Biophys Acta. 2002;1592:79–87. doi: 10.1016/s0167-4889(02)00266-5. [DOI] [PubMed] [Google Scholar]
- Thorsness PE, White KH, Fox TD. Inactivation of YME1, a member of the ftsH-SEC18-PAS1-CDC48 family of putative ATPase-encoding genes, causes increased escape of DNA from mitochondria in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:5418–5426. doi: 10.1128/mcb.13.9.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JE, Runswick MJ. The mitochondrial transport protein superfamily. J Bioenerg Biomembr. 1993;25:435–446. doi: 10.1007/BF01108401. [DOI] [PubMed] [Google Scholar]
- Wang X, Salinas K, Zuo X, Kucejova B, Chen XJ. Dominant membrane uncoupling by mutant adenine nucleotide translocase in mitochondrial diseases. Hum Mol Genet. 2008a;17:4036–4044. doi: 10.1093/hmg/ddn306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Zuo XM, Kucejova B, Chen XJ. Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat Cell Biol. 2008b;10:1090–1097. doi: 10.1038/ncb1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EH, Butler CA, Bonnefoy N, Fox TD. Translation initiation in Saccharomyces cerevisiae mitochondria: functional interactions among mitochondrial ribosomal protein Rsm28p, initiation factor 2, methionyl-tRNA-formyltransferase and novel protein Rmd9p. Genetics. 2007;175:1117–1126. doi: 10.1534/genetics.106.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]