

An improved variant of mKeima, a monomeric long Stokes shift red fluorescent protein, is introduced, called hmKeima8.5. Its increased intracellular brightness and long Stokes shift (~180 nm) make it an excellent partner with teal fluorescent protein (mTFP1) for multiphoton, multicolor applications. Here we demonstrate its utility in intracellular fluorescence correlation spectroscopy applications.
Abstract
We report an improved variant of mKeima, a monomeric long Stokes shift red fluorescent protein, hmKeima8.5. The increased intracellular brightness and large Stokes shift (∼180 nm) make it an excellent partner with teal fluorescent protein (mTFP1) for multiphoton, multicolor applications. Excitation of this pair by a single multiphoton excitation wavelength (MPE, 850 nm) yields well-separable emission peaks (∼120-nm separation). Using this pair, we measure homo- and hetero-oligomerization interactions in living cells via multiphoton excitation fluorescence correlation spectroscopy (MPE-FCS). Using tandem dimer proteins and small-molecule inducible dimerization domains, we demonstrate robust and quantitative detection of intracellular protein–protein interactions. We also use MPE-FCCS to detect drug–protein interactions in the intracellular environment using a Coumarin 343 (C343)-conjugated drug and hmKeima8.5 as a fluorescence pair. The mTFP1/hmKeima8.5 and C343/hmKeima8.5 combinations, together with our calibration constructs, provide a practical and broadly applicable toolbox for the investigation of molecular interactions in the cytoplasm of living cells.
INTRODUCTION
An individual living cell is a nonequilibrium system built up by an interplay of dynamic biomolecules and biological polymers. To understand such intracellular molecular processes and interactions in real time, optical approaches in living cells have proved powerful, in particular with the use of genetically encoded multicolor fluorescence probes (Shaner et al., 2005). Imaging modalities provide their subcellular distribution and the underlying dynamics and measures of their activity. Considering all techniques, including photobleaching and photoactivation, and recent developments in fluorophore ligation methods (HaloTags or SNAP- tags; Keppler et al., 2003; Los et al., 2008), we now can measure localization dynamics over a broad range of time scales and with high spatial resolution below the diffraction limit.
Protein interactions represent a more difficult parameter for optical methods. Methods that take advantage of Förster resonance energy transfer (FRET) can provide exquisitely specific measures of protein–protein interactions. Such FRET reporters can be difficult to generate in practice but are powerful when they work. The difficulty in designing functional FRET probes stems from the requirement that the fluorophores—often fluorescent proteins fused to the proteins of interest—have to be in close proximity (∼10–100 Å) in the complex. As a result, FRET cannot be used to rule out the presence of an interaction.
Alternative approaches to measuring cytoplasmic protein–protein complexes are methods based on fluorescence cross-correlation spectroscopy (FCCS; Bacia and Schwille, 2007). By measuring the local signal fluctuations due to the passage of fluorescently labeled molecules into and out of the excitation volume, fluorescence correlation spectroscopy (FCS) can quantitatively and selectively capture the temporal relaxation and amplitudes of these molecular motions (Magde et al., 1972; Rigler et al., 1993; Schwille et al., 1999). In turn, these fluctuations, alongside calibrated measurements of the excitation volume and appropriate models of molecular kinetics and dynamics, can provide a quantitative readout of the number, mobility, and even kinetic reactions of biomolecules. FCCS extends single-color FCS to the cross-correlation of the molecular fluctuations of two differentially labeled species (Kim et al., 2005). Statistical analysis of the covariance of the two species' intensity fluctuations provides quantitative readout of the fraction of co-diffusing species. If properly executed, FCCS experiments can even refute the presence of a biomolecular interaction, at least within the validated dynamic range, which typically is sensitive for interaction up to KD ≈ 1 μM (Maeder et al., 2007; Boeke et al., 2014).
The intracellular application of multiphoton or two-photon excitation for FCS (2PE-FCS) using pulsed, infrared (IR) lasers has several advantages compared with one-photon excitation (1PE; Schwille et al., 1999; So et al., 2000). The large separation between the excitation wavelength and emission wavelength results in excellent background rejection, since scattering (such as Raleigh and Raman scattering) can be easily filtered out, especially in the intracellular environment. 2PE occurs within the focal point, which is typically <1 fl when using high numerical objectives. As a result of this intrinsic restriction of the excitation volume, fluorophores can only be excited inside the optical focus, which also reduces the photodamage to living cells. Furthermore, IR wavelengths cause less damage to protein and DNA. Hence, 2PE is poised for use in an increasing number of live-cell applications in biophysical research.
The major difficulty for multicolor fluorescence microscopy by 2PE in intracellular application is the absence of bright fluorescent protein pair with the same optimal 2PE and well-separated emission spectra. 2PE lasers, usually femtosecond Ti:sapphire sources, are tunable over a large range of IR wavelengths (generally 650–1110 nm) but are slow to change wavelengths, making them difficult to use for multiplexed excitation. Moreover, modern auto-mode-locking femtosecond lasers are expensive, limiting the use of multiple sources. To take advantage of the features of 2PE and maintain a constant excitation volume over multiple probes, fluorophores with similar excitation maxima and separable emission spectra are required (Kawano et al., 2008; Tillo et al., 2010). Here we developed a new variant of a large Stokes shift probe, termed hmKeima8.5, evolved from the original mKeima (Kogure et al., 2006). hmKeima8.5 shares a similar excitation spectrum with the cyan fluorescent protein mTFP1 (Ai et al., 2006) in both 1PE and 2PE, but its emission peaks are significantly red shifted (492 nm for TFP vs. 612 nm for hmKeima8.5). Moreover, both fluorophores are significantly brighter than previously reported ECFP/mKeima fluorescent pairs (Kogure et al., 2008).
Here we report on the use of hmKeima8.5 and mTFP1 in intracellular FCCS applications. Our model systems highlight the measurements of protein homo/hetero interactions and protein–drug interactions in individual living cells by single-color autocorrelation brightness analysis and dual-color cross-correlation analysis. The techniques described here have broad use in the study of biomolecular interactions in the living intracellular environment.
RESULTS
Evolution of mKeima proteins for increased brightness
DNA shuffling (Crameri et al., 1996) was used to evolve improved mKeima mutants using budding yeast as a host organism (see Materials and Methods). Two brighter variants were chosen for further characterization, mKeima4.15 (from round 4) and mKeima8.5 (from round 8) (Figure 1A).
FIGURE 1:

Evolved variants of the long Stokes shift fluorescent protein mKeima. (A) Alignment of mKeima variants. (B) Normalized absorption (dashed lines) and fluorescence emission (solid lines) spectra of hmKeima8.5 (dark blue/red) and mTFP1 (blue/teal). (C) The two-photon cross sections of mTFP1 and hmKeima8.5.
We synthesized human codon–optimized versions of the two yeast mKeima variants, now referred to as hmKeimas, and compared their fluorescence properties, including extinction coefficient and maturation time (done in yeast mKeima), in comparison to other long Stokes shift red FPs (e.g., LSSmKate2; Piatkevich et al., 2010) and four cyan FPs (ECFP, Heim and Tsien, 1996; mCerulean, Rizzo et al., 2004; mTFP1, Ai et al., 2006; and mTurquoise2, Goedhart et al., 2012; Table 1). All proteins shared similar single-photon excitation properties; however, mTFP1 showed larger two-photon brightness or action cross sections (2P cross section multiplied by the quantum yield). The 1PE spectrum of hmKeima8.5 had a strong overlap with the corresponding spectra of mTFP1, and the large Stokes shift property of hmKeima8.5 separates its emission spectrum far from mTFP1 (Figure 1B). The two-photon cross sections showed similar overlap between hmKeima8.5 and mTFP1, showing the largest cross section around 870 nm (Figure 1C).
TABLE 1:
Properties of cyan fluorescent proteins and long Stokes shift red fluorescent proteins.
| One-photon property | Two- photon property | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fluorescent protein | Emission maximum (nm) | Quantum yield (QY) | Excitation maximum (nm) | Extinction coefficient ε(M−1 cm−1) | One-photon brightness, QY × ε × 10−3 | Two-photon excitation maximum (nm) | Two-photon cross section, σ2P (GM) | Two- photon brightness, σ2P (GM) × QY | Photostability half-time (s) | pKa | Maturation half-time (h) |
| ECFP (Ai et al., 2006) | 475 | 0.52a | 433 | 33,000 | 13.5 | 857 | 23a | 12a | 64 | 4.7 | n.d. |
| Cerulean (Rizzo et al., 2004) | 475 | 0.58a | 433 | 43,000 | 27.5 | 858 | 23a | 13a | 36 | 4.7 | n.d. |
| mTFP1 (Ai et al., 2006) | 492 | 0.84a | 462 | 64,000 | 54.4 | 875 | 70a | 59a | 163 | 4.3 | n.d. |
| mTurquoise2 (Goedhart et al., 2012) | 474 | 0.93 | 434 | 30,000 | 27.9 | 860 | 30 | 28a | 256 | 3.1 | n.d. |
| hmKeima (this study) | 618 | 0.21 ± 0.03 | 430 | 34,100 | 7.2 | 870 | 73 | 15 | n.d. | 5.97 | 4.0 |
| hmKeima4.15 (this study) | 612 | 0.29 ± 0.04 | 436 | 28,000 | 8.1 | 870 | 60 | 17 | n.d. | 5.18 | 3.8 |
| hmKeima8.5 (this study) | 612 | 0.34 ± 0.05 | 438 | 32,000 | 10.9 | 870 | 65 | 22 | n.d. | 5.31 | 3.8 |
Four cyan fluorescent proteins (ECFP, Cerulean, mTFP1, and mTurquoise) and three long Stokes shift red proteins (hmKeima, hmKeima4.15, and hmKeima8.5) are compared with regard to various single- and two-photon properties.
Intracellular molecular brightness comparison
To perform measurements for intracellular molecular brightness, we used a custom measurement setup (Figure 2A; Materials and Methods; Saunders et al., 2012; Gaglia et al., 2013; Kayatekin et al., 2014). Molecular brightness is a crucial parameter for FCS/FCCS. The particle brightness (molecular brightness) is calculated by division of the mean fluorescence intensity (<I(t)>, in counts per second [cps]) by the average number of particles/molecules (N) present in the observation volume (Figure 2D). The high molecular brightness of bright fluorophores provides higher signal-to-noise (S/N) ratios, especially in the intracellular environment in which autofluorescence adds to the background. In addition, brighter FPs require lower laser power and hence allow for reduced exposure of the cells to potentially harmful irradiation, which also reduces photobleaching.
FIGURE 2:

Intracellular molecular brightness of 2P-excitable FPs. (A) Schematic of 2PE-FCS/FCCS setup. A Ti:sapphire femtosecond laser (tuned to 850 nm) is focused into the cellular cytoplasm through a high–numerical aperture objective. The fluorescence from the sample is collected back through the same objective and separated by a dichroic mirror to two detectors (photomultiplier tubes [PMTs]). Two-photon absorption reduces the excitation volume to <1 fl, <0.1% of the cell volume. (B) Fluorescence images of mTFP1 and hmKeima8.5 under identical excitation in individual cells. (C) FCS calculation. The autocorrelation algorithm is applied to fluctuating fluorescence signals, generating the FCS curve (in gray), G(τ). The fitting results (in red) yield the diffusion coefficient D (derived from the characteristic decay time) and the average molecular number N (derived from the inverse of the amplitude, G(0)). The average molecular brightness is obtained by average intensity <I(t)> divided by N. (D) Comparison of intracellular molecular brightness for cyan FPs and mKeima mutants as measured by FCS. mTFP1 and hmKeima8.5 were chosen for further characterization.
To quantify the molecular brightness, the blue FPs (ECFP, mCerulean, mTFP1, and mTurquoise2) and the large Stokes shift red FPs (LssmKate2, mKeima, hmKeima4.15, and hmKeima8.5) were transiently transfected into PtK2 cells, and the molecular brightness measurements were performed at 850-nm 2PE (Materials and Methods). Cells to be used for molecular brightness analysis were identified by wide-field imaging for expression of fluorescent proteins. Representative 1PE fluorescence images mTFP1 and hmKeima8.5 in PtK2 cells were consistent with free cytoplasmic intracellular distribution (Figure 2B).
The time autocorrelation (FCS) curve G(τ) (also written as GBlue×Blue(τ) or GRed×Red(τ), indicating blue–blue or red–red correlated fluorescence emission, respectively) was derived from fluorescence intensity traces and analyzed by fitting with a time autocorrelation decay function for a three-dimensional (3D) diffusing species for the characteristic decay time τD and the amplitude G(0) as free parameters (Figure 2C, formulae; Materials and Methods). The diffusion coefficient (D) that characterizes the mobility of particles was calculated from τD (Schwille et al., 1999; Krichevsky and Bonnet, 2002; Materials and Methods). The effective particle number N (average number of labeled particles in the detection volume) is proportional to the inverse of the amplitude G(0) (Figure 2C, arrow). The molecular brightness was then calculated as described.
mTFP1 and hmKeima8.5 were found to be the brightest FPs from the blue- and red-emitting candidates and to have high photostability without photobleaching during data acquisition (Figure 2D). mTFP1 and hmKeima8.5 provide similar molecular brightness of ∼5000 counts/molecule/s, which is twice as bright as ECFP and mKeima, respectively. Therefore mTFP1 and hmKeima8.5 were further characterized as fluorescence probes in intracellular 2PE-FCS studies. Of note, the in vitro brightness measurements of hmKeima and its variants showed only a modest difference in brightness (Supplemental Figure S2) and two-photon cross section (Table 1). The origin of this difference between in vivo and in vitro brightness may be attributable to differences in pH between the intracellular environments, although all mKeima mutants showed lower pKa values than the parental mKeima protein (Supplemental Figure S3).
Calibrated measurements of intracellular homodimerization
To measure protein dimerization by single-color 2PE-FCS, we established a calibration system using an mTFP1 monomer and an mTFP1-mTFP1 tandem dimer. The tandem dimer was generated by linking two mTFP1 fragments through a (GGGS)2 linker (Figure 3A). Fluorescence images for mTFP1 and mTFP1-mTFP1 were similar by epifluorescence imaging (Figure 3B). The molecular brightness measurements via 2PE-FCS were able to resolve the difference of the molecular oligomerization state. In one example, two cells expressing either mTFP1 or mTFP1-mTFP1 exhibited similar 2PE-FCS curve G(0) amplitudes and therefore similar intracellular FP concentrations (Figure 3C). However, the mTFP1-mTFP1 cells had an ∼70% increase in mean fluorescence intensities due to the increased brightness per FP protein construct expressed (inset). Alternatively, a second set of cells exhibited similar mean fluorescence intensities (Figure 3D, inset). In this case, the mTFP1-mTFP1 cells had a time autocorrelation curve of twice the G(0) amplitude, indicating half the number of particles in the excitation volume. The average intracellular molecular brightness for dimeric mTFP1-mTFP1 was ∼50% brighter per particle than that of the monomeric mTFP1 (Figure 3E). A further increase in tandem dimer brightness was seen in cell lysates, indicating that expected doubling of brightness in the tandem dimer may not be achieved due to factors in the intracellular environment (Supplemental Figure S4). The expected molecular brightness from a mixture of monomeric (mTFP1) and dimeric (mTFP1-mTFP1) species would be between these limits.
FIGURE 3:

Intracellular single-color FCS calibration. (A) mTFP1 monomer and mTFP1-mTFP1 tandem dimer (GGGSGGGS linker) constructs. (B) Fluorescence image of mTFP1 monomer in single cells and fluorescence image of mTFP1-mTFP1 tandem dimer in single cells. (C) Representative intracellular FCS curves from cells with similar molecular number of mTFP1 and mTFP1-mTFP1. Insets show similar molecular number, but the intensity of mTFP1-mTFP1 cells is about double that of mTFP1. (D) Representative intracellular FCS curves from cells with similar fluorescence intensities of mTFP1 and mTFP1-mTFP1. Insets show similar fluorescence intensity, but the molecular number of mTFP1-mTFP1 cells is about half that of mTFP1. (E) Comparison of average intracellular molecular brightness of mTFP1 and mTFP1-mTFP1.
Measurements of induced protein homodimerization by single-color FCS
To demonstrate the practical application of molecular brightness analysis for protein dimerization studies, we constructed a set of inducible dimerization protein constructs (Figure 4A). Open reading frames of mTFP1-(GGGS)2-FKBP12 (FK506-binding protein 12) and mTFP1-(GGGS)2-FRB (FKBP12-rapamycin binding domain) were linked by a viral P2A peptide. The P2A peptide sequence acts as a translational self-cleaving site to permit nearly stoichiometric expression of the protein upstream and downstream of the sequence (Kim et al., 2011). As a result, mTFP1-FKBP12 and mTFP1-FRB were coexpressed at nearly equal levels in the same cell after transfection. The formation of FKBP12-FRB complexes was controlled by the addition of the small-molecule dimerizer rapamycin (Table 2).
FIGURE 4:

Intracellular induced dimerization measured by molecular brightness. (A) Schematic of induced heterodimerization of mTFP1-FKBP12 and mTFP1-FRB constructs by rapamycin. The two proteins are coexpressed from one vector with a viral P2A cleavage site. (B) Measurement of intracellular molecular brightness increase of mTFP1-FKBP12-P2A-mTFP1-FRB by rapamycin. Control measurements were made with a control construct (mTFP1-P2A-mTFP1) with rapamycin addition. (C) Schematic of homodimerization of FKBP12-hmKeima8.5 by a FKBP12 bifunctional binder (MAZ1889). An excess of monofunctional binder (MAZ1258) can displace the bifunctional binder. (D) Measurement of brightness increase induced by the bifunctional binder and decrease in monomer brightness by addition of excess monofunctional binder.
TABLE 2:
Chemical structures of four FKBP12 binder compounds.
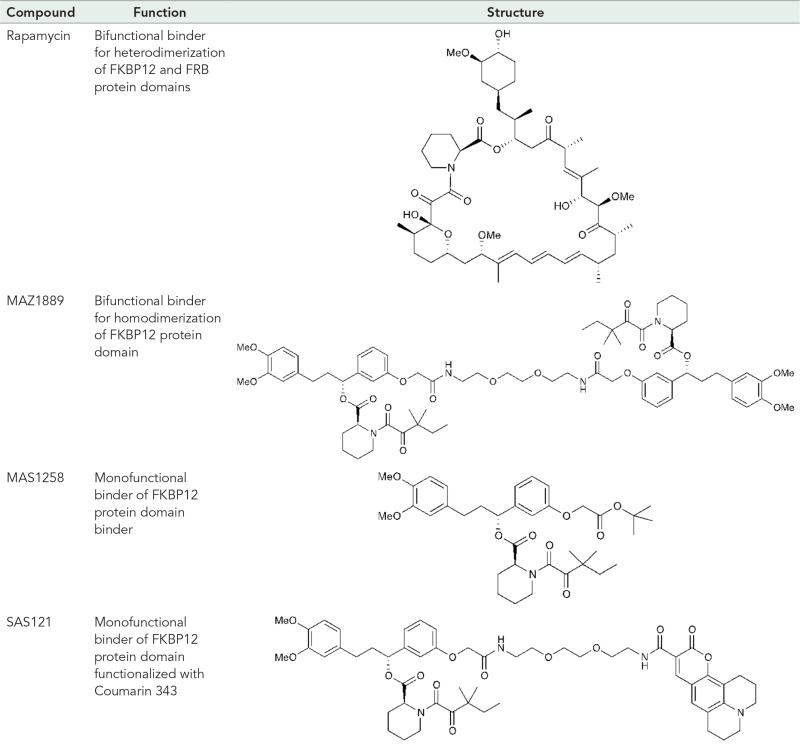 |
The molecular brightness measured by 2PE-FCS in the absence of rapamycin showed no difference between the mTFP1-FKBP12-P2A-mTFP1-FRB and the control construct mTFP1-P2A-mTFP1 (no dimerization domains; Figure 4B), and both had similar molecular brightness as monomeric mTFP1. This indicated no detectable dimerization of the mTFP1 complex in the absence of rapamycin and also demonstrated the high level of cleavage by the P2A peptide. On addition of 1 μM rapamycin, cells expressing mTFP1-FKBP12-P2A-mTFP1-FRB exhibited an increase in molecular brightness similar to that seen for the tandem mTFP1-mTFP1 dimer measured previously (Figure 4B). There was no significant change in molecular brightness in the control vector expressing mTFP1-P2A-mTFP1, indicating the specificity of the dimerization domains. These results demonstrate that intracellular dimerization events can be detected by 2PE-FCS molecular brightness analysis.
Using a bifunctional FKBP12-binding small molecule, termed here MAZ1889 (Table 2), we also measured the homodimerization of an FKBP12-hmKeima8.5 construct in cells (Figure 4C). The expression of FKBP12-(GGGS)2-hmKeima8.5 produced a protein with molecular brightness consistent with monomeric hmKeima8.5 (Figure 4D). The addition of 1.33 μM bifunctional binder MAZ1889 (Table 2) resulted in an increased molecular brightness (55%). Further addition of excess (10 μM) FKBP12 monofunctional binder MAZ1258 (Table 2) competed off the bifunctional binder, leading to decreased molecular brightness (Figure 4D). Together the heterodimerization and homodimerization (rapamycin or MAZ1889, respectively) experiments indicated that single-color 2PE-FCS with mTFP1 or hmKeima8.5 could be used to measure intracellular dimerization.
Intracellular dual-color FCCS calibration
The time cross-correlation (FCCS) function GRed×Blue(τ) (or GBlue×Red(τ)) is computed using the fluctuation traces from the two spectrally distinct fluorescently emitting species (Materials and Methods). The association between these two species can produce fluorescence emission in both channels, providing a positive cross-correlation signal. The amplitudes of FCCS curves GRed×Blue(0) (or GBlue×Red(0)) are proportional to the association (or codiffusion) between the two labeled species normalized by the amplitude of the individual autocorrelation amplitudes, or NRed×Blue/(NRedNBlue) (or NBlue×Red/(NBlueNRed)).
FCCS measurements permit the detection and quantification of hetero-oligomers between different molecular species. Taking advantage of the spectrally resolvable emission of the mTFP1/hmKeima8.5 pair, we generated expression constructs for mTFP1-(GGGS)2-hmKeima8.5 (tandem dimer) and mTFP1-P2A-hmKeima8.5 (free monomers) for 2PE-FCCS measurements (Figure 5A). Both proteins express within individual cells without obvious difference by wide-field epifluorescence microscopy (Figure 5A). The measurements from cells expressing hmKeima8.5 alone indicated no leakage of hmKeima8.5 emission into the blue channel (Figure 5B) and a small amount (∼3%) leakage of mTFP1 emission into the red channel. This level of single-color cross-talk was very small compared with the brightness of the tandem mTFP1-hmKeima8.5 construct, which showed almost equal intensity detection in each channel. The molecular brightness measurements from individual channels of mTFP1 and hmKeima8.5 were similar to monomeric values, indicating no change due to tandem dimerization (Figure 5C). These molecular brightness measurements also verify the monomeric state of mTFP1 and hmKeima8.5 in the intracellular environment.
FIGURE 5:

Intracellular dual-color FCCS calibration. (A) Comparison of tandem mTFP1-hmKeima8.5 and mTFP1-P2A-hmKeima8.5 constructs. Both vectors express mTFP1 and hmKeima8.5 with similar intracellular distributions in fluorescence images. (B) The percentage of total intensities from two channels for expression of mTFP1 alone, hmKeima8.5 alone, and mTFP1-hmKeima8.5 tandem. (C) Intracellular brightness comparison of mTFP1, hmKeima8.5, and mTFP1-hmKeima8.5. (D) Representative autocorrelation and cross-correlation curves of intracellular mTFP1-hmKeima8.5 expression from red channel. (E) Representative autocorrelation and cross-correlation curves of intracellular mTFP1-hmKeima8.5 expression from blue channel. (F) Measurement of the percentage of dual-color molecules normalized to total number of individual color molecules.
The representative autocorrelation and cross-correlation curves from PtK2 cells expressing mTFP1-hmKeima8.5 construct are shown for red (Figure 5D) and blue (Figure 5E) channels. Cross-correlation amplitudes of NRed×Blue/(NRedNBlue) for GRed×Blue(0) from the red channel and NBlue×Red/(NBlueNRed) for GBlue×Red(0) from the blue channel were multiplied by NRed and NBlue obtained from the amplitudes of autocorrelation curves. Consequently, four hetero-oligomer ratios were generated: NRed×Blue/NRed, NBlue×Red/NRed, NRed×Blue/NBlue, and NBlue×Red/NBlue. These ratios exhibited the expected increased levels for cells expressing the tandem mTFP1-hmKeima8.5 construct over cells expressing the mTFP1-P2A-hmKeima8.5 construct, where the cross signals show background levels (Figure 5F). In addition, the ratios were independent of the manner in which they were computed, by using either GRed×Blue(0) or GBlue×Red(0). Of note, there is a difference between NRed×Blue/NRed and NRed×Blue/NBlue (Figure 5F). This would indicate that for the tandem dimer, some blue signals are not correlated with a red signal. This can be accounted for by the slow maturation of hmKeima8.5 (3.8 h) compared with that of mTFP1 (0.5 h). This unmatched maturation time can accumulate mTFP1-hmKeima8.5 tandem dimers as fluorescent mTFP1 fused to a nonfluorescent hmKeima8.5.
Measurements of induced biomolecular heterodimerization by two-color FCCS
A practical intracellular application of single-excitation 2PE-FCCS was tested in cells by expressing the hmKeima8.5-(GGGS)2-FKBP12 and mTFP1-(GGGS)2-FRB with P2A linker. The schematic in Figure 6A shows how rapamycin can induce the formation of a dual-color complex between hmKeima8.5-FKBP12 and TFP-FRB. On the addition of 1 μM rapamycin, the cross-correlation signals increased over pretreatment signals when measured in the same cell (Figure 6B). There were no cross signals in the control construct of mTFP1-P2A-hmKeima8.5 before or after rapamycin treatment (Figure 6C).
FIGURE 6:

Intracellular induced dimerization measured by FCCS. (A) Schematic of induced heterodimerization of hmKeima8.5-FKBP12 and mTFP1-FRB by rapamycin. (B) Cross-correlation curves, comparison before and after addition of rapamycin. (C) Rapamycin induced dimerization measured by the percentage of dual-color molecules over total molecule number of hmKeima8.5. The binding of FKBP12 and FRB causes increase in dual-color percentage. A control construct (mTFP1-P2A-hmKeima8.5) under rapamycin addition is unchanged. (D) Time course of heterodimerization in individual cells upon addition of different concentrations of rapamycin. (E) Two cross-correlation curves from cell 1 at 45 and 75 min after treatment with 100 nM rapamycin. (F) Schematic of association of Coumarin 343–conjugated monofunctional binder (SAS121) with FKBP12-hmKeima8.5, followed by competition with excess nonfluorescent monofunctional binder (MAZ1258). (G) FCCS Measurements show the complex of SAS121 with FKBP12-hmKeima8.5 and dissociation upon excess addition of MAZ1258.
Three time courses in three individual cells with different concentrations of rapamycin (0.1, 0.33, and 1 μM) showed the increased rate of heterodimerization as a function of rapamycin concentration (Figure 6D). Two typical FCCS curves (Figure 6E) at two time points from a cell treated with 0.1 μM rapamycin indicated background and significant levels of association.
To investigate the feasibility of direct observation of small-molecule–protein interactions, we labeled a FKBP12 monofunctional binder with Coumarin 343 by chemical synthesis (SAS121; Table 2 and Supplemental Figure S1). The incubation of hmKeima8.5-FKBP12 transient transfected cells with SAS121 enabled us to detect the FCCS signal between Coumarin 343 (on the small molecule) and hmKeima8.5 (on the protein target; Figure 6F). Excess unlabeled monofunctional binder competed the dye-labeled drug off the drug–protein complex, reducing the cross-correlation to untreated levels (Figure 6, F and G).
DISCUSSION
Multicolor labeling with genetically encoded proteins in living cell is a powerful tool for investigating intracellular processes. Multicolor fluorescence microscopy requires multicolor excitation. However, for two-color FCCS, this requires optimal alignment of the two 1PE lasers in order to excite the fluorophores in the same focal volume element: imprecise alignment, as well as the wavelength dependence of the volume element size, causes systematic errors that often cannot be fully corrected (Bacia and Schwille, 2007).
Using the new hmKeima8.5-mTFP1 fluorophore combination for intracellular FCCS largely overcomes this problem. The improved hmKeima8.5 has a large Stokes shift (excitation/emission at 440/612 nm) and shares the same optimal excitation spectrum for one-photon and two-photon excitation with mTFP1 in vivo. However, both FPs also have well-separated emission spectra, which is bigger than that of the EGFP/mKeima pair that has been used before as an FP pair (Kogure et al., 2008). These well-separated emission spectra reduce the level of cross-correlation observed by channel cross-talk. The new FP pair is also brighter than the ECFP/mKeima FCCS pair (Kogure et al., 2006). In another previously proposed pair, TagRFP/mKalama1, the TagRFP was rather unstable under multiphoton excitation (Tillo et al., 2010). Together with the advantage of two-photon excitation due to lower photodamage or photobleaching, as well as reduced autofluorescence background compared with one-photon excitation, mTFP1 and hmKeima8.5 are excellent new candidates for multicolor emission detection with single-color 2P excitation.
The original mKeima was converted from its parental tetrameric form through seven mutations (L60Q, F61L, V79F, T92S, T123E, Y188R, and Y190E) that prevent the formation of homotetrameric or homodimeric complexes. In addition, the folding efficiency was increased through mutation (V191I; Kogure et al., 2006). The new improved hmKeima8.5 has kept all these sites except for R188L (R189L in our sequence; Figure 1A). As such, we expect that hmKeima8.5 will have a similar propensity for a monomeric state as mKeima itself. Moreover, if there were significant dimer formation, we would have detected an increase in the mTFP1 molecular brightness when the mTFP1-hmKeima8.5 tandem dimer was measured in living cells (Figure 5C), which was not the case. Although we cannot rule out a dimer transition at higher concentrations—for example, through nonspecific hydrophobic interactions—the data support a monomeric form for the concentrations tested in living cells for FCCS applications.
To test these FPs in FCS applications, we applied single-color autocorrelation brightness analysis (2PE-FCS) and were able to detect the rapamycin-mediated dimerization of mTFP1 constructs and the small molecule–mediated homodimerization of hmKeima8.5 with a bifunctional FKBP12 binder. We also detected the formation of mTFP1-hmKeima8.5 heterodimers through cross-correlation analysis. In addition, we were able to detect interactions between a fluorescent small molecule labeled with Coumarin 343 and its protein target labeled with hmKeima8.5 via cross-correlation analysis.
Previous approaches to intracellular FCCS methods have been difficult to implement due to complexities associated with different excitation volumes for different excitation wavelengths. Our work here highlighted the development of a new fluorescent protein, hmKeima8.5, for use in FCCS methods for detecting intracellular interactions. The long Stokes shift and overlap with mTFP1 excitation provide for robust, quantitative FCCS implementations. Our work has shown utility in detecting homodimeric, heterodimeric, and small-molecule–protein interactions. Moreover, the constructs we have developed provide a robust calibration toolbox for characterizing 2PE-FCCS implementations. We hope that together these results will reinvigorate the use of this sensitive and quantitative method for detecting biomolecular interactions in living cells.
MATERIALS AND METHODS
Directed evolution of mKeima
Directed evolution of mKeima was performed using DNA shuffling (Crameri et al., 1996) and yeast as a host for protein expression, essentially as described previously (Knop et al., 2002). Fluorescence-activated cell sorting (FACS) for the enrichment of brighter mKeima variants was performed using a MoFlo cell sorter (Beckman Coulter, Krefeld, Germany) or an Aria III from BD Bioscience (Heidelberg, Germany). Fluorescence measurements in single colonies were performed using a custom-made, camera-based fluorescence imaging light box for whole-plate imaging or a Tecan (Männedorf, Switzerland) M1000 Pro fluorescence plate reader for single-colony measurements. Fluorescence spectra of brighter mKeima variants were acquired directly from yeast cell colonies using the Tecan M1000 Pro in order to validate that the long Stokes shift characteristic of the spectra was unchanged.
Measurement of mKeima maturation kinetics
Fluorescence intensity time curves of yeast strains expressing mKeima, mKeima4.15, and mKeima8.5 from the inducible yeast Gal1 promoter were acquired as a function of promoter up-regulation as previously described for mCherry (Khmelinskii et al., 2012; Supplemental Tables S1 and S2). Briefly, yeast cells carrying the Gal1 promoter–driven mKeima variants on a low-copy plasmid were grown in synthetic complete medium (SC) containing 2% raffinose medium to mid-log-growth phase (∼107 cells/ml). Cells were then loaded on a Y04C microfluidics plate (CellAsic/Merck Millipore, Billerica, MA). After induction of mKeima expression by addition of SC medium containing 2% galactose and 2% raffinose, mKeima fluorescence was imaged for 8 h using a DeltaVision Microscope (GE Healthcare Life Sciences, Chalfont St. Giles, United Kingdom) and a 100×/1.4 numerical aperture (NA) PlanApo objective (Olympus, Tokyo, Japan) and a fluorescence filter set for mKeima imaging. Manual single-cell segmentation and tracking was performed using ImageJ (National Institutes of Health, Bethesda, MD). After background correction, a one-step maturation model (Khmelinskii et al., 2012) was fitted to the experimental data using MATLAB (MathWorks, Waltham, MA) and employing the D2D package (Raue et al., 2013). Maturation rate constants and induction time point were assigned as open parameters, whereas the dilution rate was determined from the average doubling time of 120 min, assuming an infinite half-life for protein degradation of mKeima. Maturation rate constants were fitted globally to every cell trajectory, whereas the induction time point was determined individually for each cell.
Characterization of evolved mKeima
mKeima variants were purified from Escherichia coli using pRSET-based expression (Supplemental Table S1), HIS6 affinity chromatography, and Talon resin and procedures recommended by the manufacturer (Clontech, Mountain View, CA). Individual fluorescence spectra and pKa values were measured in black 96-well glass-bottom plates (MGB096-1-2-LG-L; Brooks, Spokane, WA) using a Tecan M1000 Pro fluorescence plate reader. Wells were coated for 10 min with 150 μl 10% bovine serum albumin (BSA) and washed twice with phosphate-buffer saline (PBS; or buffer with corresponding pH) before the sample was applied into the well.
For measuring the fluorescence spectra, purified protein was applied (13 μg/well from wild-type mKeima and 3 μg/well for mKeima4.15/8.5) in 150 μl of PBS containing 1% BSA.
Emission spectra were measured from 500 to 800 nm using a fixed excitation at 440 nm. Excitation spectra were measured between 350 and 550 nm using a fixed emission at 620 nm. All fluorescence spectra were measured using a step size of 2 nm and a bandwidth of 5 nm). Background fluorescence was measured using buffer only (from PBS plus 1% BSA) and subtracted from the measurements. Smoothing of curves was done using a sliding window average of eight neighboring measurements.
pKa values were measured using buffers containing 50 mM Na2HPO4, 50 mM sodium acetate, and 50 mM glycine, with a pH range between 4 and 9, as described by Patterson et al. (1997). Purified protein was applied (1 and 5 μg/well mKeima4.15/8.5 and mKeima, respectively) in 150 μl of buffer with the corresponding pH containing 1% BSA. Measurements were recorded using excitation/emission wavelengths of 440/620 nm, respectively, and a bandwidth for the emission and detection of 10 nm. Fluorescence background from the corresponding buffer containing 1% BSA was subtracted, and Excel Solver was used to fit a curve to the data points.
Two-photon characterization of fluorescence proteins
Two-photon absorption (2PA) spectra of purified proteins (pH 8 buffer solution) and cross sections were measured using a previously described femtosecond fluorescence technique (Drobizhev et al., 2011). Briefly, an Opera Solo optical parametric amplifier (Coherent, Santa Clara, CA) was pumped with a Libra regenerative amplifier (Coherent). The second harmonic of idler was used to excite the sample in the region 800–1050 nm. The fluorescence was recorded with a Triax 550 spectrometer (Jobin Yvon, Edison, NJ) coupled with a cooled charge-coupled device detector (Spectrum One, Edison, NJ). The relative method was used to obtain corrected 2PA spectral shapes and cross sections (Makarov et al., 2008). Rhodamine B in methanol was used as a reference standard (Makarov et al., 2008) for hmKeima, hmKeima4.15, hmKeima8.5, and LSSmOrange. Fluorescein buffer solution (pH 11) was used as a reference standard (Makarov et al., 2008) for mTurquoise2.
One-photon absorption spectra were measured using a Lambda 950 UV/vis/NIR spectrometer (PerkinElmer, Waltham, MA). One-photon excited fluorescence spectra were measured using an LS 50B luminescence spectrometer (PerkinElmer). The peak extinction coefficients of hmKeima, hmKeima4.15, hmKeima8.5, and LSSmOrange were evaluated using a stepwise alkaline denaturation method (Kredel et al., 2008) and the known maximum extinction coefficient of the denatured green FP chromophore in alkaline solution (Ward, 2006). Fluorescence quantum yields were measured versus fluorescein buffer solution (pH 11) of quantum yield ϕF = 0.95 (Lakowicz, 2006).
FCS/FCCS setup
Two-photon FCS/FCCS was performed on a customized setup based on an inverted Nikon TE2000 microscope. The collimated 850-nm IR laser beam (Mai Tai, Ti:sapphire laser with 80 MHz and 100-fs pulse width; Spectra-Physics, CA) was aligned into a Nikon 100× Plan Apochromat oil immersion objective (NA 1.4) with back aperture slightly overfilled, creating a diffraction-limited focal spot. The laser power was controlled at ∼3 mW in vivo to avoid photobleaching of the fusion protein, cellular photodamage, and DNA damage. In vitro FCS power was 6 mW without photobleaching. The collected fluorescence by the same objective was passed through a dichroic filter (560dcxr; Chroma Technology Corp., Bellows Falls, VT) for splitting into two photomultiplier tubes (H7421; Hamamatsu, Hamamatsu, Japan) with individual band-pass filters (HQ485/70m-2p for mTFP1 and HQ620/60m-2P or HQ660/100m-2p for hmKeima; Chroma Technology Corp.). The HQ620/60m-2P emission filter was used for hmKeima-only measurements, whereas HQ660/100m-2p was used to minimize cross-talk between mTFP1 and hmKeima8.5 emission channels. Each autocorrelation/cross-correlation curve measured in cytoplasm was collected for 10 s using a Flex02-01D/C correlator (correlator.com, Bridgewater, NJ) and transferred to a personal computer through a high-speed USB port. For averaging purposes, at least five FCS curves were recorded at each position spatially and temporally in single cells, and the averaged curves are shown in the figures. The PMT dead time is 70 ns (∼14 MHz), and the correlator dead time is 1.56 ns. The average frequency of photon counts in experiments was kept <400 kHz, which is far from the saturation of detector. The cellular background intensity was <1 kHz and had no contribution to autocorrelation or cross-correlation signals.
Mammalian plasmid construction
Open reading frames (ORFs) for ECFP (Clontech, Mountain View, CA), hmKeima (MBL International, Woburn, MA), mCerulean (David Piston, Vanderbilt University, Nashville, TN), mTFP1 (Jeff Litchman, Harvard University, Cambridge, MA), LSSmKate2 (Addgene, Cambridge, MA), and mTurquoise2 (Theodorus W.J. Gadella, Jr., University of Amsterdam, Amsterdam, Netherlands) were amplified by PCR and cloned by restriction enzyme digest and T4 DNA ligase–mediated ligation into the pcDNA3.1(-) mammalian expression vector (Life Technologies, Carlsbad, CA). hmKeima4.15 and hmKeima8.5 were synthesized with human codon bias based on the yeast ORFs (GeneArt, Life Technologies, Regensburg, Germany; Supplemental Table S3).
mTFP1, hmKeima4.15, and hmKeima8.5 ORFs were PCR amplified as NheI-HindIII fragments and inserted into pcDNA3.1(-) with the same sites. To construct the P2A vector, we inserted the P2A sequence by oligo ligation into pcDNA3.1(-), forming an NheI-EcoR1-P2A-BamHI-HindIII frame. We used NheI/EcoRI for the insert site before P2A and BamHI/HindIII for the insert site after P2A.
ORF1-(GGGS)2-ORF2 fragments (including mTFP1-(GGGS)2-mTFP1, mTFP1-(GGGS)2-FKBP12, mTFP1-(GGGS)2-FRB, FKBP12-(GGGS)2-hmkeima8.5, mTFP1-(GGGS)2-hmKeima8.5, and hmKeima8.5-(GGGS)2-FKBP12) were generated by two-step PCR. Forward primer of ORF1 and reversed primer of ORF1 containing GGGSGGGS were used for synthesis of ORF1-GGGSGGGS. Forward primer containing GGGSGGGS and reversed primer of ORF1 were used for synthesis of GGGSGGGS-ORF2. These two fragments, ORF1-GGGSGGGS and GGGSGGGS-ORF2, were further PCR-amplified together without new primers for construction of full-length ORF1-GGGSGGGS-ORF2 fragments (Supplemental Table S4). The products were ligated into the NheI/HindIII sites of pcDNA3.1(-) or NheI/EcoRI sites before P2A and BamHI/HindIII sites after P2A.
All plasmids constructed in pcDNA3.1(-) were verified by restriction endonuclease digestion (New England Biolabs, Ipswich, MA) and DNA sequencing (Brigham and Women's Hospital DNA Core Sequencing Facility, Boston, MA).
Mammalian cell culture and transfection for FCS measurement
PtK2 (Potorous tridactylus Kidney) cells were cultured in Advanced MEM with 2% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. Cells were seeded 1 d before the transfection to a final confluency of 50–80% in glass-bottom 35-mm dishes (35-mm Petri dish, 14-mm microwell, No. 1.5 coverglass; MatTek). Transient transfection of DNA plasmid into PtK2 cells was optimized with FuGENE 6 transfection reagent (Promega, Madison, WI). The ratio of DNA:FuGENE was 1 μg:6 μl. At 24 h after transfection, cell media were replaced with CO2-independent media (Life Technologies) for image acquisition or FCS/FCCS measurements.
Cell lysate experiments were performed using PtK cells or 293T cells. PtK cells were transfected for intracellular FCS/FCCS using FuGENE 6. The transfection reagents for 293T cells were Lipofectamine LTX and PLUS Reagents (Life Technologies). The 293T cells were cultured in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. The transfected cells were incubated for 2 d and harvested, washed in PBS, and lysed in swelling buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, with KOH, 5 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol, and 1× complete protease inhibitor cocktail tablet [Roche, Basel, Switzerland]). After swelling for 30 min on ice, the cells were then disrupted by freeze-thawing. Extracts were cleaned by centrifugation at 14,000 rpm for 30 min at 4°C. The supernatant was further cleaned by centrifugation at 40,000 rpm for 30 min at 4°C. The final supernatant was used for FCS measurement in vitro on a coverslip pretreated with a coating with 1% BSA for 3 min and washout with swelling buffer.
Cell imaging
Images were acquired on a Nikon Eclipse Ti microscope system (Nikon Instruments, Melville, NY) with a humidified chamber from InVivo Scientific (St. Louis, MO) to maintain a 37°C environment. The microscope was equipped with a Mercury arc lamp, a PlanApo 60XA (NA 1.4, Ph3 DM) oil objective, mTFP1 and Keima filter cubes (Chroma Technology Corp.), and cooled charge-coupled device camera (CoolSNAP HQ; Roper Scientific, Tucson, AZ). The microscopy system was controlled by Nikon Elements software (Nikon Instruments).
FCS measurement
FCS was carried out on a custom system as previously described (Saunders et al., 2012; Gaglia et al., 2013; Kayatekin et al., 2014). Briefly, a tunable Ti:sapphire femtosecond laser (Mai-Tai; Spectra Physics) was focused into single living cells through a high–numerical aperture objective and the emitted photons were separated by a dichroic mirror (560dcxr) into blue (HQ485/70 nm) and red components (HQ620/60 nm or HQ660/100m-2p). Using a fluorescent dye solution of known diffusion constant and concentration (fluorescein in a borate, pH 11, buffer), we determined the excitation volume using a Nikon 100× Plan Apochromat oil immersion objective with NA 1.4. For FCS measurements, glass-bottom dishes were mounted on the microscope stage. The cells were focused under bright-field illumination, and fluorescent cells were selected using wide-field excitation for TFP and hmKeima8.5.
hmKeima8.5 was tested for anomalous photophysical properties by varying the excitation power during FCS measurements in living cells. Over a range of powers relevant to live-cell measurements, we were unable to detect changes in the time autocorrelation function that would be indicative of a significant change in dark-state or triplet-state contributions (Supplemental Figure S5).
FCS/FCCS data analysis
All FCS curves were analyzed by custom-written Matlab code (MathWorks, Waltham, MA) using a nonlinear least-squares fitting algorithm. The fluorescence autocorrelation function G(τ) was calculated from
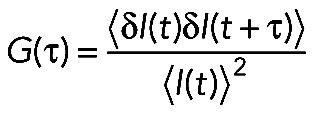 (1) (1)
|
where I(t) is the fluorescence intensity at time point t, and <I(t)> is the time average of fluorescence intensity. τ is the shift correlation time relative to time point t. The fitting formula for single-component diffusion was adapted from Krichevsky and Bonnet (2002) for autocorrelation analysis:
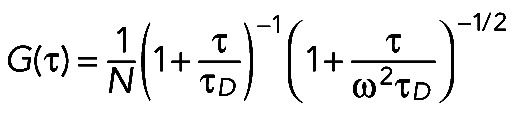 (2) (2)
|
where N is the average particle number of species in the sampling volume and τD is the residence time of species within the sampling volume, with τD = ωxy2/8D, where D is the diffusion coefficient of the species and ω = ωz/ωxy is the aspect ratio of the sampling volume.
Before the fit analysis, raw FCS autocorrelation curves were denoised by averaging the repeated collection of FCS curves. For single-species fit, each averaged FCS curve was fitted by the foregoing formula relating diffusion (D) and average particle number (N). Brightness was calculated by dividing the average fluorescence intensity by average particle number (N).
Cross-correlation Gij(τ) was calculated from
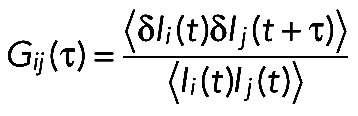 (3) (3)
|
where Ii(t) and Ij(t) are the fluorescence intensities at time point t for species i and species j, respectively, and the angle bracket is the time average of fluorescence intensity. τ is the shift correlation time relative to time point t. For cross-correlation analysis, the following fitting formulas were used for the hmKeima8.5 and mTFP1 channels:
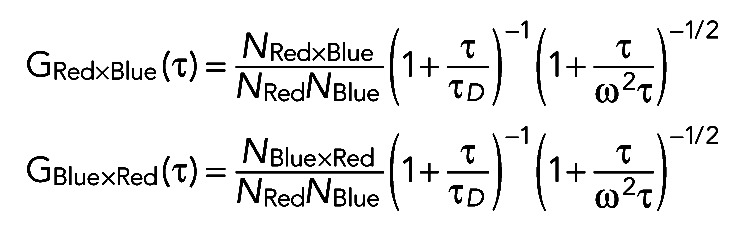 (4) (4)
|
NRed is the average particle number of all particles with color red, and NBlue is the average particle number of all particles with color blue. NRed and NBlue can be obtained by autocorrelation fitting in individual channels. NRed×Blue and NBlue×Red are the average particle numbers with dual color calculated from red and blue channels individually. The cross signals are the ratio of the number of particles with dual color to the total number of particles with single color, such as NRed×Blue/NRed, NBlue×Red/NRed, NRed×Blue/NBlue, and NBlue×Red/NBlue. Our measurements showed that NRed×Blue/NRed and NBlue×Red/NRed are very close, as are NRed×Blue/NBlue and NBlue×Red/NBlue.
Drug structure and synthesis
MAZ1258 and MAZ1889 were synthesized as reported previously (Keenan, 1998). SAS121 was synthesized following the same synthetic strategy with minor modifications. Briefly, MAZ1258 was treated with trifluoroacetic acid to yield the free carboxylic acid and coupled with excess 2,2′-(ethylenedioxy)-bis(ethylamine) to afford the amine-functionalized intermediate MAZ1266. MAZ1266 was coupled with Coumarin 343 using PyBOP and purified via reverse-phase flash chromatography (12-g C18 column [Biotage, Charlotte, NC]; linear gradient of 5−100% MeCN/H2O) to yield the desired product (Supplemental Figure S1).
Each compound was diluted into live-cell medium for the final concentration and designated time: MAZ1889 (1.33 μM for 60 min), MAZ1258 (10 μM for 60 min), and SAS121 (1.25 μM for 60 min).
Supplementary Material
Acknowledgments
We thank the Shah lab for helpful comments and also acknowledge the help of Geoffrey Wicks. This work was supported by funding from the National Institutes of Health (NS083875 to T.H., GM098083 to M.D., and GM77238 to J.V.S.), an FP7 EU Marie Curie Grant (Penelope) to M.K., a Beckman Laser SPARK Fellowship to Y.G., and SFRH/BD/80162/2011 from Fundação para a Ciência e Tecnologia, Portugal, to S.A.S.
Abbreviations used:
- FCCS
fluorescence cross-correlation spectroscopy
- FCS
fluorescence correlation spectroscopy
- FP
fluorescent protein
- mTFP
monomeric teal fluorescent protein
- 1PE
one-photon excitation
- 2PE
two-photon excitation.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-10-1473) on April 15, 2015.
*Present address: Biomedical Sciences Institute, Agency for Science, Technology and Research, Singapore 138632.
REFERENCES
- Ai H-W, Henderson JN, Remington SJ, Campbell RE. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochem J. 2006;400:531–540. doi: 10.1042/BJ20060874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacia K, Schwille P. Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat Protocols. 2007;2:2842–2856. doi: 10.1038/nprot.2007.410. [DOI] [PubMed] [Google Scholar]
- Boeke D, Trautmann S, Meurer M, Wachsmuth M, Godlee C, Knop M, Kaksonen M. Quantification of cytosolic interactions identifies Ede1 oligomers as key organizers of endocytosis. Mol Syst Biol. 2014;10:756–756. doi: 10.15252/msb.20145422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crameri AA, Whitehorn EAE, Tate EE, Stemmer WPW. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Two-photon absorption properties of fluorescent proteins. Nat Methods. 2011;8:393–399. doi: 10.1038/nmeth.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia G, Guan Y, Shah JV, Lahav G. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci USA. 2013;110:15497–15501. doi: 10.1073/pnas.1311126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, van Weeren L, Gadella TWJ, Royant A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93% Nat Commun. 2012;3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim RR, Tsien RYR. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:5–5. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Kawano H, Kogure T, Abe Y, Mizuno H, Miyawaki A. Two-photon dual-color imaging using fluorescent proteins. Nat Methods. 2008;5:373–374. doi: 10.1038/nmeth0508-373. [DOI] [PubMed] [Google Scholar]
- Kayatekin C, Matlack KES, Hesse WR, Guan Y, Chakrabortee S, Russ J, Wanker EE, Shah JV, Lindquist S. Prion-like proteins sequester and suppress the toxicity of huntingtin exon 1. Proc Natl Acad Sci USA. 2014:201412504. doi: 10.1073/pnas.1412504111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan T, Yaeger DR, Courage NL, Rollins CT, Pavone ME, Rivera VM, Yang W, Guo T, Amara JF, Clackson T, et al. Synthesis and activity of bivalent FKBP12 ligands for the regulated dimerization of proteins. Bioorg Med Chem. 1998;6:1309–1335. doi: 10.1016/s0968-0896(98)00125-4. [DOI] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol. 2012;30:708–714. doi: 10.1038/nbt.2281. [DOI] [PubMed] [Google Scholar]
- Kim SA, Heinze KG, Bacia K, Waxham MN, Schwille P. Two-photon cross-correlation analysis of intracellular reactions with variable stoichiometry. Biophys J. 2005;88:4319–4336. doi: 10.1529/biophysj.104.055319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee S-R, Li L-H, Park H-J, Park J-H, Lee KY, Kim M-K, Shin BA, Choi S-Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Barr F, Riedel CG, Heckel T, Reichel C. Improved version of the red fluorescent protein (drFP583/DsRed/RFP) BioTechniques. 2002;33 doi: 10.2144/02333rr02. 592, 594, 596–598 passim. [DOI] [PubMed] [Google Scholar]
- Kogure T, Karasawa S, Araki T, Saito K, Kinjo M, Miyawaki A. A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat Biotechnol. 2006;24:577–581. doi: 10.1038/nbt1207. [DOI] [PubMed] [Google Scholar]
- Kogure T, Kawano H, Abe Y, Miyawaki A. Fluorescence imaging using a fluorescent protein with a large Stokes shift. Methods. 2008;45:223–226. doi: 10.1016/j.ymeth.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Kredel S, Nienhaus K, Oswald F, Wolff M, Ivanchenko S, Cymer F, Jeromin A, Michel FJ, Spindler K-D, Heilker R, et al. Optimized and far-red-emitting variants of fluorescent protein eqFP611. Chem Biol. 2008;15:224–233. doi: 10.1016/j.chembiol.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Krichevsky O, Bonnet G. Fluorescence correlation spectroscopy: the technique and its applications. Rep Prog Phys. 2002;65:251–297. [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Springer; 2006. [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Maeder CI, Hink MA, Kinkhabwala A, Mayr R, Bastiaens PIH, Knop M. Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat Cell Biol. 2007;9:1319–1326. doi: 10.1038/ncb1652. [DOI] [PubMed] [Google Scholar]
- Magde D, Elson E, Webb W. Thermodynamic fluctuations in a reacting system—measurement by fluorescence correlation spectroscopy. Phys Rev Lett. 1972;29:705–708. [Google Scholar]
- Makarov NS, Drobizhev M, Rebane A. Two-photon absorption standards in the 550–1600 nm excitation wavelength range. Opt Express. 2008;16:4029–4047. doi: 10.1364/oe.16.004029. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J. 1997;73:2782–2790. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Hulit J, Subach OM, Wu B, Abdulla A, Segall JE, Verkhusha VV. Monomeric red fluorescent proteins with a large Stokes shift. Proc Natl Acad Sci USA. 2010;107:5369–5374. doi: 10.1073/pnas.0914365107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raue A, Schilling M, Bachmann J, Matteson A, Schelke M, Kaschek D, Hug S, Kreutz C, Harms BD, Theis FJ, et al. Lessons learned from quantitative dynamical modeling in systems biology. PLoS One. 2013;8:e74335. doi: 10.1371/journal.pone.0074335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigler R, Mets Ü, Widengren J, Kask P. Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur Biophys J. 1993;22:169–175. [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Saunders TE, Pan KZ, Angel A, Guan Y, Shah JV, Howard M, Chang F. Noise reduction in the intracellular pom1p gradient by a dynamic clustering mechanism. Dev Cell. 2012;22:558–572. doi: 10.1016/j.devcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P, Haupts U, Maiti S, Webb WW. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- So PTC, Dong CY, Masters BR, Berland KM. Two-photon excitation fluorescence microscopy. Annu Rev Biomed Eng. 2000;2:399–429. doi: 10.1146/annurev.bioeng.2.1.399. [DOI] [PubMed] [Google Scholar]
- Tillo SE, Hughes TE, Makarov NS, Rebane A, Drobizhev M. A new approach to dual-color two-photon microscopy with fluorescent proteins. BMC Biotechnol. 2010;10:6. doi: 10.1186/1472-6750-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WW. Biochemical and physical properties of green fluorescent protein. Methods Biochem Anal. 2006;47:39–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.