

Summary
Preservation of cell identity is necessary for homeostasis of most adult tissues. This process is challenged every time a tissue undergoes regeneration after stress or injury. In the lethal Duchenne muscular dystrophy (DMD), skeletal muscle regenerative capacity declines gradually as fibrosis increases. Using genetically engineered tracing mice, we demonstrate that, in dystrophic muscle, specialized cells of muscular, endothelial, and hematopoietic origins gain plasticity toward a fibrogenic fate via a TGFβ-mediated pathway. This results in loss of cellular identity and normal function, with deleterious consequences for regeneration. Furthermore, this fibrogenic process involves acquisition of a mesenchymal progenitor multipotent status, illustrating a link between fibrogenesis and gain of progenitor cell functions. As this plasticity also was observed in DMD patients, we propose that mesenchymal transitions impair regeneration and worsen diseases with a fibrotic component.
Highlights
-
•
Regeneration declines as fibrosis increases in Duchenne muscular dystrophy (DMD)
-
•
Myogenic, endothelial, and hematopoietic cells acquire fibrogenic plasticity in DMD
-
•
This fibrogenic plasticity in DMD is induced by TGFβ and blunts muscle regeneration
-
•
Fibrogenesis occurs through an intermediate mesenchymal progenitor multipotent state
The cellular pathways driving fibrosis in dystrophic skeletal muscle are largely unknown. Muñoz-Cánoves and colleagues found that a significant number of muscle stem cells, endothelial cells, and hematopoietic cells gain plasticity as Duchenne muscular dystrophy (DMD) progresses, and convert into fibrotic collagen-producing cells, with negative consequences on regeneration. An association between fibrogenesis and mesenchymal transitions in DMD also is demonstrated.
Introduction
Successful regeneration after tissue injury requires timely coordinated actions of diverse cell types. In skeletal muscle, in response to acute damage, the muscle stem cell (satellite cell) progeny gives rise to new regenerating myofibers, aided by the concerted action of specialized cells, such as infiltrating bone-marrow-derived inflammatory cells, which phagocytose tissue debris and provide pro-myogenic growth factors and cytokines; fibrogenic stromal cells such as fibroblasts and adipogenic progenitors (FAPs), which provide transient matrix support; and angiogenic cells that vascularize the newly formed muscle tissue (Abou-Khalil et al., 2010; Mounier et al., 2011). In chronically damaged muscle, however, this coordination is lost, leading to deficient regeneration (Serrano et al., 2011). In the yet incurable Duchenne muscular dystrophy (DMD), caused by loss of the myofiber protein dystrophin, successive cycles of tissue degeneration and regeneration lead to an eventual muscle regenerative failure and replacement of dystrophic muscle by fibrotic tissue, resulting in respiratory failure and early death (Mann et al., 2011; Stedman et al., 1991; Wallace and McNally, 2009).
Cell plasticity (i.e., the capacity of cells to change their phenotypic properties) is inherent to organismal development and is becoming increasingly associated with tissue remodeling in the adult (Medici and Kalluri, 2012; Nieto, 2013). Mesenchymal transitions (particularly epithelial- and endothelial-to-mesenchymal transitions, EMTs and EndMTs, respectively) are connected both to fibrotic pathologies and cancer progression of distinct etiologies, affecting organs such as liver, lung, heart, or kidney (Medici and Kalluri, 2012; Nieto, 2013; Nieto and Cano, 2012; Zeisberg and Kalluri, 2013). Lineage-tracing and fate-mapping strategies have precisely determined and quantified the source of fibrogenic cells in fibrotic kidney, underscoring the relevance of EMT, EndMT, and bone-marrow-derived cells to this organ’s fibrosis (LeBleu et al., 2013). Incomplete EMT also can occur in tumors, with cells acquiring mesenchymal properties without undergoing the full EMT as it also occurs in embryos, where intermediate phenotypes have been described in different contexts (Nieto, 2011, 2013; Nieto and Cano, 2012). These incomplete transitions implicate a change in cellular functions and behavior. In skeletal muscle, studies on cell plasticity during repair are emerging. In addition to resident interstitial fibroblasts and FAPs, which are considered the major producers of the collagen-rich extracellular matrix (ECM) in injured muscle and in young dystrophic muscle (Joe et al., 2010; Mann et al., 2011; Uezumi et al., 2011, 2014), perivascular progenitor cells transiently produce collagen in response to acute muscle damage, but disappear as regeneration advances (Dulauroy et al., 2012). Similarly, depletion of macrophages or age-induced Wnt signaling in acutely injured muscle can divert vascular and myogenic cell fates, respectively (Brack et al., 2007; Zordan et al., 2014). However, whether cell plasticity occurs in dystrophic muscle and how it affects disease progression have remained elusive. Recently, fibrogenesis from muscle cells has been reported in DMD (Biressi et al., 2014).
Here we demonstrate that specialized cells of muscular, endothelial, and hematopoietic origins acquire mesenchymal-fibrogenic traits in dystrophic muscle, with this cellular plasticity being particularly associated with advanced DMD stages. The mesenchymal-fibrogenic plasticity of these cells is induced by increasing TGFβ signaling in dystrophic muscle with aging, and results in the loss of cell identity, thus precluding normal regenerative functions. Together, our findings suggest that, during efficient tissue repair, specialized cells preserve their lineage identity by avoiding entrance into a mesenchymal-like/fibrogenic state. This protection is lost in chronic degenerative conditions such as DMD.
Results
The levels of TGFβ and downstream signaling mediators (activated SMAD2/3) increase in muscle of dystrophic mdx mice with age, correlating to reduced regeneration, angiogenesis and function, and higher fibrosis extent (Ardite et al., 2012; Kharraz et al., 2014; Mann et al., 2011; Vidal et al., 2008; Figure 1A; Figures S1A and S1B). Inflammatory cells and FAPs appeared as the principal sources of TGFβ in dystrophic muscle (Figure S1C). Higher levels of this pathway also were found in muscle of wild-type (WT) mice after laceration (a severe injury model that induces persistent degeneration and more sustained fibrosis) than after cardiotoxin (CTX) injury (in which collagen-rich ECM is transient and full regeneration and muscle function are achieved rapidly) (Figures S1D and S1E). In agreement with the profibrotic role of TGFβ, exogenous delivery of TGFβ to CTX-injured WT muscle or dystrophic muscle of young mdx mice delayed regeneration and vascularization, while promoting fibrogenesis. This suggests that TGFβ inhibits myogenic and angiogenic capacity of muscle stem cell (satellite cell)-derived myoblasts and endothelial cells, respectively, while promoting matrix accumulation (Figures S1F and S1G). Consistent with this, freshly isolated WT satellite cells were unable to fuse into myotubes in differentiation medium (DM) in the presence of TGFβ (Figure S2A), correlating with gain of expression of fibrogenic genes (αSma, Collagen I, Eda-Fibronectin, or Timp1) and loss of myogenic gene expression (Myf5 and Pax7) after a 10-day treatment (Figure S2B). Likewise, endothelial cells isolated from skeletal muscle could not form angiotubes in vitro after a 10-day TGFβ treatment period (Figure S2C), consistent with loss of expression of endothelial genes (Cd31 and Tie1) and de novo acquisition of fibrogenic traits (Figure S2D). These results indicate that TGFβ induces the loss of identity of muscle-resident myogenic and endothelial cells by promoting their switch into matrix-producing fibrogenic cells, thus precluding their bona fide functions.
Figure 1.
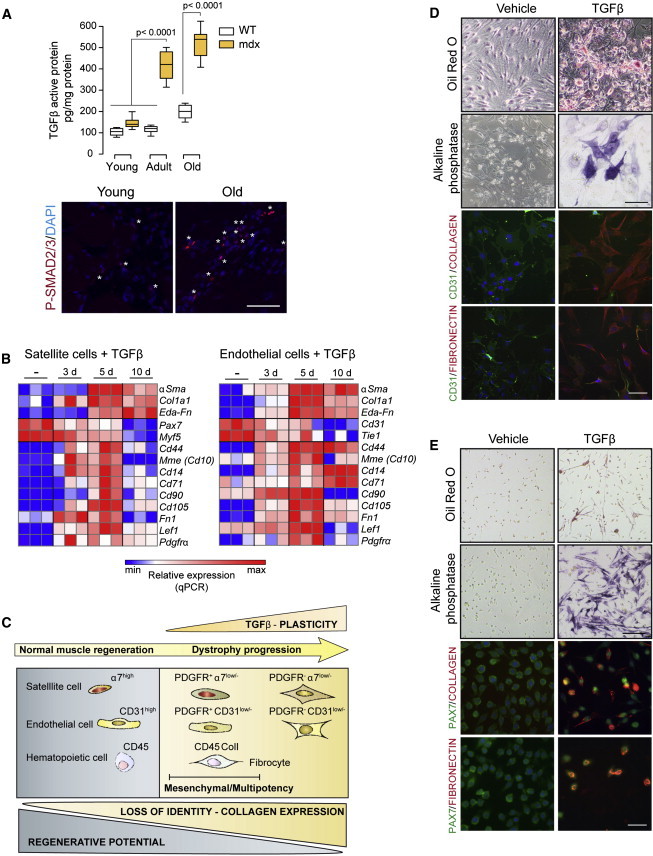
Increased TGFβ Signaling at Advanced Muscular Dystrophy Stages and TGFβ-Induced Cellular Plasticity
(A) Active TGFβ protein levels (ELISA) in limb muscles of mdx mice (C57BL/6 background) at distinct ages: young (2–3 months), adult (6–10 months), and old (18–24 months). Data correspond to the mean ± SEM values; n = 10 for each group. Non-parametric Mann-Whitney U test was used for comparison. Representative pictures of phosphorylated SMAD2/3 (P-SMAD2/3) protein immunostaining in mdx muscles are shown. ∗P-Smad2/3-positive cells. Scale bar, 50 μm.
(B) Heat map representing qRT-PCR of fibroblastic, myogenic, and mesenchymal stem cell marker expression in satellite cells or endothelial cells treated with TGFβ for the indicated time points (days). Values are mean ± SEM; n = 3 independent experiments.
(C) Summary scheme representing the process of fibrogenic plasticity of myogenic, endothelial, and hematopoietic cells in dystrophic and injured skeletal muscle. Scheme is inferred from results using satellite cells and endothelial cells obtained from skeletal muscle, with or without treatment with TGFβ. Kinetics of fibrogenic cell plasticity induced by TGFβ is represented, with cells transiting via a mesenchymal progenitor cell-like state, which in turn is endowed with multipotent capacity. Similar plastic changes occur in muscle of lineage-tracing mice for endothelial and satellite cells in mdx dystrophic background (or in bone-marrow-transplanted mdx mice for hematopoietic cell tracing) and in WT muscle after CTX injury/TGFβ administration. Endothelial and satellite cells gain fibrogenic plasticity and lose their own identities and functions in vivo (become CD31low/− or α7-integrinlow/−, and have reduced angiogenic and myogenic potential [not shown]). During this process, these cells express the mesenchymal progenitor marker PDGFRα and exhibit multipotency under adequate lineage-differentiation conditions (see D and E). We also detected fibrogenic cells of hematopoietic origin in dystrophic muscle (i.e, fibrocytes characterized by co-expression of CD45 and COLLAGEN I).
(D and E) Multipotency analysis of TGFβ pre-treated endothelial cells (D) and satellite cells (E), further cultured in adipogenic, osteogenic, and fibrogenic DM. Oil Red O (adipogenic), alkaline phosphatase (osteogenic), and COLLAGEN or FIBRONECTIN (fibrogenic) staining detect multi-lineage potential compared to non-pre-treated cells. Scale bars, 50 μm.
To further understand this cellular plasticity process induced by TGFβ, we performed a microarray gene expression analysis of satellite cells treated (or not) with TGFβ for 4 days (before achieving maximal levels of fibrogenic conversion, i.e., expression of αSma [Figure S2B]). Gene ontology functional annotation of genes upregulated in TGFβ-treated myogenic cells, compared to non-treated cells, showed enrichment in mesenchymal-fibrogenic functions (Figure S3A). Moreover, after comparison with a curated list of mesenchymal progenitor cell-specific transcripts (Kubo et al., 2009), we identified a group of mesenchymal cell-specific genes induced by TGFβ (Figure S3A). qRT-PCR analysis of a TGFβ cell-treatment kinetics experiment validated the expression of mesenchymal progenitor genes at intermediate time points (Figure 1B). These mesenchymal genes were also significantly upregulated in endothelial cells isolated from skeletal muscle in response to identical TGFβ treatment, prior to larger acquisition of fibrogenic traits (Figure 1B; see scheme in Figure 1C).
To prove whether this gain of mesenchymal gene expression translated into de novo functional cellular multipotency (i.e., potential to differentiate into distinct cellular fates), satellite cells and endothelial cells were treated with TGFβ for 3 days and further incubated with osteogenic or adipogenic medium for 7–14 days, or they continued to be treated with TGFβ for 7 extra days. Notably, cells pre-treated with TGFβ showed induced expression of adipocyte, osteoblast, or fibrogenic traits under their respective differentiation regimes (Figures 1D and 1E; Figure S3B); in contrast, cells that had not been pretreated with TGFβ did not undergo any of these conversions under identical differentiation conditions (Figures 1D and 1E). These results suggest that these two specialized cell types (myogenic and endothelial cells) gain expression of mesenchymal genes during the plastic process toward a more mature fibrogenic fate in response to TGFβ; furthermore, these cells exhibit multipotency under adequate culture conditions (see scheme in Figure 1C). Of note, TGFβ was capable of inducing the expression of transcription factors and microRNAs associated with mesenchymal transitions in myogenic and endothelial cells (Figures S3C and S3D). In particular, Mir21 induction appeared to mediate TGFβ-induced fibrogenesis in both cell types (Figure 2), reinforcing this Mir as a fibrogenic effector of TGFβ action (Acuña et al., 2014; Ardite et al., 2012; Kumarswamy et al., 2012).
Figure 2.
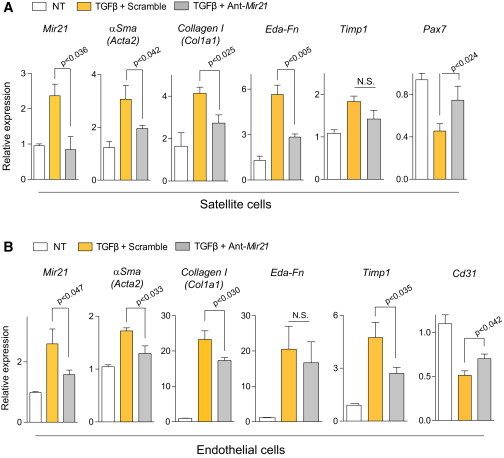
Mir-21 Mediates TGFβ-Induced Fibrogenesis of Satellite Cells and Endothelial Cells
(A and B) Satellite cells (A) and endothelial cells (B), transfected with Ant-Mir-21 or Scramble oligomiR, were treated with TGFβ for 8 days, and subjected to qRT-PCR for fibrogenic, myogenic or endothelial markers, respectively. Values are mean ± SEM; n = 3 independent experiments. Unpaired t test was used for comparison.
Since TGFβ levels and signaling are elevated in aged dystrophic muscle (Figure 1A), we next investigated whether endothelial and satellite cells also undergo plastic fibrogenesis in vivo. To this end, we generated endothelial and satellite cell-lineage-tracing mice in two mdx dystrophic backgrounds, DBA/2 and C57BL/6, which develop substantial fibrosis in limb muscles at adult (6–10 months) or old (after 18 months) age, respectively (Fukada et al., 2010; Figures S4A and S4B). First, Ve-Cad-CreER/YFP mice (obtained by intercrossing of Cdh5(Ve-Cad)-CreER and Rosa26R-YFP mice, in which the expression of Cre is induced exclusively in adult endothelial cells expressing Ve-Cadherin upon tamoxifen administration) were intercrossed with mdx/DBA/2 mice, generating Ve-Cad-CreER/YFP/mdx triple-mutant mice, which were aged until advanced fibrogenic stages (i.e., 6–10 months). A second endothelial genetic tracing line, Tie2-Cre/YFP mice (obtained by intercrossing of Tie2-Cre and Rosa26R-YFP mice, in which the Tie2 gene is expressed by endothelial cells from early vascular development), was bred into the mdx/C57BL/6 dystrophic background, generating Tie2-Cre/YFP/mdx triple-mutant mice, which were subsequently aged up to 18–24 months for fibrosis development.
Use of the CD31 endothelial cell marker in a YFP+-based sorting protocol permitted discrimination between bona fide endothelial cells (YFP+CD31high) and cells of endothelial origin that had reduced (or lost) expression of CD31 (YFP+CD31low/−). The YFP+CD31high cells represented the most prominent YFP+ cell population in Ve-Cad-CreER/YFP and Tie2-Cre/YFP (WT) muscle (Figures 3A and 3B). In fibrotic-dystrophic muscle, however, a YFP+CD31low/− cell population was induced (Figures 3A and 3B). Fibrogenic markers (Collagen I and Fibronectin) were found exclusively in the YFP+CD31low/− cell fraction (Figure 3C), which accounted for approximately 20%–25% of the endothelial cell population (Figures 3A and 3B), and this was confirmed by qRT-PCR (Figure 3D), supporting the occurrence of cell plasticity characterized by the reduction of endothelial identity traits and acquisition of the fibrogenic gene program, at advanced muscular dystrophy. This conclusion was confirmed by the detection of cells double positive for fibrogenic and endothelial cell markers in muscle at late dystrophic stages (Figure S4C; Figure 3E), which coincided with the reduced vascularization and regeneration and increased fibrosis (Figure S1B). Furthermore, delivery of TGFβ to CTX-injured muscle of Ve-Cad-CreER/YFP mice induced the presence of YFP+CD31low/− fibrogenic cells (Figure 3F), although to a lesser extent than in dystrophic muscle (Figures 3A and 3B). qRT-PCR and immunofluorescence analysis confirmed the reduction of endothelial cell markers and gain of fibrogenic ones in the YFP+/CD31low/−-sorted cells in TGFβ/CTX-injured WT muscles (Figures S4D and S4E).
Figure 3.
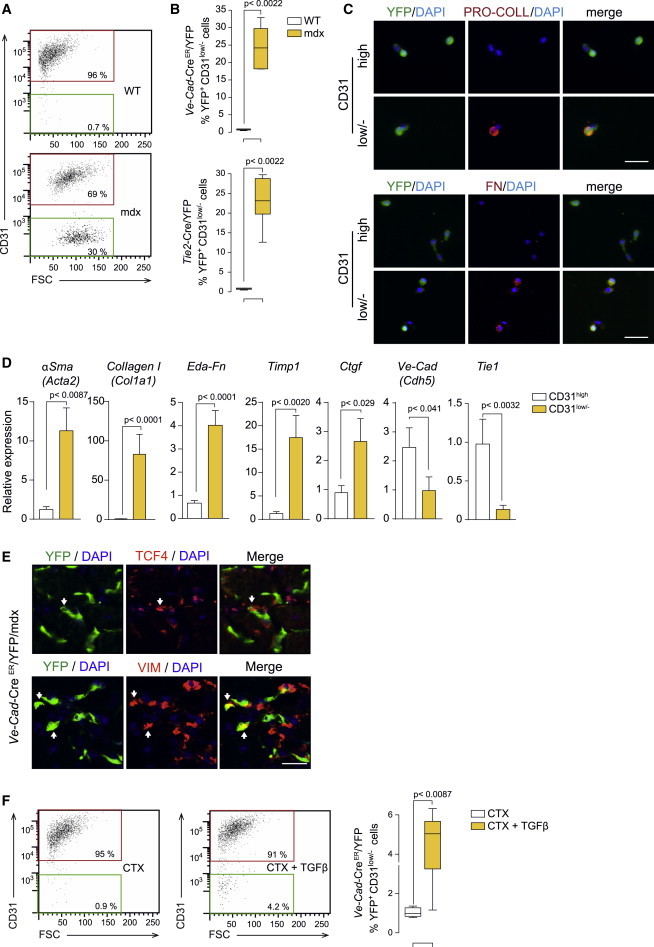
Fibrogenic Plasticity of Endothelial Cells at Advanced Muscular Dystrophy Stages and in Response to TGFβ
(A) Representative FACS plots of CD31 expression in YFP+ cells from dystrophic Ve-Cad-CreER/YFP/mdx (mdx) muscle compared to Ve-Cad-CreER/YFP (WT) muscle. Mdx mice were in the DBA2 background and analyses were performed at adult age.
(B) Quantification of YFP+ cells with reduced/lost CD31 expression in Ve-Cad-CreER/YFP/mdx mice in DBA2 background at adult age and in Tie2-Cad-Cre/YFP/mdx mice in C57BL/6 background at old age. Values are mean ± SEM; n = 6 animals for each group. Non-parametric Mann-Whitney U test was used for comparisons.
(C) Immunofluorescence analysis of FACS-isolated cells from (B): YFP+ cells that gain PRO-COLLAGEN and FIBRONECTIN expression while losing CD31 expression (CD31low/−). Nuclei are stained with DAPI. Scale bars, 50 μm.
(D) qRT-PCR in FACS-isolated YFP+CD31low/− cells from Ve-Cad-CreER/YFP/mdx mice compared to YFP+CD31high cells for the indicated fibroblastic and endothelial cell markers. Values are mean ± SEM; n = 6 independent experiments (mice) for each group. Unpaired t test was used for comparison.
(E) Representative immunostaining shows YFP+ cells co-expressing fibrogenic markers in Ve-Cad-CreER/YFP/mdx mice. Scale bars, 25 μm.
(F) Representative FACS plots of CD31 expression in YFP+ cells isolated from CTX-injured and CTX/TGFβ-injured muscle (after 14 days of the initial injury [Figure S1F]) of Ve-Cad-CreER/YFP mice, and quantification of YFP+ cells with reduced/lost CD31 expression. Values are mean ± SEM; n = 6 animals for each group. Non-parametric Mann-Whitney U test was used for comparison.
Next, using a similar lineage-tracing strategy, we generated Pax7-Cre/YFP double-transgenic mice (obtained by intercrossing Pax7-Cre and Rosa26R-YFP mice, in which the expression of Cre is induced in Pax7-expressing muscle precursor cells) that were intercrossed with mdx/C57BL/6 dystrophic mice, and the resulting Pax7-Cre/YFP/mdx triple-mutant mice were aged up to 18–24 months. We also used Pax7-CreER/YFP mice (in which Cre is induced only in adult satellite cells upon tamoxifen administration) that were intercrossed with mdx/DBA/2 mice, generating Pax7-CreER/YFP/mdx triple-mutant mice, which were aged until advanced fibrogenic stages at adult age (6–10 months). Double-labeling cell sorting for YFP and the satellite cell marker α7-INTEGRIN allowed us to discriminate between bona fide satellite cells (YFP+α7high) and cells that had reduced or lost (YFP+α7low/−) α7-INTEGRIN expression after CTX/TGFβ injury and in aged dystrophic mice. The YFP+α7low/− cell population was induced specifically in CTX/TGFβ-injured muscle (compared to CTX-injured, or non-injured, muscle) of Pax7-CreER/YFP mice (Figure 4A). This population was further increased (up to 11%–15%) in Pax7-CreER/YFP/mdx muscle of adult age (BDA/2 background) or old age (C57BL/6 background) (Figure 4B; Figure S4F). Supporting the decline in myoblast cell identity in vivo, the expression of Pax7 decreased, while fibroblastic gene transcripts increased in YFP+α7low/− cells from mdx and TGFβ/injured WT muscles (Figures 4C and 4D). Furthermore, in myofiber explants, the number of bona fide satellite cells associated with each myofiber decreased in fibrotic Pax7-Cre/YFP/mdx mice, as indicated by the lower number of YFP+ cells expressing PAX7 or MYOGENIN (markers of quiescent/proliferating or differentiated myogenic cells, respectively), compared to age-matched WT myofibers (Figure 4E). Conversely, YFP+ cells in mdx myofiber explants gained expression of fibrogenic markers (Figure 4E). Co-expression of fibrogenic and myogenic proteins (or YFP) (Figure S4G; Figure 4F) further supported satellite cell fibrogenic plasticity in aged dystrophic muscle. Together, based on quantifications of these analyses (Figures 4B and 4E; Figure S4F), 11.7% of cells of myogenic origin gained fibrogenic traits in aged dystrophic muscle.
Figure 4.
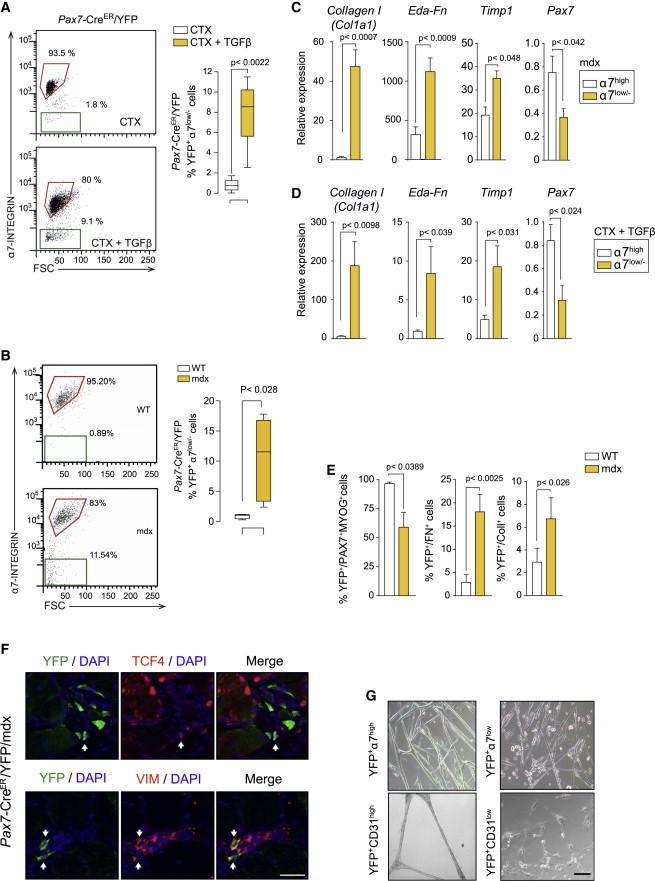
Fibrogenic Plasticity of Satellite Cells at Advanced Muscular Dystrophy Stages and in Response to TGFβ
(A) Representative FACS plots of α7-INTEGRIN expression in CTX-injured and CTX/TGFβ-injured Pax7-CreER/YFP muscles (as in Figure S1F) and quantification of YFP+ cells that have reduced/lost α7-integrin expression. Values are mean ± SEM; n = 6 animals for each group. Non-parametric Mann-Whitney U test was used for comparison.
(B) Quantification of YFP+ cells that maintain or have reduced/lost α7-INTEGRIN expression in Pax7-CreER/YFP/mdx (mdx) mice compared to Pax7-CreER/YFP (WT) mice. Values are mean ± SEM; n = 6 animals for each group. Non-parametric Mann-Whitney U test was used for comparison.
(C and D) qRT-PCR of fibroblastic and myogenic markers in YFP+α7low/− compared to YFP+α7high cells from Pax7-CreER/YFP/mdx muscle (C) and from CTX/TGFβ-injured Pax7-CreER/YFP muscle (D). Values are mean ± SEM; n = 3 independent experiments (mice) for each group. Unpaired t test was used for comparison.
(E) Percentage of YFP+ cells that express myogenic and fibrogenic markers in single fibers from Pax7-Cre/YFP/mdx (mdx) mice compared to Pax7-Cre/YFP (WT) mice. Values are mean ± SEM; n = 5 animals each group. Non-parametric Mann-Whitney U test was used for comparison.
(F) Representative immunostaining shows YFP+ cells co-expressing fibrogenic markers in Pax7-CreER/YFP/mdx mice. Scale bars, 25 μm.
(G) Myogenic potential of FACS-isolated YFP+α7high cells from aged Pax7-Cre/YFP/mdx mice, compared to YFP+α7low/− cells, after culture in DM for 4 days. The angiogenic potential of CD31high versus CD31low/− endothelial cells was similarly determined in angiogenic-promoting conditions (see Experimental Procedures). Scale bars, 50 μm.
We subsequently tested the functional consequences of this fibrogenic plasticity in dystrophic muscle. The fluorescence-activated cell sorting (FACS)-isolated cells of endothelial and myogenic origins (YFP+) that had reduced (or lost) expression of CD31 or α7-INTEGRIN (YFP+CD31low/− or YFP+α7low/−) showed a severely impaired capacity to form angiotubes and myotubes in pro-angiogenic and pro-myogenic differentiation conditions, respectively (Figure 4G). Despite the significant fraction of both endothelial and myogenic cells undergoing fibrogenic plasticity in dystrophic muscle, surprisingly, each cell type only constituted about 2% of the bona fide active collagen-expressing cell population (i.e., active fibroblasts), based on intracellular collagen protein staining of the YFP+CD31low/− and YFP+α7low/− cell populations, respectively (Figure S5A). Similarly, using a ColI-GFP reporter mice (in which GFP expression is under the control of the Collagen I promoter), only a low percentage of CD31+ and α7-INTEGRIN+ cells were found within the fibroblastic (GFP+) cell population (Figure S5B). These findings strongly suggest that, unlike other fibrotic organs, such as kidney (LeBleu et al., 2013), these fibrogenic changes do not lead to full and ample transformation into collagen-producing cells. Instead, this fibrogenic plasticity mainly precludes efficient myogenesis and angiogenesis and it impairs tissue repair.
We next investigated if endothelial and satellite cells also transit through intermediate mesenchymal progenitor states during the process of fibrogenesis in diseased muscle. To this end, we set up a FACS protocol based on the use of the cell surface mesenchymal progenitor marker PDGFRα (Chong et al., 2013; Pinho et al., 2013; Uezumi et al., 2010, 2014). A subpopulation of YFP+ cells that were low for CD31 or α7-INTEGRIN expression appeared positive for PDGFRα (YFP+PDGFRα+) in fibrotic muscles of dystrophic lineage-tracing mice (Figures 5A and 5B). The YFP+PDGFRα+CD31low/− and YFP+PDGFRα+α7low/− populations represented 14.6% and 16.3% of the fibrogenic YFP+CD31low/− and YFP+α7low/− cell fractions in adult mdx muscle, respectively (Figures 5A and 5B), and these percentages even increased in older mice (Figure S5C). As for the YFP+CD31low/− and YFP+ α7low/− cell fractions, the PDGFRα+-expressing cell subpopulations were incapable of forming myotubes or angiotubes under appropriate differentiation conditions (not shown). The qRT-PCR analysis confirmed induction of mesenchymal progenitor markers in the freshly isolated YFP+PDGFRα+CD31low/− and YFP+PDGFRα+α7low/− subpopulations from dystrophic muscle (Figure 5C). Thus, during the process of endothelial and satellite cell plasticity toward fibrogenesis, a fraction of cells shows mesenchymal progenitor traits. This was consistent with a subpopulation of YFP+ endothelial or myogenic cells also gaining PDGFRα+ expression in WT muscle subjected to CTX/TGFβ injury (Figure 5D). Consistent with the notion that TGFβ signaling is a driving cause for these plastic mesenchymal transitions, we could detect co-expression of activated SMAD2/3 in PDGFRα+/YFP+ cells in muscle of the distinct lineage-tracing mice (in various muscle degeneration/fibrosis paradigms) (Figure 5E; Figure S5D; data not shown) and in human DMD (see below).
Figure 5.
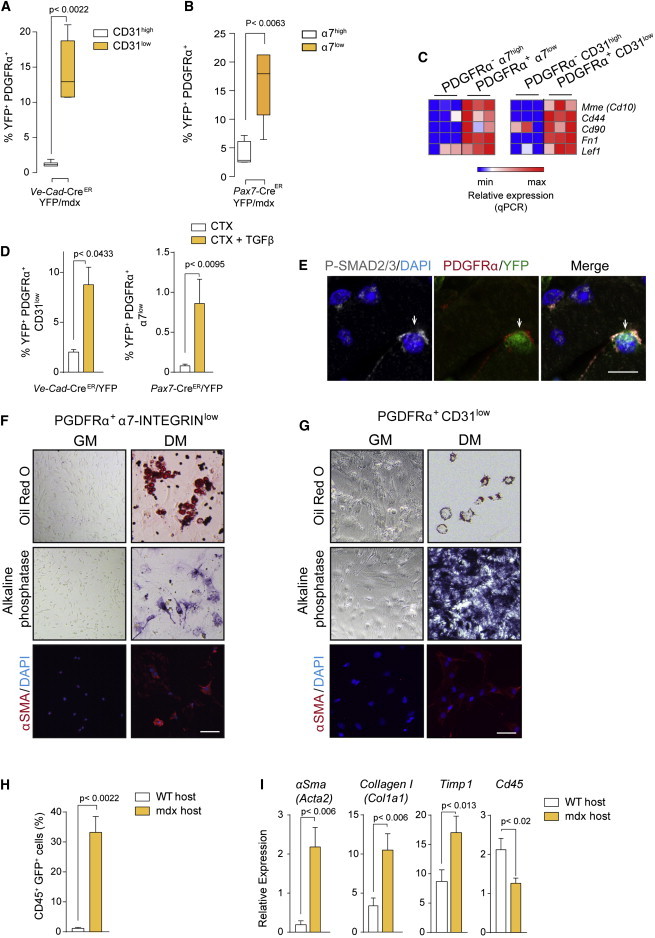
Mesenchymal Progenitor Cells of Endothelial and Myogenic Origins from Dystrophic Muscle Exhibit Multipotent Capacity
(A and B) Percentage of double-positive YFP+ PDGFRα+ cells from muscles of Ve-Cad-CreER/YFP/mdx (that were CD31high or CD31low) mice (A) and Pax7-CreER/YFP/mdx (that were α7-INTEGRINhigh or α7-iINTEGRINlow) mice (B). Values are mean ± SEM relative to age-matched lineage-tracing mice in non-dystrophic background; n = 6 animals for each group. Non-parametric Mann-Whitney U test was used for comparison.
(C) qRT-PCR of mesenchymal stem cell markers in YFP+/PDGFRα+ cells with either α7-INTEGRINhigh or α7-INTEGRINlow expression from mice in (B), or CD31high or CD31low expression from mice in (A, left). Values are mean ± SEM; n = 3 independent experiments (mice) for each group.
(D) Percentage of YFP+ cells that express PDGFRα in CTX/TGFβ-injured versus CTX-injured muscles from the indicated mouse genotypes. Values are mean ± SEM; n = 4–6 for each group. Non-parametric Mann-Whitney U test was used for comparison.
(E) Representative immunostaining shows P-SMAD2/3 in PDGFR+/YFP+ cells in Ve-Cad-CreER/YFP/mdx muscle. Scale bars, 10 μm.
(F and G) Multipotency analysis of FACS-isolated YFP+PDGFRα+α7low and YFP+PDGFRα+CD31low from Pax7-Cre/YFP/mdx muscle (F) and Ve-Cad-CreER/YFP/mdx muscle (G), cultured in adipogenic, osteogenic, and fibrogenic differentiation-promoting conditions (DM), compared to growth medium (GM)-cultured cells. Oil Red O, alkaline phosphatase, and αSMA staining are shown (n = 3 biological replicates). Scale bars, 50 μm.
(H) Percentage of GFP+ cells in muscles of 22-month-old mdx mice, after transplantation of bone marrow from ColI-GFP mice. Values are mean ± SEM; n = 3 animals for each group. Non-parametric Mann-Whitney U test was used for comparison.
(I) qRT-PCR for fibrogenic markers and CD45 of GFP+-sorted cells after bone marrow transplantation (H). Values are mean ± SEM; n = 3 independent experiments (mice) for each group. Unpaired t test was used for comparison.
To finally prove that these subpopulations of YFP+ PDGFRα+ cells (from endothelial or myogenic origin) are indeed mesenchymal in nature, we tested their multipotency (the capacity to be coaxed to differentiate into distinct terminal fates: fat, bone, cartilage, or scar/fibrous) if exposed to adequate conditions. YFP+PDGFRα+ cells were FACS isolated from muscle of lineage-tracing dystrophic mice and subsequently cultured with osteogenic, adipogenic, or chondrogenic differentiation media or with TGFβ (for fibrogenic differentiation). In response to these treatments, sorted cells that had gained PDGFRα+ expression (YFP+PDGFRα+ cells), but not YFP+PDGFRα− cells, were positive for oil red staining (adipocyte), alkaline phosphatase staining (osteoblast), or collagen staining (fibroblast) (Figures 5F and 5G). In contrast, YFP+ cells, which were PDGFRα−CD31low/− or PDGFRα−α7low/−, did not show multipotency (not shown), indicative of a more differentiated fibrogenic state. These results suggest that the process of fibrogenic plasticity of endothelial and myogenic cells within dystrophic muscle, as with the plastic response to TGFβ in vitro (see Figure 1), involves multipotent progenitor cell intermediate states. Of note, in vivo interference with PDGFRα signaling with imatinib (a tyrosine kinase inhibitor), which has been shown to target PDGFRα-expressing mesenchymal progenitor cells (Ito et al., 2013; Uezumi et al., 2014), prevented the loss of cell identity in injured muscle in response to TGFβ (Figure S5E).
Mesenchymal cells in the bone marrow are multipotent in nature. Since fibrocytes (defined as bone-marrow-derived cells expressing the hematopoietic marker Cd45 and Collagen I) contribute to fibrosis in organs such as kidney, liver, lung, and heart (Duffield et al., 2013; Kisseleva and Brenner, 2012; Krenning et al., 2010), we hypothesized that fibrocytes also could contribute to fibroblast heterogeneity and fibrosis in dystrophic muscle. To test this possibility, we transplanted bone marrow from ColI-GFP reporter mice into young (3-month-old) mdx/C57BL/6 mice, and the transplanted mice were aged for fibrosis development for 18 extra months. A significantly increased number of GFP+ cells was found in the muscle of transplanted dystrophic mice of 22 months of age (as an indication of collagen-producing cells derived from the donor bone marrow) compared to muscle of similarly transplanted non-dystrophic mice (Figure 5H). Consistent with this, compared to GFP+ cells from transplanted WT muscles, the FACS-isolated GFP+ cell population in aged dystrophic muscle showed reduced expression of the Cd45 hematopoietic cell marker, while the expression of fibrogenic markers increased (Figure 5I), and this was in agreement with the detection of cells co-expressing fibrogenic and hematopoietic markers in advanced muscle disease stages (Figure S5F). As for endothelial and satellite cells, only 2%–3% of fibrocytes appeared to contribute to the overall population of collagen-expressing cells in dystrophic muscle (Figures S5A and S5B). Together, these results demonstrate the acquisition of fibrogenic traits by hematopoietic cells at advanced fibrotic states of muscular dystrophy, and further illustrate the existence of mesenchymal plasticity of distinct specialized cells in dystrophic muscle, with deleterious consequences on disease progression.
As in dystrophic mice, we found that fibrosis was specifically induced in muscles of human DMD patients (compared to healthy individuals), being more prominent in affected patients between 6 and 8 years of age than in younger 2- to 4-year-old children (Figure 6A), and this correlated with disease severity and physical incapacitation (not shown). We also detected fibrogenic (TCF4+) cells in fibrotic muscle of DMD patients that increased with age (Figure 6A). In DMD muscles, we also identified fibrogenic cells co-expressing vWF (a marker of endothelial cells), CD56 (a marker of human satellite cells), and CD45 (a hematopoietic marker) (Figure 6B), demonstrating cell plasticity in the human pathology, as in mdx dystrophic mice (Figures S4C, S4G, and S5F; Figures 3, 4, and 5). Because active TGFβ levels and signaling (P-SMAD2/3) also were increased in human DMD muscles, as fibrosis and disease severity progressed with age (Figure 6C), and because activated SMAD2/3 was specifically associated with cells expressing markers of fibrogenic and specialized cells in human and murine dystrophic muscle (Figures 6C and 6D; Figure S5D), these results, taken together, reinforce the idea of TGFβ being a plasticity-promoting factor in DMD. Consistent with this, DMD muscles contained cells expressing the mesenchymal progenitor cell marker PDGFRα together with markers of endothelial, myogenic, and hematopoietic cells, respectively (Figure 6E), and PDGFRα-expressing cells that were double positive for P-SMAD2/3 and TCF4 (Figure 6D). Thus, in muscle of DMD patients, as in dystrophic mice, fibrogenic cells are a heterogeneous population, and part of these cells may arise from plastic events within the TGFβ-enriched dystrophic milieu. Consistent herewith, human myoblasts also showed fibrogenic plasticity in vitro in response to TGFβ treatment (Figure 6F). Moreover, in vivo inhibition of TGFβ signaling, via the administration of LY2157299 (a specific inhibitor of the TGFβ receptor type 1 kinase) (Zhou et al., 2011) in old dystrophic mdx mice, reduced the presence of collagen-producing cells co-expressing markers of the specialized lineages (Figure 6G), while reducing fibrosis and restoring dystrophic muscle regeneration and vascularization (Figure 6G). Altogether, these results unveil TGFβ as a driver of fibrogenic cell plasticity both in human and mouse dystrophic and severely damaged muscle.
Figure 6.
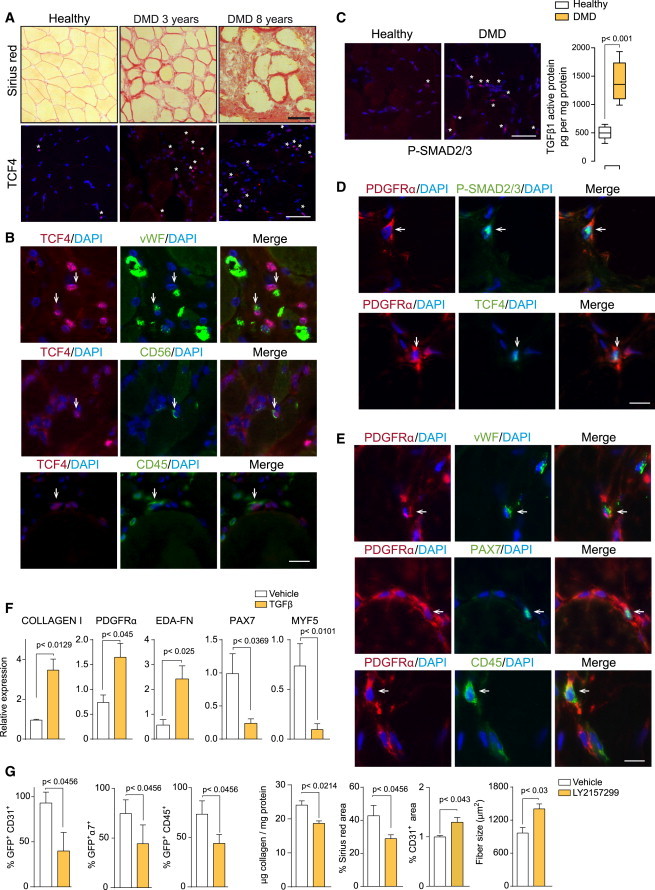
Fibrogenic Plasticity in Human Muscle of DMD Patients
(A) Representative Sirius Red and TCF4 staining in human muscle biopsies from healthy individuals and DMD patients 3 and 8 years of age are shown.
(B) Representative picture of co-staining of TCF4 with endothelial, myogenic, and hematopoietic cell markers, respectively, in DMD muscles. Nuclei are stained with DAPI.
(C) Representative picture of P-SMAD2/3 staining and active TGFβ protein levels in muscle biopsies from healthy individuals and DMD patients 6–8 years of age. Data correspond to the mean ± SEM; n = 10 in each group. Non-parametric Mann-Whitney U test was used for comparison.
(D) Representative immunostaining shows human cell co-expressing PDGFRα and P-SMAD2/3 or TCF4 in DMD muscle.
(E) Representative immunostaining shows human cells expressing PDGFRα and vWF, PAX7, or CD45 in DMD muscle. Nuclei are stained with DAPI.
(F) Human myoblasts were treated with TGFβ or vehicle for 6 days and analyzed for the expression of the indicated genes. Values are mean ± SEM; n = 3 independent experiments for each group. Unpaired t test was used for comparison.
(G) Reduction of GFP+ cells co-expressing CD31, α7-INTEGRIN, or CD45 in adult ColI-GFP/mdx muscle after treatment with the TGFβ signaling inhibitor LY2157299 (or vehicle) for 1 month. Quantification shows fibrosis reduction and increased regeneration and vascularization. Data correspond to the mean ± SEM; n = 3 for each group. Non-parametric Mann-Whitney U test was used for comparison. Scale bars, 50 μm.
Discussion
Our study provides insights into the mechanisms underlying the loss of regenerative potential and increasing fibrogenesis with age in DMD. We report that a proportion of specialized cells, which are critical for muscle regeneration, cannot maintain their identifying functions in aged dystrophic muscle of humans and mice, and acquire instead the capacity to produce matrix proteins. This is in agreement with a recent report from the Rando group (Biressi et al., 2014). Fibrogenesis in dystrophic muscle does not appear to be an all-or-nothing but rather a partial transition, as most cells share original as well as new fibroblastic traits, resembling the partial EMTs occurring in carcinosarcomas and fibrotic liver (Nieto, 2013; Sarrió et al., 2008; Zeisberg et al., 2007). This cellular plasticity, however, hampers muscle tissue repair potential. Mechanistically, we show that the loss of cell identity toward fibrogenesis in dystrophic muscle may involve the acquisition of mesenchymal traits, triggered by increasing TGFβ as disease progresses. This link between skeletal muscle fibrogenesis and mesenchymal-like transitional states, resulting in the loss of regenerative potential, was a striking finding. Because adipose tissue also accumulates in muscle of DMD patients, we postulated that the loss of identity of specialized cells concomitantly with the acquisition of mesenchymal-like (multipotent) characteristics might be a general feature for fibrogenesis as for adipogenesis in DMD, and by extension in other pathological conditions coursing with high TGFβ signaling and tissue damage. Indeed, TGFβ2- or BMP4-induced EndMT into multipotent stem-like cells was proposed to be the origin of heterotopic cartilage and bone in individuals with fibrodysplasia ossificans progressiva (FOP) lesions (Medici et al., 2010). A distinct muscle-resident multipotent progenitor additionally was proposed as an origin for FOP, based on its osteogenic potential in response to BMP2 (Wosczyna et al., 2012).
Recent studies have shown that, in addition to fibroblasts, tissue-resident FAPs and perivascular cells, which express PDGFRα, can differentiate to a fibroblastic fate and contribute to collagen accumulation in acutely injured muscle (Dulauroy et al., 2012; Joe et al., 2010; Uezumi et al., 2010). FAPs also accumulate in young dystrophic muscle prior to maximal fibrosis (Uezumi et al., 2011), being the main source of the fibrogenic progeny leading to collagen production at early dystrophy stages. Our results demonstrate that at advanced DMD stages there are additional reservoirs of fibrogenic cells derived from myogenic, endothelial, and hematopoietic cells. However, unlike organs like kidney, where a great proportion of the fibrotic cells arise from the bone marrow (and to a lesser extent from EndMT or EMT) (LeBleu et al., 2013), in dystrophic skeletal muscle, the net contribution of these cells to the actual population of collagen-producing cells (the key cells for fibrosis development) is modest. Instead, our results reveal that it is the loss of myogenic and endothelial cells’ biochemical and phenotypic identities, through plastic mesenchymal transitions (characterized by the expression of PDGFRα), that causes severe deficits in myogenesis and angiogenesis, thus exacerbating the regenerative impairment in dystrophic muscle (see scheme in Figure 1). Therapeutically, a unifying model of loss of cell fate and acquisition of mesenchymal traits centered on the PDGFRα-expressing cell could be envisioned. How to restrict deleterious PDGFRα-dependent functions while preserving the beneficial ones is likely to be more challenging.
Our findings support the notion that, in chronic degenerative conditions, the ability of specialized cells (such as myogenic, endothelial, and inflammatory cells) to undergo mesenchymal transitions is inversely correlated to the degree of tissue regeneration, while facilitating fibrogenesis. This has implications for regenerative medicine, as our findings unravel a physiological form of plasticity that can be co-opted toward disease-associated tissue degeneration and aging.
Experimental Procedures
Mice
Mdx mice were maintained in C57BL/10 or DBA/2 backgrounds. Lineage-tracing mice have been described previously as follows: Cdh5-CreER (Wang et al., 2010), Tie2-Cre transgenic (Kisanuki et al., 2001), Pax7-Cre and Pax7-CreER (Nishijo et al., 2009), R26R-EYFP (Srinivas et al., 2001), and Col1a1-3.6GFP (Coll-GFP) (Krempen et al., 1999). Lineage-tracing mice were intercrossed with C57BL/10 or DBA/2 dystrophic mdx mice (Fukada et al., 2010). When needed, Cre activity was induced by intraperitoneal injection (one injection per day for 4 days) with 5 mg/25 g body weight tamoxifen (TAM) (Sigma; 10 mg/ml in corn oil). Bone marrow transplantation experiments were performed as previously described (Perdiguero et al., 2011), using Coll-GFP mice as bone marrow donor and mdx/C57BL/10 at 3 months of age as recipient mice; mice were aged for 18 months for fibrosis development.
Induction of Muscle Regeneration and Fibrosis
Regeneration of skeletal muscle was induced by intramuscular injection of 50 μl 10−5 M CTX (Latoxan). Muscles were collected at the indicated times on each set of experiments, which was usually 2 weeks after myotoxin injection. Contralateral control muscles were left un-injured. To induce a more severe fibrotic injury, tibialis anterior (TA) muscles of WT mice were subjected to laceration as previously described (Ardite et al., 2012). To exacerbate fibrotic muscle damage, 50 ng TGFβ (recombinant TGFβ1 and TGFβ2, R&D Systems) was injected into previously injured (or dystrophic) TA and gastrocnemius muscle in a volume of 50 μl PBS, as previously described (Pessina et al., 2014). For the inhibition of TGFβ, LY2157299 (a specific inhibitor of the TGFβ receptor type 1 kinase) (Zhou et al., 2011) was administered to old mdx mice (in the Coll-GFP background) for 30 days at a 25 mg/kg daily dose, and different parameters were analyzed thereafter. For the inhibition of PDGFRα signaling, imatinib (a tyrosine kinase inhibitor), which has been shown to target PDGFRα-expressing mesenchymal progenitor cells (Ito et al., 2013; Uezumi et al., 2014), was administered to 7-day-CTX-injured mice for an additional 7-day period (50 mg/kg daily), coinciding with the administration of TGFβ, and analyzed subsequently.
Cell Culture and Differentiation
Primary myoblasts were obtained from mouse skeletal muscle, grown in Ham’s F10 medium (BioWest) supplemented with 20% fetal bovine serum (FBS) and basic fibroblast growth factor (bFGF, 0.025 μg ml−1), as described previously (Perdiguero et al., 2011). Primary endothelial cells were grown in high-glucose DMEM (BioWest) supplemented with 10% FBS and 10 ng/ml vascular endothelial growth factor (VEGF), as described previously (Ieronimakis et al., 2008). Human myoblasts were purchased from Cook Myosite and cultured following the provided instructions. When indicated, cells were treated with TGFβ1 (myoblasts) or TGFβ2 (endothelial cells) (10 ng/ml). To induce differentiation into distinct lineages, cells were grown in StemXVivo osteogenic and adipogenic culture media (R&D Systems), after 4 days of TGFβ treatment. Alkaline phosphatase staining to detect osteoblasts was performed with the alkaline phosphatase kit (Sigma-Aldrich) on cultures grown in osteogenic medium for 14 days. Oil red O (Sigma-Aldrich) staining to detect adipocytes was performed on cultures grown in adipogenic medium for 14 days. Alternatively, for fibrogenic differentiation, cells were cultured in TGFβ-containing medium for an additional 7-day period.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism software using the nonparametric Mann-Whitney U test or unpaired t test for independent samples, with a confidence level of 95% being considered statistically significant. The results are expressed as mean ± SEM. The number of samples analyzed per group is detailed in each figure.
Acknowledgments
We are indebted to V. Ruiz-Bonilla, V. Lukesova, S. Gutarra, M. Raya, B. Ampudia, and members of the Cell Biology Group for their contributions to this study. J. Martín-Caballero (PRBB Animal Facility), O. Fornas (CRG/UPF FACS unit), and CRG Genomic Unit. We also thank M. Reyes and N. Ieronimakis for help in isolation of muscle endothelial cells, S. Biressi and T. Rando for the generous offer to provide samples and for information exchange, C. Keller for Pax7-Cre lines, R.H. Adams and J.L de la Pompa for Ve-CadER-Cre and Tie2-Cre lines, D. Brenner for ColI-GFP reporter mice, and D. Medici for advice on cell plasticity studies. The authors acknowledge funding from the Ministry of Economy and Competitiveness (MINECO)-Spain (SAF2012-38547, PI13/02512, and PLE2009-0124), Association Française Myopathies (AFM), E-Rare, Fundació Marató TV3, Muscular Dystrophy Association (MDA), European Commission Research and Innovation funding EU-FP7 (Myoage, Optistem, and Endostem), and Duchenne PP-NL. P.P. and Y.K. were partly supported by postdoctoral fellowships from AFM.
Published: May 14, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Accession Numbers
The accession number for the microarray data reported in this paper is GEO GSE67687.
Supplemental Information
References
- Abou-Khalil R., Mounier R., Chazaud B. Regulation of myogenic stem cell behavior by vessel cells: the “ménage à trois” of satellite cells, periendothelial cells and endothelial cells. Cell Cycle. 2010;9:892–896. doi: 10.4161/cc.9.5.10851. [DOI] [PubMed] [Google Scholar]
- Acuña M.J., Pessina P., Olguin H., Cabrera D., Vio C.P., Bader M., Muñoz-Canoves P., Santos R.A., Cabello-Verrugio C., Brandan E. Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-β signalling. Hum. Mol. Genet. 2014;23:1237–1249. doi: 10.1093/hmg/ddt514. [DOI] [PubMed] [Google Scholar]
- Ardite E., Perdiguero E., Vidal B., Gutarra S., Serrano A.L., Muñoz-Cánoves P. PAI-1-regulated miR-21 defines a novel age-associated fibrogenic pathway in muscular dystrophy. J. Cell Biol. 2012;196:163–175. doi: 10.1083/jcb.201105013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biressi S., Miyabara E.H., Gopinath S.D., Carlig P.M., Rando T.A. A Wnt-TGFβ2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med. 2014;6:267ra176. doi: 10.1126/scitranslmed.3008411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack A.S., Conboy M.J., Roy S., Lee M., Kuo C.J., Keller C., Rando T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Chong J.J., Reinecke H., Iwata M., Torok-Storb B., Stempien-Otero A., Murry C.E. Progenitor cells identified by PDGFR-alpha expression in the developing and diseased human heart. Stem Cells Dev. 2013;22:1932–1943. doi: 10.1089/scd.2012.0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield J.S., Lupher M., Thannickal V.J., Wynn T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulauroy S., Di Carlo S.E., Langa F., Eberl G., Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012;18:1262–1270. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- Fukada S., Morikawa D., Yamamoto Y., Yoshida T., Sumie N., Yamaguchi M., Ito T., Miyagoe-Suzuki Y., Takeda S., Tsujikawa K., Yamamoto H. Genetic background affects properties of satellite cells and mdx phenotypes. Am. J. Pathol. 2010;176:2414–2424. doi: 10.2353/ajpath.2010.090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieronimakis N., Balasundaram G., Reyes M. Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS ONE. 2008;3:e0001753. doi: 10.1371/journal.pone.0001753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Ogawa R., Uezumi A., Ohtani T., Watanabe Y., Tsujikawa K., Miyagoe-Suzuki Y., Takeda S., Yamamoto H., Fukada S. Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-marker expression in muscle mesenchymal progenitors. Neuromuscul. Disord. 2013;23:349–356. doi: 10.1016/j.nmd.2012.10.025. [DOI] [PubMed] [Google Scholar]
- Joe A.W., Yi L., Natarajan A., Le Grand F., So L., Wang J., Rudnicki M.A., Rossi F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010;12:153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharraz Y., Guerra J., Pessina P., Serrano A.L., Muñoz-Cánoves P. Understanding the process of firbrosis in Duchenne muscular dystrophy. Biomed. Res. Int. 2014;2014:965631. doi: 10.1155/2014/965631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki Y.Y., Hammer R.E., Miyazaki J., Williams S.C., Richardson J.A., Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kisseleva T., Brenner D.A. The phenotypic fate and functional role for bone marrow-derived stem cells in liver fibrosis. J. Hepatol. 2012;56:965–972. doi: 10.1016/j.jhep.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempen K., Grotkopp D., Hall K., Bache A., Gillan A., Rippe R.A., Brenner D.A., Breindl M. Far upstream regulatory elements enhance position-independent and uterus-specific expression of the murine alpha1(I) collagen promoter in transgenic mice. Gene Expr. 1999;8:151–163. [PMC free article] [PubMed] [Google Scholar]
- Krenning G., Zeisberg E.M., Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo H., Shimizu M., Taya Y., Kawamoto T., Michida M., Kaneko E., Igarashi A., Nishimura M., Segoshi K., Shimazu Y. Identification of mesenchymal stem cell (MSC)-transcription factors by microarray and knockdown analyses, and signature molecule-marked MSC in bone marrow by immunohistochemistry. Genes Cells. 2009;14:407–424. doi: 10.1111/j.1365-2443.2009.01281.x. [DOI] [PubMed] [Google Scholar]
- Kumarswamy R., Volkmann I., Jazbutyte V., Dangwal S., Park D.H., Thum T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012;32:361–369. doi: 10.1161/ATVBAHA.111.234286. [DOI] [PubMed] [Google Scholar]
- LeBleu V.S., Taduri G., O’Connell J., Teng Y., Cooke V.G., Woda C., Sugimoto H., Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann C.J., Perdiguero E., Kharraz Y., Aguilar S., Pessina P., Serrano A.L., Muñoz-Cánoves P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D., Kalluri R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012;22:379–384. doi: 10.1016/j.semcancer.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D., Shore E.M., Lounev V.Y., Kaplan F.S., Kalluri R., Olsen B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier R., Chrétien F., Chazaud B. Blood vessels and the satellite cell niche. Curr. Top. Dev. Biol. 2011;96:121–138. doi: 10.1016/B978-0-12-385940-2.00005-X. [DOI] [PubMed] [Google Scholar]
- Nieto M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- Nieto M.A. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- Nieto M.A., Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin. Cancer Biol. 2012;22:361–368. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Nishijo K., Hosoyama T., Bjornson C.R., Schaffer B.S., Prajapati S.I., Bahadur A.N., Hansen M.S., Blandford M.C., McCleish A.T., Rubin B.P. Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J. 2009;23:2681–2690. doi: 10.1096/fj.08-128116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E., Sousa-Victor P., Ruiz-Bonilla V., Jardí M., Caelles C., Serrano A.L., Muñoz-Cánoves P. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol. 2011;195:307–322. doi: 10.1083/jcb.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessina P., Cabrera D., Morales M.G., Riquelme C.A., Gutiérrez J., Serrano A.L., Brandan E., Muñoz-Cánoves P. Novel and optimized strategies for inducing fibrosis in vivo: focus on Duchenne Muscular Dystrophy. Skelet. Muscle. 2014;4:7. doi: 10.1186/2044-5040-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S., Lacombe J., Hanoun M., Mizoguchi T., Bruns I., Kunisaki Y., Frenette P.S. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrió D., Rodriguez-Pinilla S.M., Hardisson D., Cano A., Moreno-Bueno G., Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- Serrano A.L., Mann C.J., Vidal B., Ardite E., Perdiguero E., Muñoz-Cánoves P. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr. Top. Dev. Biol. 2011;96:167–201. doi: 10.1016/B978-0-12-385940-2.00007-3. [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.S., William C.M., Tanabe Y., Jessell T.M., Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- Uezumi A., Fukada S., Yamamoto N., Takeda S., Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010;12:143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- Uezumi A., Ito T., Morikawa D., Shimizu N., Yoneda T., Segawa M., Yamaguchi M., Ogawa R., Matev M.M., Miyagoe-Suzuki Y. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011;124:3654–3664. doi: 10.1242/jcs.086629. [DOI] [PubMed] [Google Scholar]
- Uezumi A., Fukada S., Yamamoto N., Ikemoto-Uezumi M., Nakatani M., Morita M., Yamaguchi A., Yamada H., Nishino I., Hamada Y., Tsuchida K. Identification and characterization of PDGFRα+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014;5:e1186. doi: 10.1038/cddis.2014.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal B., Serrano A.L., Tjwa M., Suelves M., Ardite E., De Mori R., Baeza-Raja B., Martínez de Lagrán M., Lafuste P., Ruiz-Bonilla V. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–1752. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace G.Q., McNally E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009;71:37–57. doi: 10.1146/annurev.physiol.010908.163216. [DOI] [PubMed] [Google Scholar]
- Wang Z., Storb R., Lee D., Kushmerick M.J., Chu B., Berger C., Arnett A., Allen J., Chamberlain J.S., Riddell S.R., Tapscott S.J. Immune responses to AAV in canine muscle monitored by cellular assays and noninvasive imaging. Mol. Ther. 2010;18:617–624. doi: 10.1038/mt.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosczyna M.N., Biswas A.A., Cogswell C.A., Goldhamer D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012;27:1004–1017. doi: 10.1002/jbmr.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M., Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013;304:C216–C225. doi: 10.1152/ajpcell.00328.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M., Yang C., Martino M., Duncan M.B., Rieder F., Tanjore H., Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- Zhou L., McMahon C., Bhagat T., Alencar C., Yu Y., Fazzari M., Sohal D., Heuck C., Gundabolu K., Ng C. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res. 2011;71:955–963. doi: 10.1158/0008-5472.CAN-10-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan P., Rigamonti E., Freudenberg K., Conti V., Azzoni E., Rovere-Querini P., Brunelli S. Macrophages commit postnatal endothelium-derived progenitors to angiogenesis and restrict endothelial to mesenchymal transition during muscle regeneration. Cell Death Dis. 2014;5:e1031. doi: 10.1038/cddis.2013.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.