

A genome-wide analysis is given of DNA methylation in mouse embryonic stem cells in which both Gsk-3α and Gsk-3β have been genetically deleted. DNA methylation patterns are compared to those of wild-type cells. More than 75% of known imprinted loci have reduced DNA methylation in the Gsk-3–knockout cells.
Abstract
Glycogen synthase kinase-3 (Gsk-3) is a key regulator of multiple signal transduction pathways. Recently we described a novel role for Gsk-3 in the regulation of DNA methylation at imprinted loci in mouse embryonic stem cells (ESCs), suggesting that epigenetic changes regulated by Gsk-3 are likely an unrecognized facet of Gsk-3 signaling. Here we extend our initial observation to the entire mouse genome by enriching for methylated DNA with the MethylMiner kit and performing next-generation sequencing (MBD-Seq) in wild-type and Gsk-3α−/−;Gsk-3β−/− ESCs. Consistent with our previous data, we found that 77% of known imprinted loci have reduced DNA methylation in Gsk-3-deficient ESCs. More specifically, we unambiguously identified changes in DNA methylation within regions that have been confirmed to function as imprinting control regions. In many cases, the reduced DNA methylation at imprinted loci in Gsk-3α−/−;Gsk-3β−/− ESCs was accompanied by changes in gene expression as well. Furthermore, many of the Gsk-3–dependent, differentially methylated regions (DMRs) are identical to the DMRs recently identified in uniparental ESCs. Our data demonstrate the importance of Gsk-3 activity in the maintenance of DNA methylation at a majority of the imprinted loci in ESCs and emphasize the importance of Gsk-3–mediated signal transduction in the epigenome.
INTRODUCTION
Glycogen synthase kinase-3 (Gsk-3) enzymes Gsk-3α and Gsk-3β are intracellular kinases that together regulate the activity of numerous signal transduction pathways (Kockeritz et al., 2006). The genetic deletion of Gsk-3α and Gsk-3β (referred to hereafter as Gsk-3 double knockout [Gsk-3 DKO]; Doble et al., 2007) in mouse embryonic stem cells (ESCs) revealed the importance of Gsk-3 activity in the regulation of pluripotency; Gsk-3 DKO ESCs are unable to undergo differentiation, even in the absence of leukemia inhibitory factor (Doble et al., 2007). A recently identified novel role for Gsk-3 activity is the regulation of DNA methylation via N-myc and the de novo DNA methyltransferase 3a, isoform 2 (Dnmt3a2), in mouse ESCs (Popkie et al., 2010). Although initially identified in Gsk-3 DKO ESCs, further experimentation showed that treating wild-type ESCs with a small-molecule inhibitor of Gsk-3 activity also reduced DNA methylation, as did constitutive activation of phosphatidylinositol 3-kinase (PI3K), which also inhibits Gsk-3 activity (Popkie et al., 2010). It remains an open question as to whether reduced DNA methylation contributes to the regulation of pluripotency in mouse ESCs.
Imprinted loci are distinct from other alleles due to their retention of DNA methylation, which is inherited from either the mother (maternal) or father (paternal; Lawson et al., 2013). De novo methyltransferase activity has been shown to be required for the establishment and maintenance of DNA methylation at imprinted loci (Chen et al., 2003; Kaneda et al., 2004), and, as predicted, DNA methylation in Gsk-3 DKO ESCs was reduced at two imprinted loci, H19/Igf2 and Igf2r/Air (Igf2 = insulin-like growth factor 2; Popkie et al., 2010). There are ∼150 imprinted genes identified in mice (Williamson et al., 2013), and we sought to determine whether the reduced DNA methylation in Gsk-3 DKO ESCs extended to other imprinted loci. The development of DNA methylation capture based on affinity to the methyl-binding domain of human methyl-CpG binding domain 2 (MBD2; Baubec et al., 2013), combined with next-generation DNA sequencing technology (MBD-Seq; Mardis, 2009), allowed us to ask which regions of the genome display DNA hypomethylation in Gsk-3 DKO ESCs.
Here we present the genome-wide analysis of regions with reduced DNA methylation in Gsk-3 DKO ESCs. Consistent with our earlier study, we find DNA hypomethylation at a majority of the known imprinted genes in the mouse genome. Moreover, the MethylMiner–Sequencing by Oligonucleotide Ligation and Detection (SOLiD) workflow allowed us to demonstrate that reduced DNA methylation in Gsk-3 DKO ESCs occurred precisely at differentially methylated regions (DMRs), which were previously shown to contain the experimentally defined imprinting control regions (ICRs) for multiple imprinted genes (Fitzpatrick et al., 2002; Yoon et al., 2002; Coombes et al., 2003). We provide extensive experimental validation of bioinformatically identified DMRs by performing bisulfite sequencing. In addition, we quantitatively measure gene expression for 91 imprinted genes in Gsk-3 DKO ESCs and correlate the changes in gene expression with changes in DNA methylation. Furthermore, we find that many Gsk-3–dependent DMRs map precisely to DMRs identified in uniparental ESCs (Hiura et al., 2010). In addition, we show that many nonimprinted genes, such as the pluripotency-related transcription factor Klf4 (Takahashi and Yamanaka, 2006) and the entire Hoxa locus (Soshnikova et al., 2013), have reduced DNA methylation in Gsk-3 DKO ESCs. Finally, we compare the DMRs obtained from this study with genes identified as having parent-of-origin bias in the mouse brain (Gregg et al., 2010) and find an extensive correlation between the data sets. The data presented here offer a global view of the effects on DNA methylation in Gsk-3 DKO ESCs and provide a framework for an integrative analysis of gene expression and DNA methylation status that should be useful for better understanding the effect of signal transduction pathways on epigenetic modifications.
RESULTS
MBD-Seq in wild-type and Gsk-3 DKO ESCs
We previously observed decreased DNA methylation at the ICRs for H19/Igf2 and Igf2r/Air in Gsk-3 DKO ESCs due to reduced expression of the de novo DNA methyltransferase Dnmt3a2 (Popkie et al., 2010). On the basis of these findings, we reasoned that decreased Dnmt3a2 expression might also be affecting the maintenance of DNA methylation at other imprinted loci in a Gsk-3xdependent manner. To investigate this possibility, we chose an unbiased approach to locating all of the regions of the mouse genome that have reduced DNA methylation in ESCs. MethylMiner uses a biotinylated recombinant fragment of the human MBD2 protein to enrich for fragments of methylated DNA. Therefore MethylMiner was used to obtain methylated genomic DNA from wild type (WT) and Gsk-3 DKO ESCs. After shearing high–molecular weight genomic DNA into 200– to 250–base pair fragments, we captured methylated DNA on biotinylated recombinant human MBD2 bound to streptavidin-coated Dynabeads. After washing away unmethylated DNA, consecutive elutions were performed under low-salt (600 mM NaCl) and high-salt (2 M NaCl) conditions to liberate DNA from the MBD2 protein. The 2 M fraction is expected to contain the DNA fragments that are most highly methylated, since the lower salt will elute DNA that is less tightly bound to the beads, that is, less methylated. Using 15 μg of input genomic DNA, we recovered ∼9% of the DNA in the 600 mM and 2 M fractions combined (Supplemental Figure S1).
The eluted DNA was then used to prepare a library for next-generation sequencing using a SOLiD instrument. We prepared one library each of the WT and Gsk-3 DKO 600 mM fraction and two libraries from each of the WT and Gsk-3 DKO 2M fraction. In addition, we also created libraries from unenriched input DNA from each cell line. SOLiD sequencing yielded >630,000,000 reads, averaging almost 80,000,000 reads per library. After removing clonal reads, the number of unique starts ranged from 2 × 107 to 2.5 × 107 in the 600 mM fraction and from 6 × 106 to 8 × 106 in the 2 M fraction (Supplemental Table S1). This yielded ∼4–5% coverage of the genome having >1x unique starts (Supplemental Figure S2).
Peak detection by Model-based Analysis for ChIP-Seq (MACS) was used to identify genomic regions where DNA methylation was significantly reduced in the Gsk-3 DKO ESCs compared with WT ESCs. Focusing on the 2 M fraction, we identified 29,029 DMRs, which had a mean size of 439 base pairs and together comprised 12.7 Mb, or 0.50% of the mouse genome (Supplemental Table S2). The mean score for these peaks was 128, and the SD was 121. The DMRs were further subdivided into those that were 1 SD or higher than the mean (1SD; i.e., scoring >249) and 2 SDs or higher than the mean (2SD; i.e., scoring >370). Using these metrics, we obtained a total of 2545 peaks (8.8% of all peaks identified) that were 1SD (total size 1.6 Mb). Of these, 993 peaks were 2SD (total size 683 kb; Supplemental Table S2). As expected, the same analysis of DMRs identified from the 600 mM fraction yielded an even larger number of DMRs: 3929 peaks that were 1SD, and, of these, 1540 peaks that were 2SD (Supplemental Table S2). Given the smaller number of peaks and the greater enrichment for the greatest amount of DNA methylation, we focused on the 1SD and 2SD peaks from the 2 M faction for the remainder of our analyses.
Examination of imprinted loci for Gsk-3-dependent DMRs
We next set out to analyze bioinformatically the MethylMiner-SOLiD data. Because we previously demonstrated a role for Gsk-3 in the maintenance of DNA methylation at the imprinted loci H19/Igf2 and Igf2r/Air (Popkie et al., 2010), we first asked whether DNA methylation was reduced at additional imprinted loci. ICRs are known to function both locally and at a distance (Lewis and Reik, 2006; Edwards and Ferguson-Smith, 2007), and therefore we determined whether there were any Gsk-3–dependent DMRs within 250 kb of a known imprinted gene. Of the 102 imprinted genes we examined, we found that 77% (78 of 102) had Gsk-3–dependent DMRs nearby (Supplemental Table S3). Both maternally imprinted and paternally imprinted loci were essentially equally affected (36 paternally imprinted genes and 42 maternally imprinted genes), which suggested that Gsk-3 activity maintains DNA methylation at imprinted loci regardless of the parent-of-origin imprints.
We were particularly satisfied to find that this unbiased approach to analyzing DNA methylation revealed hypomethylation in Gsk-3 DKO ESCs at the H19/Igf2 and Igf2r/Air DMRs that we had analyzed previously (Popkie et al., 2010). In fact, we saw remarkable concordance between the location of experimentally defined ICRs and Gsk-3-dependent DMRs, including the Kcnq1/Kcnq1ot1 (Fitzpatrick et al., 2002), Gnas/Nespas (Coombes et al., 2003), and Rasgrf1 (Plass et al., 1996) loci. Raw data showing the mapping of individual sequencing reads from WT and Gsk-3 DKO ESCs at the Gnas/Nespas locus, along with those regions called peaks, are shown in Figure 1A.
FIGURE 1:

MBD-Seq in WT and Gsk-3 DKO mouse embryonic stem cells. (A) Bam files visualized in Integrative Genome Viewer (IGV) showing aligned sequence reads from WT and Gsk-3α−/−;Gsk-3β−/− (DKO) ESCs at the Gnas/Nespas locus, whose exon/intron structure is also shown. Vertical red bars represent differentially methylated peaks as determined by MACS. (B) Cartoon schematic of the Gnas/Nespas locus, showing the DMR locations as identified by MACS (in red), along with the region validated by bisulfite sequencing (in blue). Bisulfite sequencing results for this region in WT and Gsk-3 DKO ESCs are represented by lollipop diagrams. Each circle represents a CpG dinucleotide; horizontal rows represent individual clones, and vertical columns show the position of each CpG. Filled-in circles represent methylated cytosines, and open circles represent unmethylated cytosines. (C) qPCR data showing relative quantification of Gnas and Nespas gene expression in WT and Gsk-3 DKO ESCs compared with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Each experiment shown was performed in triplicate.
Independent validation of MBD-Seq data via bisulfite sequencing
Given that Gsk-3-dependent DMRs were identified computationally at the end of our MethylMiner-SOLiD workflow, it was critical to use an independent method to confirm the validity of these DMRs. To do this, we used conventional bisulfite conversion of genomic DNA, followed by PCR, cloning, and Sanger sequencing (Frommer et al., 1992). We selected 20 DMRs for validation, corresponding to 14 distinct genomic loci, which were obtained via MethylMiner-Seq. We included the H19 DMR, previously shown to be Gsk-3 dependent (Popkie et al., 2010), as a positive control. Of the remaining 19 DMRs, 15 were from the 2 M fraction and four were from the 600 mM fraction (Table 1). Several known imprinted genes—Blcap/Nnat, Gnas, Mest/Copg2, Peg3, Kcnq1ot1, and Begain—were analyzed. We also examined additional genes of interest that contained Gsk-3-dependent DMRs—Pax3, En1, Adar, Klf4, Cdc42, Fkbp6, and Mycn. Our goal was to obtain at least 12 nonidentical clones from both WT and Gsk-3 DKO ESCs for each DMR. Representative data showing the locations of Gsk-3-dependent DMRs at the Gnas/Nespas (Coombes et al., 2003), Mest/Copg2 (Lefebvre et al., 1997), Blcap/Nnat, Kcnq1ot1 (Fitzpatrick et al., 2002), and Klf4 (Takahashi and Yamanaka, 2006) loci, as well as the regions that were validated by bisulfite sequencing, are shown in Figures 1B and 2, A–D. For all of the Gsk-3-dependent DMRs tested, we found statistically significant differences in DNA methylation between WT and Gsk-3 DKO ESCs (Table 1, p > 0.05, two-tailed Student's t test). Further increasing our confidence that DMRs identified in Gsk-3 DKO ESCs accurately denoted genomic regions involved in imprinting, two Gsk-3-dependent DMRs were found ∼30 kb upstream of the Rasgrf1 gene (Figure 3), a region previously identified experimentally to contain a differentially methylated ICR (Plass et al., 1996). (The Rasgrf1 DMR was unable to be validated by bisulfite sequencing due to the presence of repetitive elements that prevented successful PCR.)
TABLE 1:
Summary of Gsk-3–dependent DMR validation by bisulfite sequencing.
| Locus | DMR genomic coordinates | WT% CpG | DKO% CpG | Fraction |
|---|---|---|---|---|
| Pax3 | chr1:78191696-78192257 | 51.8 | 15 | 2 M |
| En1 | chr1:122496266-122497436 | 45.3 | 18.7 | 2 M |
| Blcap/Nnat | chr2:157385054-157386455 | 77.4 | 8.1 | 2 M |
| Gnas | chr2:174120416-174122205 | 63.6 | 5.8 | 2 M |
| Gnas | chr2:174122344-174123811 | 43.2 | 18.7 | 2 M |
| Gnas | chr2:174123824-174124999 | 36.8 | 8.7 | 2 M |
| Gnas | chr2:174125041-174126654 | 67.3 | 17.5 | 2 M |
| Adar | chr3:89471020-89471670 | 80.4 | 15.6 | 2 M |
| Klf4 | chr4:55540960-55541232 | 86.3 | 13.0 | 2 M |
| Cdc42 | chr4:136927927-136928474 | 69.0 | 35.6 | 2 M |
| Fkbp6 | chr5:135725089-135726550 | 88.8 | 48.7 | 600 mM |
| Mest/Copg2 | chr6:30682552-30683326 | 80.7 | 13.0 | 2 M |
| Mest/Copg2 | chr6:30685155-30685990 | 86.3 | 19.1 | 2 M |
| Peg3 | chr7:6680997-6682243 | 44.4 | 1.1 | 600 mM |
| H19 | chr7:149825745-149826024 | 47.2 | 24.4 | 600 mM |
| Kcnq1 | chr7:150480888-150482586 | 90.7 | 35.1 | 2 M |
| Kcnq1 | chr7:150491439-150492052 | 97.1 | 87.2 | 600 mM |
| Kcnq1 | chr7:150526408-150527088 | 89.0 | 75.3 | 600 mM |
| Mycn | chr12:12943555-12944660 | 54.2 | 13.0 | 2 M |
| Begain | chr12:110311312-110311914 | 81.2 | 24.3 | 2 M |
Genomic loci and chromosomal coordinates of each DMR. Also shown is the percentage of DNA methylation at each validated DMR in WT and Gsk-3 DKO ESCs and the fraction in which the DMR was identified (2 M or 600 mM). All p < 0.05.
FIGURE 2:

Reduced DNA methylation at imprinted loci in Gsk-3 DKO mouse ESCs. (A) Schematic of the Mest/Copg2 locus, showing the DMR locations as identified by MACS (in red), along with the region validated by bisulfite sequencing (in blue). Bisulfite sequencing results for this region in WT and Gsk-3 DKO ESCs are represented by lollipop showing relative quantification of Mest and Copg2 gene expression in WT and Gsk-3 DKO ESCs compared with GAPDH. Each experiment shown was performed in triplicate. (B) Schematic of the Blcap/Nnat locus, showing the DMR locations as identified by MACS (in red), along with the region validated by bisulfite sequencing (in blue). Bisulfite sequencing results for this region in WT and Gsk-3 DKO ESCs are represented by lollipop diagrams as described for Figure 1. qPCR data showing relative quantification of Blcap and Nnat gene expression in WT and Gsk-3 DKO ESCs compared with GAPDH. Each experiment shown was performed in triplicate. (C) Schematic of the Kcnq1 locus, showing the DMR locations as identified by MACS (in red), along with the region validated by bisulfite sequencing (in blue). Bisulfite sequencing results for this region in WT and Gsk-3 DKO ESCs are represented by lollipop diagrams as described for Figure 1. qPCR data showing relative quantification of Kcnq1 gene expression in WT and Gsk-3 DKO ESCs compared with GAPDH. Each experiment shown was performed in triplicate. (D) Schematic of the Klf4 locus, showing the DMR locations as identified by MACS (in red), along with the region validated by bisulfite sequencing (in blue). Bisulfite sequencing results for this region in WT and Gsk-3 DKO ESCs are represented by lollipop diagrams as described for Figure 1. qPCR data showing relative quantification of Klf4 gene expression in WT and Gsk-3 DKO ESCs compared with GAPDH. Each experiment shown was performed in triplicate.
FIGURE 3:
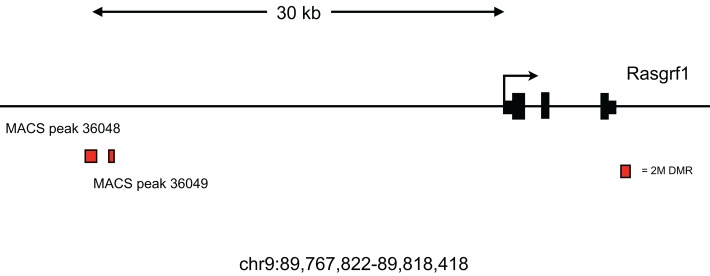
Rasgrf1 DMR identified in Gsk-3 DKO ESCs. Schematic representation of the murine Rasgrf1 genomic locus (chr9:89,767,822-89,818,418) and location of Gsk-3–dependent DMRs located ∼30 kb upstream of the gene in the vicinity of the ICR for Rasgrf1.
Functional enrichment analysis of Gsk-3–dependent DMRs
Because the 2 M DMRs had a greater level of statistical significance, we focused on the 2503 DMRs identified in this fraction to determine whether there were underlying functional relationships among the genes that were located near Gsk-3-dependent DMRs. We used ToppFun, within the ToppGene Suite, to perform this functional enrichment (Table 2 and Supplemental Table S4; Chen et al., 2009). The most common Gene Ontology (GO) Molecular Function was nucleic acid binding transcription factor activity (p = 8.99 × 10−22; Table 2). Of the 1011 terms (genes) associated with this function in the genome, 251 were represented in the 2 M DMR fraction. In fact, the top 9 GO terms all describe DNA-binding activity (only top 5 shown). More specifically, transcription factors containing a homeodomain were found to be highly enriched among the genes associated with Gsk-3–dependent DMRs (91 of 314 terms, p = 7.7 × 10−12), with the Fox (F-box) and Pax (Paired homeobox) gene families highly represented (14 of 43, p = 7.86 × 10−6, and 5 of 9, p = 7.11 × 10−3, respectively). The greatest enrichments for mouse phenotypes associated with the 2 M DMRs were abnormal nervous system development and abnormal heart morphology (195 of 779 terms, p = 6.6 × 10−6; 186 of 771 terms, p = 3.75 × 10−4, respectively; Table 2). Not surprisingly, given these two predicted categories of phenotypes, the most enriched biological process was embryo development (216 of 830 terms, p = 4.18 × 10−26). Of interest, enrichment analysis also revealed that Gsk-3–dependent DMRs were highly represented among the genes whose expression is affected by the pesticide vinclozolin, whose effects have been associated with transgenerational epigenetic effects in male rats (250 of 1263, p = 2.15 × 10−10; Anway et al., 2005). Similar gene enrichment results were found among the Gsk-3–dependent DMRs in the 600 mM fraction (Supplemental Table S4).
TABLE 2:
Summary of ToppFun functional enrichment analysis/2M fraction.
| Category | ID | p value |
|---|---|---|
| GO: Molecular function | ||
| Nucleic acid–binding transcription factor activity | GO:0001071 | 8.99 × 10−22 |
| Sequence-specific DNA-binding transcription factor activity | GO:0003700 | 1.59 × 10−21 |
| Sequence-specific DNA binding | GO:0043565 | 1.05 × 10−17 |
| Regulatory-region DNA binding | GO:0000975 | 3.89 × 10−11 |
| Regulatory-region nucleic acid binding | GO:0001067 | 3.89 × 10−11 |
| GO: Biological process | ||
| Embryo development | GO:0009790 | 6.93 × 10−19 |
| Regulation of developmental process | GO:0050793 | 3.64 × 10−18 |
| Organ morphogenesis | GO:0009887 | 5.52 × 10−17 |
| Regulation of multicellular organismal development | GO:2000026 | 8.69 × 10−17 |
| GO: Cellular component | ||
| Cell junction | GO:0030054 | 2.46 × 10−10 |
| Cell projection | GO:0042995 | 1.43 × 10−9 |
| Cell–cell junction | GO:0005911 | 1.89 × 10−8 |
| Neuron projection | GO:0043005 | 6.43 × 10−8 |
| Mouse phenotype | ||
| Abnormal nervous system development | MP:0003861 | 4.18 × 10−5 |
| Perinatal lethality | MP:0002081 | 1.35 × 10−4 |
| Domain | ||
| Homeodomain-like | IPR009057 | 7.70 × 10−12 |
| Homeodomain-rel | IPR012287 | 2.00 × 10−10 |
| Homeobox | IPR001356 | 8.68 × 10−10 |
| Interaction | ||
| SMAD3 interactions | int:SMAD3 | 3.08 × 10−5 |
| SMAD2 interactions | int:SMAD2 | 5.08 × 10−5 |
| MAPK1 interactions | int:MAPK1 | 7.13 × 10−5 |
| CTNNB1 interactions | int:CTNNB1 | 3.20 × 10−4 |
| Transcription factor–binding site | ||
| RTAAACA_V$FREAC2_01 | 3.06 × 10−6 | |
| RTTTNNNYTGGM_UNKNOWN | 1.60 × 10−5 | |
| TGGAAA_V$NFAT_Q4_01 | 3.82 × 10−5 | |
| Gene family | ||
| CD molecules | CD | 2.48 × 10−6 |
| Forkhead box genes | FOX | 7.86 × 10−6 |
| Drug | ||
| Vinclozolin | C025643 | 2.15 × 10−10 |
| 2-(1′H-Indolo-3′-carbonyl)thiazole-4-carboxylic acid methyl ester | C548651 | 2.40 × 10−10 |
| Mustard gas | D009151 | 4.46 × 10−10 |
| Calcitriol | D002117 | 1.86 × 10−9 |
Enrichment analysis of 2245 of 2503 genes located near a Gsk-3–dependent DMR from the 2 M fraction.
Comparison of Gsk-3-dependent DMRs with brain imprinted genes
Gregg et al. (2010) used RNA-Seq to identify ∼1300 mRNA transcripts in the mouse brain that exhibit a parent-of-origin bias, suggesting that these represent potential new imprinted genes. This study did not, however, examine DNA methylation, the primary epigenetic mark associated with genomic imprinting. Nonetheless, we performed a meta-analysis of their data and ours to determine whether there were Gsk-3–dependent changes in DNA methylation that accompanied the parent-of-origin changes in gene expression at these putative imprinted loci. As we had done previously, we determined which brain imprinted genes had Gsk-3–dependent DMRs within 250 kb of the gene body in mouse ESCs. Of the 822 protein-coding putative imprinted genes identified in the RNA-Seq study, 424 (52%) had a Gsk-3–dependent DMR in the proximity. Similarly, 61% (166 of 274) genes for noncoding RNAs had a Gsk-3–dependent DMR nearby (Figure 4). Taken together with our identification of 75% of previously known imprinted genes having Gsk-3–dependent DMRs, the correlation we see between Gsk-3–dependent DMRs in ESCs and putative brain imprinted genes strongly supports our hypothesis that Gsk-3 activity is critical for maintaining DNA methylation at a majority of loci subjected to imprinting. It is also worth noting the high concordance between the parent-of-origin bias in gene expression in the brain and the presence of DMRs at the same loci in ES cells. These data suggest that despite significant differences in cell types, the correlation between Gsk-3–dependent DMRs and imprinting is quite strong.
FIGURE 4:
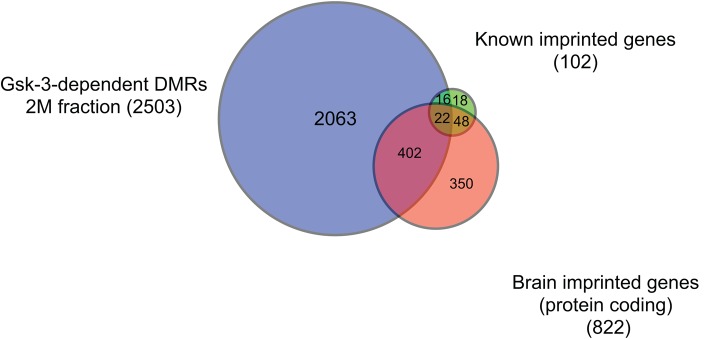
DMR comparison. Venn diagram showing the overlap between known imprinted genes (green), Gsk-3–dependent DMRs (blue), and brain imprinted genes (orange).
Comparison of Gsk-3-dependent DMRs with DMRs from uniparental ESCs
Obviously the most appropriate and informative comparison regarding imprinted genes would be to analyze DNA methylation differences in mouse ESCs. Fortunately, an experiment similar to ours was performed using methylated DNA immunoprecipitation-on-chip (meDIP-on-chip) in uniparental mouse ESCs (Hiura et al., 2010). Because imprints arise from inheriting DNA methylation of an allele from only one parent, deriving ESCs from embryos containing only the maternal (parthenogenetic [PG]) or paternal (androgenetic [AG]) genome is quite useful for the study of genomic imprinting (Mann et al., 1990). Using meDIP-on-chip with PG and AG ESCs, Hiura et al. (2010) identified 458 DMRs—141 paternally methylated DMRs and 317 maternally methylated DMRs—many of which corresponded exactly to known imprinted genes. Therefore we compared the location of uniparental DMRs with our Gsk-3-dependent DMRs. We were quite surprised to find that 96 of 458 mapped precisely to DMRs identified in Gsk-3 DKO ESCs (Supplemental Table S5). Furthermore, contrary to what was observed with our initial survey of known imprinted genes, the greatest degree of concordance between DMRs from the two data sets was seen with the maternal DMRs—83 of 317 DMRs (26.2%) overlap, whereas only 13 of 141 paternal DMRs (9.2%) overlap. Of the maternal overlapping DMRs, 17 are known to be at imprinted loci (Supplemental Table S5). Thus 66 DMRs that are shared between uniparental ESCs and Gsk-3 DKO ESCs map to the exact same genomic region but have not been identified as imprinted. Examples of overlapping DMRs are shown for the Gnas/Naspas and Nkx6-2 loci (Figure 5). The comparison of the uniparental DMRs and GSK-3–dependent DMRs provides an additional level of independent validation of our MBD-Seq approach and significantly strengthens our conclusion that we identified biologically significant DMRs in Gsk-3 DKO ESCs.
FIGURE 5:

Examples of overlapping DMRs from Gsk-3 DKO ESCs and uniparental ESCs. Gsk-3–dependent DMRs from the 2 M fraction are in red and from the 600 mM fraction are in blue, and the DMRs identified by Hiura et al. (2010), are in green. (A) Gnas/Nespas locus. (B) Tcf21 locus. (C) Nkx6-2 locus.
Quantitative analysis of imprinted gene expression in Gsk-3 DKO ESCs
Previous analysis of microarray gene expression data indicated that the expression of many imprinted genes was altered in Gsk-3 DKO ESCs relative to WT ESCs (Popkie et al., 2010; Bartman et al., 2014). We set out to validate the observed changes in gene expression by performing quantitative PCR (qPCR). Relative levels of gene expression was determined for 91 murine imprinted genes, with expression levels normalized to Gapdh. A majority of the genes (68 of 91) were down-regulated in Gsk-3 DKO ESCs, and of these, 37 were decreased in expression twofold or more compared with WT ESCs (Table 3). The greatest reductions in gene expression were for Kcnq1, Cmah, Htr2a, Grb10, Slc22a2, Cdkn1c, Slc22a3, Wt1, Dlx5, and Drd1a (Figure 2C and Table 3). The remainder (23 of 91) were up-regulated in Gsk-3 DKO ESCs, including seven that showed at least twofold increase in expression compared with WT ESCs (Table 3). The genes showing the largest increase in expression in Gsk-3 DKO ESCs were Peg10, Dhcr7, Klrb1f, Nespas, Xlr3c/Xlr3b/Xlr3a, Plagl1, and Xlr4b/Xlr4c/Xlr4a (Table 3). These qPCR data confirm that the deletion of Gsk-3 isoforms in mouse ESCs results in widespread changes in the expression of imprinted genes.
TABLE 3:
Summary of TaqMan qPCR data for imprinted genes in WT and Gsk-3 DKO ESCs.
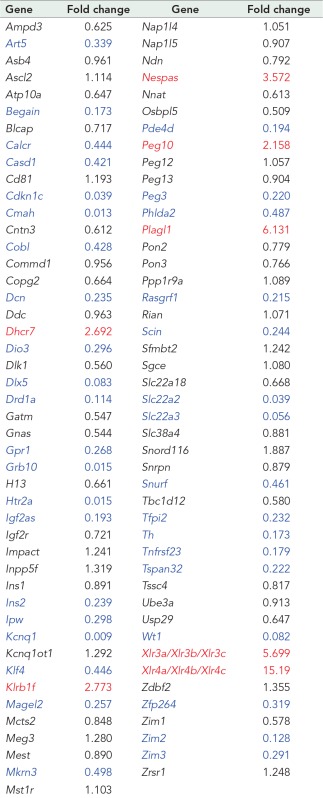 |
Values represent the fold change in Gsk-3 DKO ESCs vs. WT ESCs. For all assays, n = 9 (3 biological replicates and 3 technical replicates) for each gene. Genes in red are increased twofold or more; genes in blue are decreased twofold or more.
Analysis of the Hoxa cluster in Gsk-3 DKO ESCs
In addition to identifying DMRs associated with imprinted genes, we found a number of nonimprinted genes that exhibit Gsk-3–dependent reductions in DNA methylation, exemplified by the Hoxa gene cluster. Located on the distal arm of chromosome 6, the Hoxa cluster contains 11 homeobox genes within an ∼125-kb region. Strikingly, this region contained a relatively high number of Gsk-3-dependent DMRs—nine from the 2 M fraction and four from the 600 mM fraction (Figure 6A). We were curious to learn whether the presence of these DMRs correlated with changes in gene expression. Therefore we examined the expression of the genes in the Hoxa cluster in WT and Gsk-3 DKO ESCs by quantitative reverse transcription PCR (qRT-PCR) using TaqMan probes (three technical replicates on each of four biological samples; n = 12). Expression of all genes in the Hoxa cluster was decreased in Gsk-3 DKO ESCs compared with WT ESCs, and, with the exception of Hoxa1, expression of all of the Hoxa genes was decreased by at least 60% (Figure 6B). Taken together with the changes in gene expression and reduction in DNA methylation at imprinted loci, these data suggest that reductions in DNA methylation affect the transcription of genes in the Hoxa cluster and that this effect is influenced by Gsk-3 activity.
FIGURE 6:

Gsk-3-dependent DNA methylation and gene expression at the Hoxa locus. (A) Schematic representation of the Hoxa locus, along with the position of Gsk-3–dependent DMRs (2 M fraction in red, 600 mM fraction in blue). (B) qPCR data for all of the genes in the Hoxa cluster in WT and Gsk-3 DKO ESCs. For all results shown, RQ is relative quantification (all values were normalized to a GAPDH endogenous control); error bars represent SEM between biological replicates (n = 3) and technical replicates (n = 3; n = 9 total for each gene in each cell line).
Correlation between gene expression and DNA methylation in Gsk-3 DKO ESCs
Because we had both microarray gene expression data and MBD-Seq data from Gsk-3 DKO ESCs, we sought to address how much correlation there is between reduced DNA methylation and changes in gene expression. Of the 2503 genes from the 2 M fraction that showed Gsk-3–dependent reduction in DNA methylation and the 1536 genes whose expression changed twofold or more in Gsk-3 DKO ESCs versus WT ESCs, 299 were in common between the two studies (Figure 7). Therefore there appears to be a correlation between DNA methylation and gene expression for ∼10–15% of genes affected in Gsk-3 DKO ESCs. When we focus specifically on imprinted genes, Gsk-3xdependent DMRs are found near 64% (28 of 44) of imprinted genes whose expression increased or decreased at least twofold in the Gsk-3 DKO ESCs (Supplemental Table S6).
FIGURE 7:
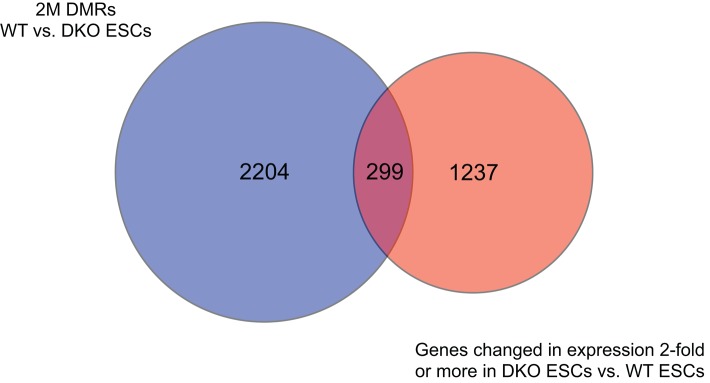
Integrating DNA methylation and gene expression. Venn diagram showing overlap between genes with Gsk-3–dependent DMRs (blue circle) and those genes whose expression is changed twofold or more in Gsk-3 DKO ESCs (orange circle).
DISCUSSION
The advent of new DNA sequencing technology (Mardis, 2009), combined with enrichment of methylated DNA (Harris et al., 2010), has provided a genome-wide view of the epigenetic landscape in mammalian cells (Meissner et al., 2008; Ball et al., 2009; Lister et al., 2009; Hawkins et al., 2010; Serre et al., 2010; Nair et al., 2011). Here we used that technology to examine the state of DNA methylation in mouse ESCs in which both Gsk-3α and Gsk-3β have been genetically deleted (Doble et al., 2007). Focusing specifically on regions of the mouse genome that were hypomethylated in Gsk-3 DKO ESCs compared with WT ESCs, we unambiguously identified Gsk-3–dependent DMRs in the immediate proximity of three-fourths of known imprinted genes, extending our previous observation of reduced DNA methylation at the imprinted H19/Igf2 and Igf2r/Airn loci in Gsk-3 DKO ESCs (Popkie et al., 2010). Because the DNA methylation at imprinted loci was already established by the time ESCs were isolated from mouse embryos, we believe that the reduction in DNA methylation is due not to a loss of imprinting but instead to an impairment in the maintenance of DNA methylation at these genomic regions. Furthermore, reexpression of human Gsk-3β in Gsk-3 DKO ESCs fully restores DNA methylation deficits observed at the H19/Igf2 locus (Supplemental Figure S3). In addition, treating WT ESCs with a small-molecule inhibitor of Gsk-3 activity results in a decrease in the activity of a reporter gene driven by the Dnmt3a2 promoter (Popkie et al., 2010; Supplemental Figure S4). Taken together, these data demonstrate the importance of Gsk-3 activity in the maintenance of DNA methylation at imprinted loci, which is likely due to reduced Dnmt3a2 expression (Popkie et al., 2010).
Of interest, not all imprinted genes exhibited Gsk-3-dependent DNA methylation. Notably absent from the list of Gsk-3–dependent DMRs is the imprinted cluster on proximal chromosome 7 that includes Atp10a, Ube3a, Ipw, Snurf, and Snrpn, as well as the Mirg cluster of miRNAs on chromosome 12 (Lewis and Reik, 2006; Edwards and Ferguson-Smith, 2007). This suggests that maintenance of DNA methylation at these ICRs is regulated by a mechanism that is independent of Gsk-3 activity, adding an additional layer of complexity to the process of maintaining DNA methylation at imprinted loci.
In addition, the sex chromosomes were surprisingly devoid of Gsk-3–dependent DMRs. The X chromosome showed only seven 2 M fraction DMRs (all 1SD), and eleven 600 mM fraction DMRs (five 2SD DMRs), whereas there were no Gsk-3–dependent DMRs identified on the Y chromosome. Of the DMRs identified on the X chromosome, none were in close proximity to the gene cluster containing Xlr3a, Xlr3b, Xlr3c, Xlr4a, Xlr4b, and Xlr4c, genes whose expression was found to be substantially up-regulated in Gsk-3 DKO ESCs compared with WT ESCs.
An initial genome-wide survey of DMRs drew our attention to the Hoxa locus because of the presence of 13 Gsk-3-dependent DMRs within a 125-kb genomic region. The identification of significant hypomethylation at the Hoxa gene cluster was particularly intriguing, given the functional importance of Hoxa genes in development and disease. Of particular relevance to this study, altered Hoxa DNA methylation and/or gene expression has been seen in pediatric acute lymphoblastic leukemia (ALL; Starkova et al., 2010) and chronic lymphocytic leukemia (CLL; Pei et al., 2012). Furthermore, mutations in DNMT3A have been shown to cause acute myeloid leukemia (AML; Ley et al., 2010), suggesting a role for aberrant DNA methylation in AML. Of interest, Gsk-3 inhibitors have been found to be candidate therapeutics for the treatment of mixed lineage leukemia (MLL; Wang et al., 2008). Although all of these leukemias are molecularly distinct and have different etiologies, it is of interest to note the connection between Gsk-3 activity, DNA methylation, and Hoxa gene expression that is common to ALL, CLL, MLL, and AML. Recent mechanistic studies on MLL suggest that regulation of MEIS1 activity via Gsk-3–dependent control of CREB, TORC, and CBP could represent a common molecular link between these disparate cancers (Wang et al., 2010).
Although our data provide evidence that maintenance of DNA methylation at ICRs previously identified for several imprinted genes is Gsk-3 dependent (Plass et al., 1996; Fitzpatrick et al., 2002; Coombes et al., 2003), the most compelling data come from the comparison of Gsk-3–dependent DMRs with DMRs identified in uniparental ESCs (Hiura et al., 2010). Whereas both maternal and paternal contributions to the mammalian genome have been shown to be required for embryogenesis (McGrath and Solter, 1984; Surani et al., 1984), PG and AG mice survive long enough for ESCs to be derived (Allen et al., 1994; Szabo and Mann, 1994), providing a valuable resource for the study of uniparental epigenetics and the process of imprinting. Although the Arima group used a slightly different technique (meDIP-on-chip; Hiura et al., 2010), the principle is similar to our MBD-Seq approach—enrichment of methylated DNA from different samples, followed by quantitative measurement of differences in DNA methylation between samples. We found a remarkable degree of overlap between the DMRs identified in these independent studies. In addition to a number of previously characterized imprinted genes, these studies suggest the possibility of additional imprinted genes. Using bisulfite sequencing, we validated the DMRs at one putatively imprinted gene, Engrailed 1 (En1), a transcription factor that is known to be an important regulator of embryonic development (Simon and Alavian, 2009). Further studies need to be performed to determine how many of the genes listed in Supplemental Table S5 are indeed imprinted.
Gsk-3 activity regulates multiple signal transduction pathways, such as Wnt and insulin signaling, and therefore is important for numerous biological processes (Kockeritz et al., 2006). We now provide conclusive evidence that ESCs deficient in Gsk-3α and Gsk-3β have reductions in DNA methylation throughout the genome, with particular enrichment at imprinted loci. Of interest, there appears to be a difference between inhibition of Gsk-3 via insulin or Wnt signaling. We previously showed that constitutive activation of PI3K results in reduced DNA methylation at the H19/Igf2 locus (Popkie et al., 2010), but this same effect is not seen in WT ESCs expressing a constitutively active form of β-catenin (Kelly et al., 2011; Supplemental Figure S5). It will be interesting to see whether other signaling pathways that also modulate Gsk-3α/β activity via phosphorylation of N-terminal serine residues (Cross et al., 1995) have a similar effect on the DNA methylation of imprinted genes. When DNA methylation levels are integrated with microarray gene expression data, we observe that changes in gene expression are correlated with reduced DNA methylation. Our findings should serve as a platform for further study of Gsk-3–dependent biological functions. Of particular note, the mood stabilizer lithium directly inhibits Gsk-3 activity (Klein and Melton, 1996; Stambolic et al., 1996) and was shown to affect DNA methylation at the H19/Igf2 ICR (Popkie et al., 2010). Given that psychiatric illnesses such as bipolar disorder and schizophrenia have been suggested to have alterations in genomic DNA methylation patterns (Petronis, 2010; Labrie et al., 2012), our data raise the possibility that the effectiveness of lithium in the treatment of bipolar disorder could be through influencing DNA methylation. Further studies on cells from patients with bipolar disorder, especially neuronal cells, would help to address this possibility.
MATERIALS AND METHODS
Methylated DNA enrichment, SOLiD library construction, and SOLiD sequencing
Genomic DNA was isolated from wild-type and Gsk-3 DKO ESCs and then sheared to 150– to 200–base pair fragments using a Covaris sonicator. The MethylMiner kit (Invitrogen, Carlsbad, CA) was used to enrich for methylated DNA. DNA was eluted from beads using both low-salt (600 mM NaCl) and high-salt (2 M NaCl) conditions. Eluted DNA was blunt-end repaired, and adaptors were ligated onto the DNA fragments. Emulsion PCR and bead enrichment were then performed, followed by DNA sequencing on a SOLiD instrument (Applied Biosystems, Foster City, CA) with 10 ligation cycles, using five primer rounds (McKernan et al., 2009). Fifty base pairs of each fragment were interrogated for sequence analysis.
Analysis of MethylMiner/SOLiD reads
Reads were mapped to reference genome mm9 using Bowtie (Langmead et al., 2009). Clonal reads were removed to obtain nonredundant starts. MACS software was used to find peaks in the mapped reads (Zhang et al., 2008). High-scoring peaks with significant differences in read pileups between WT and Gsk-3 DKO samples were considered as putative DMRs. Hypomethylated DMRs in Gsk-3 DKO ESCs were samples with fewer reads than WT ESCs for the same region. The mean and SD of significance scores (based on values) were determined, and the scoring peaks 1 or 2 SDs higher than the average score for the whole set were used to select for the most significant DMRs (Supplemental Table S2). To validate DMRs bioinformatically, the top scoring peaks were compared with true genome target regions (known imprinted loci) and false target regions. The numbers of hits in each class were calculated and compared with the numbers of hits expected by chance, given the size of the genome, the genomic footprint of each class, and the number of peaks.
Functional enrichment analysis
DMR functional enrichment studies were performed using ToppFun within the ToppGene Suite (toppgene.cchmc.org; Chen et al., 2009). ToppGene then performs functional enrichments, looking for sets of coregulated genes. Calculations are performed using false discovery rate for correction and a p-value cut-off of <0.05.
Bisulfite PCR primers
PCR primers for the amplification of the Gsk-3-dependent DMRs from bisulfite-converted genomic DNA were designed using the BiSearch primer design tool (bisearch.enzim.hu; Tusnady et al., 2005). The selected primers yielded a single amplified product for each desired region. Primer sequences are given in Supplemental Table S7.
Bisulfite sequencing
High–molecular weight genomic DNA was fragmented by rapid freezing on dry ice and thawing at 42°C for five repetitions, and 500 ng of fragmented genomic DNA was bisulfite-converted with the Cells-to-CpG Bisulfite Conversion Kit (Life Technologies, Carlsbad, CA) per the manufacturer's instructions. Genomic regions were amplified from bisulfite-converted DNA with Platinum Taq (Life Technologies) using primers shown in Supplemental Table S7 under the following conditions: 94°C for 10 min, 5 cycles of 94°C for 45 s/54°C for 45 s/72°C for 1 min, followed by 30 cycles of 94°C for 45 s/59°C for 45 s/72°C for 1 min) followed by 72°C for 7 min. PCR products were TA cloned into pCR2.1 (Life Technologies). Plasmid clones were sequenced with M13 Reverse primer at Eton Bioscience (San Diego, CA) and the Molecular and Cellular Imaging Center at Ohio State University (Wooster, OH). Resulting sequences were analyzed using MacVector (Cary, NC) software. Identical clones and clones containing partially bisulfite-converted DNA were easily identified by analyzing patterns of non-CpG DNA methylation, which was still present in WT and Gsk-3 DKO ESCs, and subsequently removed from analyses.
RNA isolation, cDNA synthesis, and quantitative PCR
TaqMan probes for 91 mRNAs (Williamson et al., 2013) were subjected to a two-step RT-PCR protocol. For each assay,100 ng of total RNA was used, and reactions were run on an ABI HT7900 real-time PCR instrument. RNA was isolated from 5 × 105 to 1 × 106 WT and Gsk-3 DKO ESCs using the MirVana Total RNA Isolation Kit (Applied Biosystems) according to the manufacturer's instructions. Then 100 ng of RNA was used for cDNA synthesis using the High Capacity cDNA reverse transcription kit (Applied Biosystems), following the manufacturer's protocol. Quantitative RT-PCR was performed on an ABI HT7900 real-time PCR instrument using TaqMan master mix and TaqMan assays (Applied Biosystems). TaqMan probe IDs are listed in Supplemental Table S8. Three biological replicates and three technical replicates were used for WT and Gsk-3 DKO ESCs. All threshold cycle (Ct) values were normalized to a mouse Gapdh endogenous control (Applied Biosystems), and relative quantification was calculated from the median Ct value (Schmittgen and Livak, 2008).
Microarray and statistical analysis of microarray data
RNA was measured in an ND-1000 UV-Vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA), and a 2100 Bioanalyzer Lab-On-A-Chip 6000 Series II chip (Agilent, Santa Clara, CA) was used to determine the integrity of the samples. RNA from each cell line was hybridized onto a SurePrint G3 Mouse GE 8 × 60K Microarray, AMADID 028005 (Agilent). Hybridization was performed overnight at 45ºC. We performed arrays for each cell line using RNA that was isolated in biological triplicate (n = 3). For Gsk-3 DKO ESCs, we used n = 4 biological replicates. SurePrint arrays were scanned with a G2505C Microarray Scanner (Agilent). Additional details can be found in Bartman et al. (2014).
Luciferase assay
Wild-type ESCs stably transfected with Dnmt3a2-luciferase (Popkie et al., 2010) were treated with 10 μM SB-415,286 for 3 d. After each day, cells were collected in luciferase lysis buffer and stored at −20°C. At the end of the time course, firefly luciferase assays were performed according to the manufacturer's protocol (Biotium, Hayward, CA) and measured in a Perkin Elmer-Cetus EnSpire Multimode Plate Reader. Three replicates were performed for each treatment group.
Supplementary Material
Acknowledgments
We thank Jeff Kuret for critical reading of the manuscript and John McLaughlin and Sigrid Eckardt for helpful suggestions and discussions. This work was supported in part by National Institute on Aging Grant R01AG031883 to C.J.P.
Abbreviations used:
- DMR
differentially methylated region
- Dnmt3a2
DNA methyltransferase 3a, isoform 2
- Gsk-3
glycogen synthase kinase-3
- Gsk-3 DKO
Gsk-3α and Gsk-3β double knockout
- Igf2
insulin-like growth factor 2
- ICR
imprinted control region.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-01-0013) on April 1, 2015.
Present addresses: *NanoString Technologies, Seattle, WA 98109
†Duke University, Durham, NC 27710
‡Van Andel Research Institute, Grand Rapids, MI 49503;
§Harvard University, Cambridge, MA 02138
‖Genentech, South San Francisco, CA 94080.
REFERENCES
- Allen ND, Barton SC, Hilton K, Norris ML, Surani MA. A functional analysis of imprinting in parthenogenetic embryonic stem cells. Development. 1994;120:1473–1482. doi: 10.1242/dev.120.6.1473. [DOI] [PubMed] [Google Scholar]
- Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman CM, Egelston J, Kattula S, Zeidner LC, D'Ippolito A, Doble BW, Phiel CJ. Gene expression profiling in mouse embryonic stem cells reveals glycogen synthase kinase-3-dependent targets of phosphotidylinositol 3-kinase and Wnt/beta-catenin signaling pathways. Front Endocrinol (Lausanne) 2014;5:133. doi: 10.3389/fendo.2014.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T, Ivanek R, Lienert F, Schubeler D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. 2013;153:480–492. doi: 10.1016/j.cell.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes C, Arnaud P, Gordon E, Dean W, Coar EA, Williamson CM, Feil R, Peters J, Kelsey G. Epigenetic properties and identification of an imprint mark in the Nesp-Gnasxl domain of the mouse Gnas imprinted locus. Mol Cell Biol. 2003;23:5475–5488. doi: 10.1128/MCB.23.16.5475-5488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D, Alessi D, Cohen P, Andjelkovich M, Hemmings B. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CA, Ferguson-Smith AC. Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol. 2007;19:281–289. doi: 10.1016/j.ceb.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32:426–431. doi: 10.1038/ng988. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura H, Sugawara A, Ogawa H, John RM, Miyauchi N, Miyanari Y, Horiike T, Li Y, Yaegashi N, Sasaki H, et al. A tripartite paternally methylated region within the Gpr1-Zdbf2 imprinted domain on mouse chromosome 1 identified by meDIP-on-chip. Nucleic Acids Res. 2010;38:4929–4945. doi: 10.1093/nar/gkq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- Kelly KF, Ng DY, Jayakumaran G, Wood GA, Koide H, Doble BW. beta-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell. 2011;8:214–227. doi: 10.1016/j.stem.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockeritz L, Doble B, Patel S, Woodgett JR. Glycogen synthase kinase-3—an overview of an over-achieving protein kinase. Curr Drug Targets. 2006;7:1377–1388. doi: 10.2174/1389450110607011377. [DOI] [PubMed] [Google Scholar]
- Labrie V, Pai S, Petronis A. Epigenetics of major psychosis: progress, problems and perspectives. Trends Genet. 2012;28:427–435. doi: 10.1016/j.tig.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14:609–617. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Surani MA. Genomic structure and parent-of-origin-specific methylation of Peg1. Hum Mol Genet. 1997;6:1907–1915. doi: 10.1093/hmg/6.11.1907. [DOI] [PubMed] [Google Scholar]
- Lewis A, Reik W. How imprinting centres work. Cytogenet Genome Res. 2006;113:81–89. doi: 10.1159/000090818. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann JR, Gadi I, Harbison ML, Abbondanzo SJ, Stewart CL. Androgenetic mouse embryonic stem cells are pluripotent and cause skeletal defects in chimeras: implications for genetic imprinting. Cell. 1990;62:251–260. doi: 10.1016/0092-8674(90)90363-j. [DOI] [PubMed] [Google Scholar]
- Mardis ER. New strategies and emerging technologies for massively parallel sequencing: applications in medical research. Genome Med. 2009;1:40. doi: 10.1186/gm40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37:179–183. doi: 10.1016/0092-8674(84)90313-1. [DOI] [PubMed] [Google Scholar]
- McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, Robinson MD, Clark SJ. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics. 2011;6:34–44. doi: 10.4161/epi.6.1.13313. [DOI] [PubMed] [Google Scholar]
- Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, Speir E, Awan F, Tae H, Arthur G, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7:567–578. doi: 10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- Plass C, Shibata H, Kalcheva I, Mullins L, Kotelevtseva N, Mullins J, Kato R, Sasaki H, Hirotsune S, Okazaki Y, et al. Identification of Grf1 on mouse chromosome 9 as an imprinted gene by RLGS-M. Nat Genet. 1996;14:106–109. doi: 10.1038/ng0996-106. [DOI] [PubMed] [Google Scholar]
- Popkie AP, Zeidner LC, Albrecht AM, D'Ippolito A, Eckardt S, Newsom DE, Groden J, Doble BW, Aronow B, McLaughlin KJ, et al. Phosphatidylinositol 3-kinase (PI3K) signaling via glycogen synthase kinase-3 (Gsk-3) regulates DNA methylation of imprinted loci. J Biol Chem. 2010;285:41337–41347. doi: 10.1074/jbc.M110.170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–399. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HH, Alavian KN. Transcriptional regulation of their survival: the Engrailed homeobox genes. Adv Exp Med Biol. 2009;651:66–72. doi: 10.1007/978-1-4419-0322-8_6. [DOI] [PubMed] [Google Scholar]
- Soshnikova N, Dewaele R, Janvier P, Krumlauf R, Duboule D. Duplications of hox gene clusters and the emergence of vertebrates. Dev Biol. 2013;378:194–199. doi: 10.1016/j.ydbio.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett J. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Starkova J, Zamostna B, Mejstrikova E, Krejci R, Drabkin HA, Trka J. HOX gene expression in phenotypic and genotypic subgroups and low HOXA gene expression as an adverse prognostic factor in pediatric ALL. Pediatric Blood Cancer. 2010;55:1072–1082. doi: 10.1002/pbc.22749. [DOI] [PubMed] [Google Scholar]
- Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308:548–550. doi: 10.1038/308548a0. [DOI] [PubMed] [Google Scholar]
- Szabo P, Mann JR. Expression and methylation of imprinted genes during in vitro differentiation of mouse parthenogenetic and androgenetic embryonic stem cell lines. Development. 1994;120:1651–1660. doi: 10.1242/dev.120.6.1651. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tusnady GE, Simon I, Varadi A, Aranyi T. BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005;33:e9. doi: 10.1093/nar/gni012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Iwasaki M, Ficara F, Lin C, Matheny C, Wong SH, Smith KS, Cleary ML. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell. 2010;17:597–608. doi: 10.1016/j.ccr.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TC, Cleary ML. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature. 2008;455:1205–1209. doi: 10.1038/nature07284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson CM, Blake A, Thomas S, Beechey CV, Hancock J, Cattanach BM, Peters J. MRC Harwell, Oxfordshire. World Wide Web Site—Mouse Imprinting Data and References. http://www.har.mrc.ac.uk/research/genomic_imprinting/ (accessed 22 October 2014) 2013 [Google Scholar]
- Yoon BJ, Herman H, Sikora A, Smith LT, Plass C, Soloway PD. Regulation of DNA methylation of Rasgrf1. Nat Genet. 2002;30:92–96. doi: 10.1038/ng795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.