

Abstract
In an earlier study, β3-puromycin was used for the selection of modified ribosomes, which were utilized for the incorporation of five different β-amino acids into Escherichia coli dihydrofolate reductase (DHFR). The selected ribosomes were able to incorporate structurally disparate β-amino acids into DHFR, in spite of the use of a single puromycin for the selection of the individual clones. In this study, we examine the extent to which the structure of the β3-puromycin employed for ribosome selection influences the regio- and stereochemical preferences of the modified ribosomes during protein synthesis; the mechanistic probe was a single suppressor tRNACUA activated with each of four methyl-β-alanine isomers (1–4). The modified ribosomes were found to incorporate each of the four isomeric methyl-β-alanines into DHFR but exhibited a preference for incorporation of 3(S)-methyl-β-alanine (β-mAla; 4), i.e., the isomer having the same regio- and stereochemistry as the O-methylated β-tyrosine moiety of β3-puromycin. Also conducted were a selection of clones that are responsive to β2-puromycin and a demonstration of reversal of the regio- and stereochemical preferences of these clones during protein synthesis. These results were incorporated into a structural model of the modified regions of 23S rRNA, which included in silico prediction of a H-bonding network. Finally, it was demonstrated that incorporation of 3(S)-methyl-β-alanine (β-mAla; 4) into a short α-helical region of the nucleic acid binding domain of hnRNP LL significantly stabilized the helix without affecting its DNA binding properties.
The ribosome mediates the incorporation of amino acids into protein with exceptionally high fidelity.1,2 Nevertheless, the mechanisms that impart high fidelity to protein biosynthesis also place constraints on the structures of the amino acids that can be incorporated into proteins. Engineering proteins with noncanonical amino acids has been a subject of interest for both chemists and biologists; the strategy remains challenging because of constraints imposed by the ribosome.3 β-Amino acids are interesting analogues and have the ability to introduce regio- and stereochemical diversity into the current set of amino acid residues available for protein modifications. β-Amino acids are not found in ribosomally synthesized proteins even though they are present in cells.4
The bias of the ribosome against β-amino acids is well-established.5−9 It has been demonstrated that β-aminoacyl-tRNAs are poor substrates for the bacterial ribosome5−8 and β-aminoacyl-tRNA cannot participate effectively in peptide bond formation at the peptidyltransferase center (PTC) (Figure 1).9 The strong bias of the ribosome against β-amino acids has limited their incorporation into proteins using cell free or cellular protein biosynthesizing systems. Puromycin (Figure 2) is an aminonucleoside antibiotic with a structure that resembles the 3′-end of the aminoacyl-tRNA, rendering it a putative aminoacyl-tRNA mimic.10,11 It binds to the A-site of the PTC and takes part in peptide bond formation with the nascent polypeptide attached to the peptidyl-tRNA in the P-site, leading to premature chain release.10,11 Recently, we described a set of modified ribosomes selected using β3-puromycin (Figure 2),12 which allowed the site-specific incorporation of five different β-amino acids into full length Escherichia coli dihydrofolate reductase (DHFR) in varying yields in a cell free translation system.13
Figure 1.
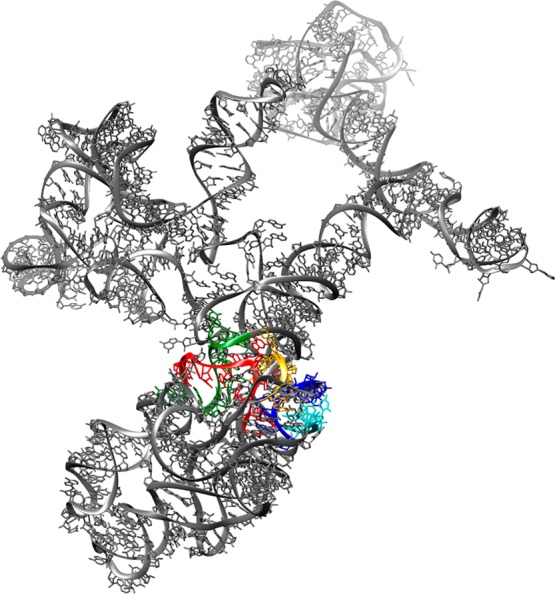
Structure of domain V of 23S rRNA of the E. coli strain K12 70S ribosome (Protein Data Bank entry 2WWQ). Peptidyltransferase center (PTC) loop regions are highlighted as follows: yellow, nucleotides 2057–2063; green, nucleotides 2447–2456; red, nucleotides 2496–2507; blue, nucleotides 2582–2588; cyan, nucleotides 2606–2611.
Figure 2.

Structures of puromycin and β-puromycin derivatives.
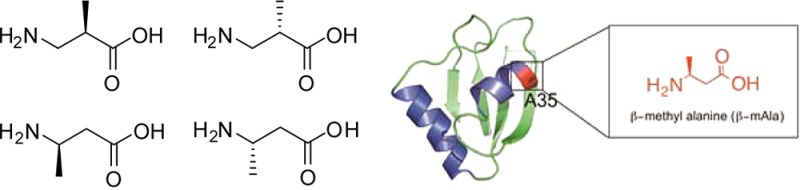
The foregoing studies12,13 did not directly address the question of the scope of β-amino acid structures that might be recognized by ribosomes selected using a single puromycin. The β3-puromycin used for the selection of the modified ribosomes12,13 had its β-amino acid moiety as a single regio- and stereoisomer (3-substituted S-isomer). It seemed logical to anticipate that the modified ribosomes might display a preference during protein synthesis for aminoacyl-tRNAs having region- and stereochemistry analogous to that of the β-amino acid moiety in the β3-puromycin used for ribosome selection. Therefore, we assessed the regio- and stereochemical preference of the modified ribosomes toward the incorporation of β-amino acids. We utilized methyl-β-alanine isomers 1–4 for that purpose (Figure 3A).
Figure 3.

(A) Structures of β-amino acids studied. (B) Strategy employed for their incorporation into DHFR using modified ribosomes.
To further verify the ability of the puromycin derivative to select modified ribosomes having an A-site architecture complementary to the regio- and stereochemistry of the puromycin, we selected a new set of modified ribosomes using a β2-puromycin (Figure 2). This puromycin derivative has regio- and stereochemistry opposite from that of the β3-puromycin. Methyl-β-alanine isomers 1–4 were again employed to evaluate the regio- and stereoselectivity of the new set of modified ribosomes in protein synthesis, and a model of the key regions of the 23S rRNAs of two ribosomal clones is presented.
Finally, we also exemplify the utility of the ribosomes capable of utilizing β-amino acids by incorporating methyl-β-alanine 4 into position 35 of a nucleic acid binding domain (RRM1) of heterogeneous nuclear ribonucleoprotein L-like (hnRNP LL), which has been shown to bind to a DNA i-motif in the promoter region of BCL2.14 Position 35, which normally contains Ala, is in a short and relatively unstable α-helical region of an RNA recognition motif (RRM1) of hnRNP LL; the introduction of 4 (β-mAla) in lieu of Ala was found to significantly stabilize the α-helical structure, while retaining its i-motif DNA binding properties.
Materials and Methods
Tris, acrylamide, bis-acrylamide, urea, ammonium persulfate, N,N,N′,N′-tetramethylenediamine (TEMED), dihydrofolic acid, glycerol, ampicillin, pyruvate kinase, lysozyme, erythromycin, isopropyl β-d-thiogalactopyranoside (IPTG), dithiothreitol (DTT), and 2-mercaptoethanol were purchased from Sigma Chemicals (St. Louis, MO). [35S]Methionine (10 μCi/μL) was obtained from Amersham (Pitscataway, NJ). BL-21(DE-3) competent cells and T4 RNA ligase were from Promega (Madison, WI). The Plasmid MaxiKit (Life Science Products, Inc., Frederick, CO) and the GenEluteHP plasmid miniprep kit (Sigma) were used for plasmid purification.
Phosphorimager analysis was performed using a Molecular Dynamics 400E PhosphorImager equipped with ImageQuant version 3.2. Ultraviolet and visible spectral measurements were taken using a PerkinElmer lambda 20 spectrophotometer. Circular dichroism spectra were recorded using a Jasco-810 spectropolarimeter.
Preparation of β-Aminoacyl-tRNACUAs
The activation of suppressor tRNACUAs was conducted as described previously.15,16 Briefly, a 100 μL reaction mixture of 100 mM Na Hepes (pH 7.5) contained 1.0 mM ATP, 15 mM MgCl2, 100 μg of suppressor tRNACUA-COH, 0.5 A260 unit of N-pentenoyl-protected β-aminoacyl-pdCpA, 15% DMSO, and 100 units of T4 RNA ligase. The reaction mixture was incubated at 37 °C for 1.5 h and the reaction quenched by the addition of 0.1 volume of 3 M NaOAc (pH 5.2). The N-protected aminoacylated tRNA was precipitated with 3 volumes of cold ethanol. The efficiency of ligation was estimated by 8% polyacrylamide–7 M urea gel electrophoresis (pH 5.0).
The N-pentenoyl-protected aminoacyl-tRNACUAs were deprotected by being treated with 5 mM aqueous I2 at 25 °C for 15 min. The solution was centrifuged, and the supernatant was adjusted to 0.3 M NaOAc and treated with 3 volumes of cold ethanol to precipitate the aminoacylated tRNA. The tRNA pellet was collected by centrifugation, washed with 70% aqueous EtOH, air-dried, and dissolved in 20 μL of RNase free water.
Preparation of S-30 Extracts from Cells Having Modified Ribosomes
Aliquots (5–10 μL) from liquid stocks of E. coli BL-21(DE-3) cells, harboring plasmids with a wild-type or modified rrnB gene, were plated on LB agar supplemented with 100 μg/mL ampicillin and grown at 37 °C for 16–18 h. One colony was picked from each agar plate and transferred into 3 mL of LB medium supplemented with 100 μg/mL ampicillin and 0.5 mM IPTG. The cultures were grown at 37 °C for 3–6 h in a thermostated shaker until the OD600 reached ∼0.15–0.3, diluted with LB medium supplemented with 100 μg/mL ampicillin, 1 mM IPTG, and 3 μg/mL erythromycin (for selectively enhancing the modified ribosome fraction) until the OD600 reached 0.01, and then grown at 37 °C for 12–18 h. The optimal concentration of the final cultures was an OD600 of 0.5–1.0. Cells were harvested by centrifugation (5000g, 4 °C, 10 min) and washed three times with S-30 buffer [10 mM Tris-OAc (pH 8.2) containing 1.4 mM Mg(OAc)2, 6 mM KOAc, and 0.1 mM DTT] supplemented with β-mercaptoethanol (0.5 mL/L) and once with S-30 buffer having 0.05 mL/L β-mercaptoethanol. The weight of the wet pellet was estimated, and 1.27 mL of S-30 buffer was added to suspend each gram of cells. The volume of the suspension was measured and used for estimating the amount of other components. The preincubation mixture (0.3 mL) [0.29 M Tris (pH 8.2) containing 9 mM Mg(OAc)2, 13 mM ATP, 84 mM phosphoenolpyruvate, 4.4 mM DTT, and 5 μM amino acids mixture], 15 units of pyruvate kinase, and 10 μg of lysozyme were added per milliliter of cell suspension, and the resulting mixture was incubated at 37 °C for 30 min. The incubation mixture was then frozen at −80 °C (∼30 min), thawed (37 °C, 30 min), and again frozen and thawed at room temperature (∼30 min). Ethylene glycol tetraacetic acid (EGTA) was then added to a final concentration of 2.5 mM, and the cells were incubated at 37 °C for 30 min; CaCl2 was added to a final concentration of 2.5 mM and the mixture again frozen (−80 °C, 30 min). The frozen mixture was centrifuged (15000g, 4 °C, 1 h), and the supernatant was stored in aliquots at −80 °C.
In Vitro Protein Translation
Protein translation reactions were conducted in 12–1200 μL of incubation mixture containing 0.2–0.4 μL/μL of S-30 system, 100 ng/μL plasmid, 35 mM Tris acetate (pH 7.4), 190 mM potassium glutamate, 30 mM ammonium acetate, 2 mM DTT, 0.2 mg/mL total E. coli tRNA, 3.5% PEG 6000, 20 μg/mL folinic acid, 20 mM ATP and GTP, 5 mM CTP and UTP, 100 μM amino acid mixture, 0.5 μCi/μL of [35S]methionine, and 1 μg/mL rifampicin. In the case of plasmids having a gene with a TAG codon, β-aminoacyl-tRNACUAs were added to a concentration of 0.6–1.0 μg/μL. Reactions were conducted at 37 °C for 1 h and terminated when the mixtures were chilled on ice. Aliquots from in vitro translation mixtures were analyzed by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis followed by quantification of the radioactive bands by phosphorimager analysis.
“In-Gel” Trypsin Digestion17
Samples to be digested in the gel were run in three or four lanes of a 12% SDS–polyacrylamide gel, stained with Coomassie R-250, and destained until the background was clear. The band containing DHFR was cut from the gel and washed with 0.1 M ammonium bicarbonate (1 h, room temperature). The solution was discarded, and 0.1–0.2 mL of 0.1 M ammonium bicarbonate and 10–30 μL of 0.045 mM DTT were added. Gel pieces were incubated at 60 °C for 30 min, cooled to room temperature, and incubated at room temperature for 30 min in the dark after the addition of 10–30 μL of 0.1 M iodoacetamide. Gel pieces were washed with a 1:1 acetonitrile/0.1 M ammonium bicarbonate mixture until they became colorless. After the solution had been discarded, the gel pieces were incubated in 0.1–0.2 mL of acetonitrile (10–20 min at room temperature) and, after removal of solvent, were reswelled in 50–100 μL of 25 mM ammonium bicarbonate containing 0.02 μg/μL trypsin. After incubation at 37 °C for 4 h, the supernatant was removed to a new tube and the peptides were extracted with 60% acetonitrile in 0.1% CF3COOH (20 min at room temperature). The combined fractions were dried and reconstituted in a minimal amount of 60% acetonitrile in 0.1% CF3COOH, all of which was used for MALDI-MS analysis.
Selection of Modified Ribosomes Using β2-Puromycin
The selection experiments were conducted as described previously.12 Briefly, a mini library (96-well master plate) was prepared from 62 clones, which previously demonstrated some, but <40% inhibition of cell growth in the presence of β3-puromycin, by adding 100 μL of a glycerol stock of each clone in each well. Three new plates were prepared by transferring 2 μL of culture from each well of the master plate to the corresponding well of a new plate, and 98 μL of the assay solution was added to each well. Assay solutions were prepared from LB medium (pH 8.4) containing 100 μg/mL ampicillin, 1 mM IPTG, and 100 μg/mL β2- or β3-puromycin. A solution without any puromycin was used for a control plate. All plates were incubated at 37 °C for 16–18 h in a temperature-controlled shaker, and cell growth was estimated by measuring the optical density at 600 nm (OD600). Inhibition of cell growth by β2- and β3-puromycin was estimated as described previously.12 Clones with an inhibition value of >50% were used for verification experiments. Cultures were diluted with an assay solution (LB medium supplemented with 100 μg/mL ampicillin and 1 mM IPTG) until the OD600 value was ∼0.01 and placed in eight wells of a 96-well plate, having six different dilutions of β2- or β3-puromycin (dilution from 200 to 6.25 μg/mL). Plates were incubated at 37 °C for 16–18 h in a temperature-controlled shaker, and cell growth was estimated by measuring the optical density at 600 nm. IC50 data were calculated for β2- and β3-puromycin for each clone.
Plasmids from cultures showing a 1.5–2-fold difference in IC50 values between β2- and β3-puromycin were isolated and retransformed into BL-21(DE-3) cells. The sensitivity of new cultures to β2- and β3-puromycin in the presence of erythromycin was determined as described above. In this case, 2.5 μg/mL erythromycin was also added to the assay solution.
Modeling of the Structures of the Modified 23 rRNAs
The structure of E. coli 23S rRNA [Protein Data Bank (PDB) entry 2WWQ] was used as the reference structure.18 PTC loop regions were defined as reported.19 Three-dimensional structure visualization was conducted with Chimera 1.9.20 Hydrogen bonds were predicted using constraints relaxed by 0.4 Å and 20.0°.
Expression and Study of an α-Helix in hnRNP LL RRM1
Mutagenesis and Subcloning
The PCR (polymerase chain reaction) site-directed mutagenesis procedure was conducted using the New England Biolabs Q5 Mutagenesis kit. The NEB base changer web-tool was used to design the primers, to incorporate an amber codon (TAG) at position A35 of RRM1. PCR was performed in a 25 μL reaction mixture, following the procedure described in the kit manual. Each reaction mixture contained 25 ng of wild-type RRM1 template, encoded in a pET28a vector, 125 ng of forward and reverse primers, 10 nmol of dNTPs, 2.5 units of DNA polymerase in 35 mM Tris-HCl (pH 8.0) containing 12 mM potassium acetate, 5 mM DTT, and 0.05% Triton X-100 in 0.05 mM EDTA. The PCR products were ligated with the Q5 ligase master mix and transformed into DH5α E. coli cells. Purified plasmids were verified by sequencing.
In Vitro RRM1 Translation
Proteins for CD spectral analysis were prepared by in vivo (RRM1wt) and in vitro (RRM1wt and RRM 1β-mAla) translation and purified by using the Strep-tag@ protein purification system (IBA Inc.). Final samples were stored (10 μM protein concentration) in elution buffer (100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA and 2.5 mM desthiobiotin).
Circular Dichroism (CD)
CD spectra were recorded on a Jasco-810 spectropolarimeter equipped with a temperature controller using 1 mm quartz cells. Spectra were obtained at scan rates of 50 nm/min (protein) and 100 nm/min (DNA). Three scans for each sample were performed at 25 °C over a wavelength range of 195–260 nm, and the average means were calculated. The noise in the data was smoothed using JASCO-810 software. The ellipticity data were estimated in millidegrees and converted to molar ellipticity according to a standard protocol.21 The melting points (Tm) of RRM1wt and RRM1β-mAla were estimated on the basis of CD spectral data obtained at different temperatures (from 4 to 94 °C with 5 °C intervals). The signal intensities at 222 nm were plotted against the corresponding temperature value and smoothed with a nonlinear sigmoidal data fit program.
DNA–protein binding studies were performed on the basis of a published protocol.14 The samples, containing 5 μM i-motif oligonucleotide, which is involved in binding by RRM1, and varying (0–7.5 μM) amounts of RRM1wt and RRM1β-mAla were prepared in 50 mM Tris-HCl (pH 6.6) containing 100 mM NaCl. Before CD spectral measurements, all samples were incubated for 15 min at room temperature. Each sample was scanned over a wavelength range of 240–350 nm, averaged, smoothed, and baseline corrected. The molar ellipticity [θ] was estimated and plotted versus wavelength. Sigmoidal curves (the CD signal at 286 nm on the Y-axis vs the molar protein:DNA ratio on the X-axis) were prepared with use of prism data plotting tools. Kd estimations for both samples were done with the highest R2 values by using the Hill slope option for data points, which were fitted using a predefined equation for 1:1 nonlinear binding saturation.
The equation Y = Bmax × Xh/(Kdh + Xh) was employed, where Y is the determinant of specific binding, X = [protein]/[DNA], Bmax is maximal specific binding, and h is the Hill slope.
End Labeling of Oligonucleotides
The BCL2 i-motif was 5′-32P end labeled with [γ-32P]ATP and T4 polynucleotide kinase. Ten picomoles of DNA was 5′-32P end labeled by incubation with 20 units of T4 polynucleotide kinase and 0.06 mCi of [γ-32P]ATP [specific activity of 6000 Ci (222 TBq)/mmol] in 50 μL (total volume) of 70 mM Tris-HCl (pH 7.6) containing 10 mM MgCl2 and 5 mM DTT. The reaction mixture was incubated at 37 °C for 1 h followed by purification of DNA by 16% polyacrylamide gel electrophoresis at 1800 V for 2.5 h.
Electrophoretic Mobility Shift Assay
The BCL2 i-motif was 5′-32P end labeled as noted above. These constructs were prepared in 50 mM Tris buffer (pH 7.0) containing 100 mM NaCl. One microliter (10000 cpm) of the DNA was mixed with 10 μM unlabeled oligonucleotide. Ten picomoles of the DNAs was then incubated with 250 pmol of RRM1wt or RRM1β-mAla for 15 min at room temperature in the same buffer. The DNA/protein mixtures were subjected to 12% nondenaturing gel electrophoresis at 80 V for 1.5 h. The resulting bands were visualized using a phosphorimager.
Results
Suppressor tRNACUAs misacylated with β-amino acids were prepared from individual β-aminoacyl-pdCpAs as described previously.12,13 Scheme 1 illustrates the preparation of a suppressor tRNACUA misacylated with β-amino acid 4. The preparation of the remaining β-aminoacyl-tRNACUAs starting from free β-amino acids is described in the Supporting Information (Schemes S1–S3). For the incorporation of β-amino acids 1–4 into DHFR, S-30 systems were prepared from E. coli colonies harboring different modified ribosomes.12,13 Their ability to synthesize full length DHFR in the presence of β-aminoacyl-tRNACUA and a DHFR mRNA transcript having a UAG codon at position Val10 was measured. The suppression efficiency for each β-aminoacyl-tRNACUA was expressed relative to the synthesis of full length wild-type DHFR using the S-30 systems prepared from different modified ribosomes. As a negative control, DHFR synthesis in the presence of nonacylated tRNACUA was evaluated. All S-30 preparations produced very low levels of nonspecific read-through of the UAG codon in the presence of nonacylated suppressor tRNACUA. The amounts of DHFR produced by each S-30 system were quantified with a phosphorimager, which monitored the incorporation of [35S]methionine into DHFR.
Scheme 1. Preparation of 8, tRNACUA Activated with β-Amino Acid 4.

In Vitro Translation Using Methyl-β-alanine Derivatives 1–4 with Modified Ribosomes Selected Using β3-Puromycin
Table 1 compares the efficiencies of incorporation of methyl-β-alanine derivatives 1–4 into DHFR at position 10, relative to the wild-type DHFR synthesis using S-30 systems prepared from four different ribosomal clones (Figure 3B). These clones were identified previously, on the basis of a selection employing β3-puromycin.12,13 Among the four methyl-β-alanine isomers examined, the best incorporation results were obtained with 4 (up to 19% suppression yield) using clone 040217. The incorporation efficiency of 3(S)-methyl-β-alanine (4) was 2–4 times higher than that of 3(R)-methyl-β-alanine (3). Furthermore, the incorporation of 3-substituted methyl-β-alanine derivatives produced up to 5-fold greater amounts of modified DHFR, when compared with those of the 2-substituted methyl-β-alanine derivatives. These ribosomal clones were unable to discriminate between the stereoisomers of 2-substituted methyl-β-alanines, where β-amino acids 1 (R-isomer) and 2 (S-isomer) were incorporated in similar yields (∼2–4% suppression yields). Ribosomal clone 040321 demonstrated the best regio- and stereoselectivity, giving up to 4-fold greater amounts of full length DHFR in the presence of β-aminoacyl-tRNACUA bearing 4 (∼8% suppression yield) relative to those of other methyl-β-alanine derivatives (∼2% suppression yields). To provide direct evidence of the incorporation of methyl-β-alanine derivatives into DHFR, we prepared a modified DHFR having 4 at position 10 on a larger scale. The modified DHFR was purified by the use of Ni-NTA chromatography followed by SDS–polyacrylamide gel electrophoresis. The purified DHFR sample present in the SDS–polyacrylamide gel was treated with trypsin according to a published protocol,17 and the tryptic digest was subjected to matrix-assisted laser desorption ionization (MALDI) mass spectrometry (MS) analysis. A tryptic fragment encompassing amino acids 1–12 (4 at position 10) was anticipated to have a molecular mass of 1259 Da. As shown in Figure 4, there was an ion peak observed m/z 1259.17. This ion was not present in the tryptic digest of DHFR V10F, but the anticipated ion at m/z 1321.49 (reflecting the presence of Phe10) was readily apparent (Figure S1 of the Supporting Information).
Table 1. Incorporation of β-Amino Acids 1–4 into Position 10 of E. coli Dihydrofolate Reductase (DHFR) by Using S-30 Systems Having Different Modified Ribosomes Selected Using β3-Puromycin.
| sequence
in 23S rRNA of the modified ribosome |
suppression efficiency in different S-30 systems,
having modified ribosomes (%)a |
||||||
|---|---|---|---|---|---|---|---|
| clone | region 1 | region 2 | –b | 1 | 2 | 3 | 4 |
| 0403x4 | 2057AGCGUGA2063 | 2502AGCCAG2507 | 0.07c ± 0.05 | 2.7 ± 1.1 | 3 ± 1.3 | 6.7 ± 1.1 | 11.8 ± 1.5 |
| 040321 | 2057AGCGUGA2063 | 2502AGAUAA2507 | 0.12 ± 0.04 | 2.1 ± 1 | 3.4 ± 1.9 | 1.8 ± 0.8 | 8.3 ± 1.5 |
| 040329 | 2057AGCGUGA2063 | 2502UGGCAG2507 | 0.06 ± 0.03 | 1.8 ± 0.4 | 2.8 ± 0.6 | 5.3 ± 1.5 | 9.8 ± 1.1 |
| 040217 | 2057AGCGUGA2063 | 2496AUAGAA2501 | 0.2 ± 0.1 | 3.6 ± 0.5 | 4.2 ± 1.1 | 10.6 ± 3.3 | 17.8 ± 1.2 |
| wild typed | 2057GAAAGAC2063 | 2496CACCUC2501 | |||||
| 2502GAUGUC2507 | |||||||
The amount of wild-type DHFR translated using S-30 systems having the corresponding modified ribosome was arbitrarily assigned a value of 100. The suppression efficiency for each β-amino acid was calculated relative to the amount of wild-type DHFR.
Nonspecific read-through of the amber stop codon relative to the amount of wild-type DHFR synthesis.
Each number represents the average of three independent experiments ± the standard deviation.
Incorporation of representative β-amino acids by an S-30 system prepared from wild-type ribosomes affected suppression to the extent of <1%.
Figure 4.

MALDI mass spectrum of tryptic fragments of modified DHFR having 3(S)-methyl-β-alanine (βA; 4) at position 10 (MISLIAALAβADR). Mass range of 1000–1600 Da (the asterisk denotes an estimated value in daltons).
Additionally, clone 040329 was found to exhibit particularly good regioselectivity (3 and 4 vs 1 and 2) and was used for direct comparison with clones selected using β2-puromycin.
Selection of the Modified Ribosomes Using β2-Puromycin
To further establish that the regio- and stereochemical control shown by modified ribosomes reflects the regio- and stereochemistry of the amino acid moiety of the puromycin derivative used in the selection, we selected modified ribosomes using a puromycin derivative having the opposite regio- and stereochemistry compared with those of β3-puromycin. Figure 2 shows the structure of this β2-puromycin derivative; its preparation is outlined in Scheme 2. During our first selection experiments using β3-puromycin, clones showing some (but <40%) inhibition of cell growth in the presence of β3-puromycin were not used for any further experiments.12 It seemed logical to think that this pool of clones might be a good source of clones more sensitive to β2-puromycin. Therefore, we used this pool of clones for a cell growth inhibition assay in the presence of β2-puromycin; six clones were found to exhibit >40% inhibition of cell growth (Figure 5A). The sensitivity to β2-puromycin was verified for these six selected clones by means of an IC50 assay, using both β-puromycin derivatives (Figure 5B). Three clones (010335, 010374, and 070215) demonstrated good sensitivity to β2-puromycin (IC50 values 1.5–2-fold lower than those of β3-puromycin) and were chosen for isolation and sequencing.
Scheme 2. Synthesis of β2-Puromycin (11).

Figure 5.
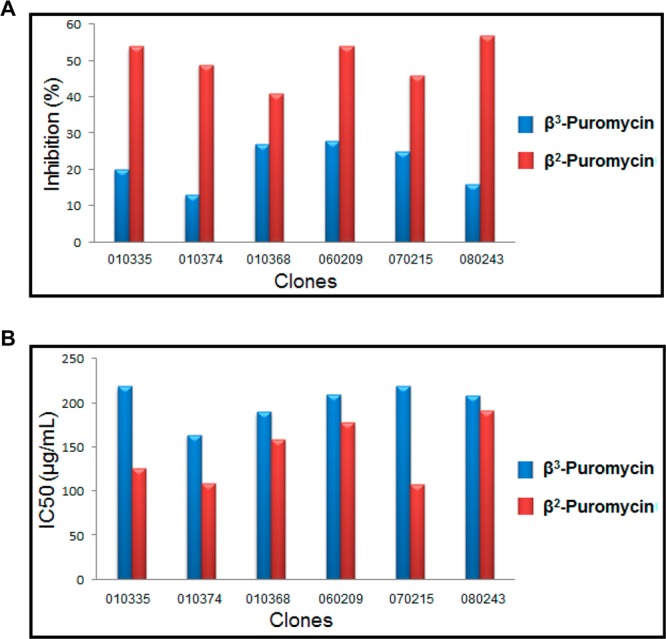
(A) Cell growth inhibition assay in the presence of β3-puromycin and β2-puromycin for the selection of modified ribosomes. (B) IC50 assay for measuring the sensitivity of modified ribosomal clones to β3-puromycin and β2-puromycin.
Two of these clones (010335 and 010374) had modifications in both crucial regions (nucleotides 2057–2063 and 2502–2507) of the PTC in the 23S rRNA, while clone 070215 had the wild-type sequence in the second mutated region. Plasmids from these two clones were transformed in BL-21(DE-3) cells to further verify the sensitivity to β-puromycin derivatives in the presence of erythromycin (Table 2). Cell cultures having modified ribosomal clones 010335 and 010374 demonstrated >2-fold greater sensitivity to β2-puromycin as compared to β3-puromycin in the presence of erythromycin (2.5 μg/mL). These two clones were used for translation experiments with β-amino acids 1–4.
Table 2. IC50 Assay for Verifying the Sensitivity of the Modified Ribosomes to β-Puromycin Derivatives in the Presence of Erythromycin (2.5 μg/mL).
| sequence
in 23S rRNA of the modified ribosome |
IC50 (μg/mL)a |
|||
|---|---|---|---|---|
| clone | region 1 | region 2 | β3-puromycin | β2-puromycin |
| 010335 | 2057UGCGUGG2063 | 2502AGAUGA2507 | 163 ± 17 | 71 ± 10 |
| 010374 | 2057UGCGUGG2063 | 2502CGCUCG2507 | 125 ± 7 | 59 ± 5 |
Each number represents the average of three independent experiments ± the standard deviation.
In Vitro Translation Using Methyl-β-alanine Derivatives 1–4 with Modified Ribosomes Selected Using β2-Puromycin
After successful selection of two modified ribosomes displaying >2-fold enhanced sensitivity to β2-puromycin relative to β3-puromycin, their regio- and stereochemical preferences during protein translation were examined. The amino acid moiety in β2-puromycin has a benzyl group at position 2 with R-stereochemistry (Figure 2). It was anticipated that these clones would favor 2-methyl-β-alanine derivatives 1 and 2, possibly with enhanced selectivity for R-isomer 1. In accordance with the regioisomerism of the amino acid moiety in β2-puromycin used in the selection experiments, both ribosomal clones incorporated 2-methyl-β-alanine isomers 1 and 2 with moderately increased yields over the 3-methyl-β-alanine derivatives; this is illustrated in Figure 6 for the incorporation of the four β-amino acids into position 10 of DHFR by clone 010335. Also illustrated in Figure 6 for comparison is the incorporation of the same four β-amino acids into position 10 of DHFR by clone 040329, which was selected using β3-puromycin (Table 1). As anticipated, β-amino acid 4 was incorporated with the greatest efficiency.
Figure 6.

Translation of DHFR from a wild-type and modified DHFR (lanes 1–5) mRNA (UAG codon at position 10) by the use of S-30 systems prepared from ribosomal clones 010335 and 040329 in the presence of different suppressor tRNAs (lanes 1–5): lane 1, nonacylated tRNACUA; lane 2, tRNACUA activated with β-amino acid 1; lane 3, tRNACUA activated with β-amino acid 2; lane 4, tRNACUA activated with β-amino acid 3; lane 5, tRNACUA activated with β-amino acid 4.
Partial Structures of the Peptidyltransferase Center of Clones 040329 and 010335 at the Level of 23S rRNA Depicting the Regions at Which the Modifications Were Introduced
Extrapolation from the study on the Haloarcula marismortui 50S ribosomal subunit suggests that the putative catalytic function22−24 of A2451 as a general base during the nucleophilic attack by the α-amino group of the A-site substrate is supported by interactions among G2061, G2447, A2450, and A2451.25 In this context, the hydrogen bond nets in loop regions 2057–2063 and 2496–2507 were compared for the two clones used in the incorporation experiments shown in Figure 6 (Figure 7 and Table S1 of the Supporting Information). Both clones had a G2061U replacement that alters the space around A2451 and likely affects the 2451–2061 hydrogen bond length. Of particular interest is the fact that all four clones selected using β3-puromycin also had uridine at position 2061. The presence of U2061 may be critical for the recognition of substituted β-amino acids but cannot solely account for the differences in regio- and stereoselectivity observed between clones 040329 and 010335. Most likely, the combination of G2061U replacement and the densely mutated region of nucleotides 2502–2507, essentially nonidentical between the clones and also compared to the wild type, contributes to the specific characteristics of the modified ribosomes.
Figure 7.
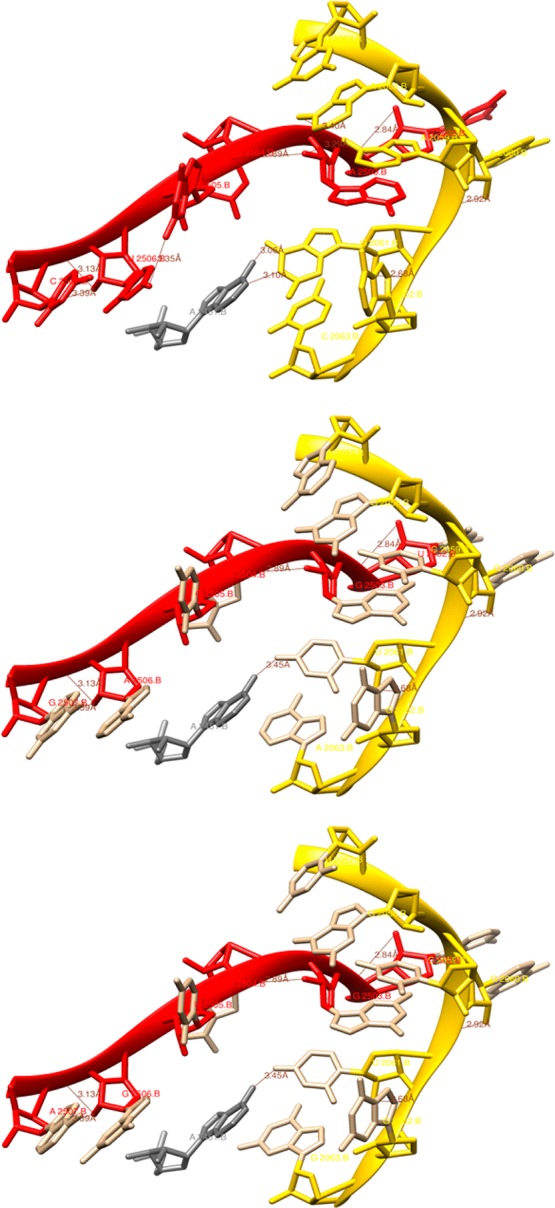
Partial structure of PTC of the E. coli strain K12 70S ribosome at the level of 23S rRNA depicting the nucleotide regions 2057–2063 (yellow) and 2496–2507 (red) where the mutations were introduced: top panel, wild-type ribosome; middle panel, clone 040329; bottom panel, clone 010335. The catalytically important nucleotide A2451 is colored gray along with possible hydrogen bonding interactions.
Stabilization of an α-Helical Structure by Replacement of Alanine with 3(S)-Methyl-β-alanine (4)
The wild-type RRM1 domain of human hnRNP LL protein was cloned into the pET28a vector and expressed using E. coli BL21(DE-3) cells. The domain structure was determined by sequence homology interpretation based on other well-characterized homologous proteins of the family (N-terminus of mouse protein BAB28521, PDB entry 1WEX) using Modeller.26,27 The DNA binding residues in RRM1 were found using DP-bind.28,29 The predicted DNA binding residues were R26, G27, F55, K56, R57, Q58, Q86, F89, N91, and Y92, all of which reside on the loop structure of RRM1 (Figure S2A of the Supporting Information). The modified RRM1 protein containing β-amino acid 4 at position 35 was synthesized in a cell free in vitro expression system in the presence of an S-30 preparation derived from ribosomal clone 040329; as shown in Figure 8, the suppression yield was 12–13%. Unmodified RRM1 was prepared using an in vivo expression system and had a CD spectrum identical with that of the same protein (RRM1wt) made by using the plasmid construct encoding RRM1 in a wild-type cell free protein-synthesizing system (Figure 9B). As is evident from the CD spectrum, the RRM1 domain was largely unstructured, evident by a negative peak centered at 205 nm in the far-UV region corresponding to random coil.
Figure 8.
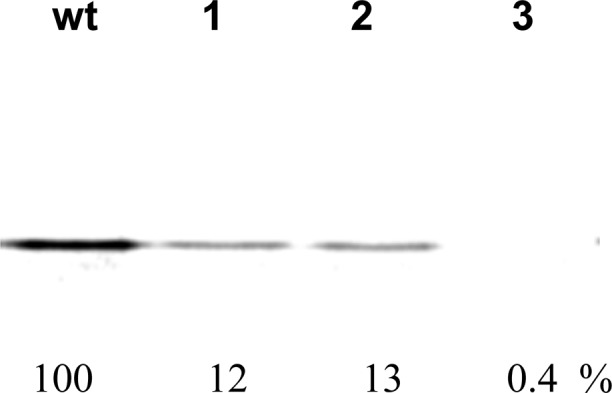
Translation of RRM1 from wild-type (wt) and modified RRM1 mRNA (TAG codon at the position corresponding to Ala35) by using an S-30 system prepared from ribosomal clone 040329 in the presence of 3(S)-methyl-β-alanyl-tRNACUA (β-mAla; 4) (lanes 1 and 2) or in the absence of any tRNACUA (lane 3).
Figure 9.

Structural change in RRM1 caused by the incorporation of β-mAla 4. (A) Three-dimensional structure of RRM1, highlighting A35, modeled with reference to the solution structure of N-terminal mouse protein BAB28521 (PDB entry 1WEX). (B) CD spectra of RRM1 (in vivo) and RRM1 (in vitro) showed identical patterns of random coil structure, whereas that of RRM1β-mAla showed a stable α-helix. (C) Melting points for RRM1wt and RRM1β-mAla.
As noted above, a modified RRM1 domain of i-motif DNA binding protein hnRNP LL was prepared using modified ribosomes for the incorporation of 3(S)-methyl-β-alanine (β-mAla). The β-amino acid was used to activate a suppressor tRNACUA, the latter of which was used to suppress a UAG codon at the position corresponding to alanine 35 (A35) of wild-type RRM1. Alanine 35 is situated at the beginning of the smaller α-helix of RRM1 (α2) (Figure 9A) which is thought to be a critically important region for helix stability. Interestingly, the CD spectrum (Figure 9B) of RRM1β-mAla exhibited two minima at 222 and 208 nm, characteristic of α-helical secondary structures. To probe the stability of this construct, thermal denaturation of the proteins was performed, revealing a Tm of 65.9 °C (R2 = 0.97) while RRM1wt showed little spectral change over the same temperature range (Tm of 62.8 °C; R2 = 0.91), as expected from an unstructured protein (Figure 9C).
Further, the wild-type RRM1 protein (prepared in vitro) and RRM1β-mAla were compared with respect to the DNA binding activity using an i-motif DNA construct (Figure 10A and Figure S2B of the Supporting Information). As shown in a previous study, the i-motif has a characteristic CD peak at 286 nm, and the intensity of the peak diminishes with unwinding of the DNA fold, with an increase in pH or because of the binding of hnRNP LL.14 As demonstrated in Figure 10, the intensity of the CD peak at 286 nm gradually decreased while 5 μM i-motif DNA was titrated with wild-type RRM1 (Figure 10B) or RRM1β-mAla (Figure 10C). When plotted at the varying concentrations of RRM1wt and RRM1β-mAla, the normalized CD signal intensity at 286 nm revealed dissociation constants (Kd) of 0.27 μM (R2 = 0.97) and 0.26 μM (R2 = 0.98), respectively (Figure 10D). i-Motif protein binding was also confirmed by employing an electrophoretic mobility shift assay (EMSA), as shown in Figure 10E.
Figure 10.

Binding interaction between the i-motif DNA and RRM1 containing Ala or β-mAla (4) at position 35. (A) Schematic representation of i-motif and RRM1 binding resulting in the unwinding of i-motif DNA. (B) CD spectra of i-motif DNA upon its titration with RRM1wt until saturation. (C) Curves of normalized molar ellipticity ([θ]DNA − [θ]DNA + protein) at 286 nm as a function of protein-DNA molar ratio for RRM1wt and RRM1β-mAla. (D) Dissociation constant (Kd) for DNA protein binding in the presence of Ala or β-mAla at position 35. (E) Electrophoretic mobility shift assay (EMSA) with radiolabeled i-motif and RRM1wt derived from in vivo and in vitro expression, or RRM1β-mAla expressed in vitro.
Discussion
Methods of incorporation of noncanonical amino acids into proteins to improve function or create novel functionalities have applications in protein therapeutics,30 bioimaging,31 protein folding and self-assembly,32 and organic synthesis.33 Successful ribosome-mediated incorporation of a large number of noncanonical amino acid residues into peptides,34−36 proteins,37,38 and molecular libraries39,40 has been reported. These studies reflect the broad acceptance of a variety of side chains of α-l-amino acids by the peptidyltransferase center (PTC) of the ribosome. However, the set of amino acid building blocks available for ribosomal synthesis of modified proteins is presently limited to α-l-amino acids. β-Amino acids are interesting amino acid analogues and have been studied extensively as alternatives to α-amino acids.41 Synthetic peptide analogues having β-amino acids have been shown to have helical properties, some of which are not dissimilar to the overall structures of peptides formed from α-amino acids.42−45 Additionally, peptide analogues having β-amino acids often show enhanced resistance to proteolysis, which make them ideal candidates to include in the growing list of unnatural amino acids incorporated into proteins.46−49 The applications of β-amino acids as β-peptides are not limited to peptide therapeutics but are also gaining importance in the development of novel biomaterials. Recently, β-peptide analogues were reported to have proteinlike properties,50 which were capable of substrate binding and catalysis.51
At present, peptide analogues having β-amino acids are generally prepared by synthetic or semisynthetic methods, which limit their size and thus their range of potential uses. Therefore, their utilization in modified proteins is presently unusual. In a previous study, we reported the selection of a set of modified ribosomes having modifications in two regions of the 23S rRNA (nucleotides 2057–2063 and 2502–2507 or 2496–2501), which were sensitive to β3-puromycin (Figure 2). Mutations in the 23S rRNA region were able to restructure the PTC architecture and allow the modified ribosomes to synthesize full length DHFR containing β-alanine.12 Critically, it was shown that recognition of β-amino acids was not achieved by diminishing the fidelity of protein synthesis with α-amino acids.12 In a follow-up study, five modified ribosomes were employed to incorporate each of five different β-amino acids into DHFR; two of the modified DHFRs having single β-amino acid substitutions were characterized using an “in-gel” trypsin digestion followed by MALDI-TOF mass spectrometry.13 These β-amino acids differed in the nature, position, and stereochemistry of their side chains, yet all were incorporated to some extent. Thus, the extent to which the selected ribosomes would contain architectures that mimicked the regio- and stereochemistry of the puromycin “template” or the consequences of such architectures for the efficiency of incorporation of specific β-amino acids into protein were not clear from this study.
It seemed logical to anticipate that the substrate preferences of the modified ribosomes should reflect the regio- and stereochemistry of the puromycin analogues used for their selection. On the basis of the structure of the β-amino acid moiety in β3-puromycin (Figure 2), we predicted that the modified ribosomes would show reasonable preference toward 3-substituted β-amino acids. We also thought that these modified ribosomes might be able to discriminate between two spatial orientations of the side chain at position 3.
Clones 040329, 040321, 0403x4, and 040217 were selected using β3-puromycin (Table 1).12 When subsequently employed to support the incorporation of β-amino acids 1–4 into DHFR, the modified ribosomes displayed good regioselectivity, preferentially accepting 3-substituted methyl-β-alanines 3 and 4 (up to 5-fold) better than 2-substituted β-amino acids 1 and 2 (Table 1). The β-tyrosine moiety in β3-puromycin has a 4-methoxybenzyl group at position 3 (Figure 2), consistent with the observed preference displayed by the modified ribosomes for 3-substituted methyl-β-alanine isomers. Clearly, all four modified ribosomes preferred stereoisomer 3(S)-methyl-β-alanine (4) relative to 3(R)-methyl-β-alanine (3); this accurately reflected the stereochemistry of the β-tyrosine moiety in β3-puromycin. The S-30 system prepared from clone 040217 synthesized full length DHFR containing β-amino acid 4 in suppression yields of up to 19%. Nonetheless, the ratios of efficiency of incorporation of 3 and 4 varied from one clone to another. For a direct verification of the incorporation of β-amino acids into DHFR, we prepared modified DHFR having 4 at position 10 on a larger scale. As a control, we utilized DHFR V10F having phenylalanine at position 10. A MALDI MS spectrum (mass range of 1000–1600 Da) of the peptides generated from “in-gel” trypsin digestion of modified DHFR is shown in Figure 4. Peptide MISLIAALAβADR having 4 at position 10 gave an ion peak at m/z 1259.17, which correlated well with the expected ion peak at 1259 Da. This ion peak was absent in the MALDI MS spectrum of the tryptic digest of DHFR V10F (Figure S1 of the Supporting Information). As seen from the MALDI mass spectrum for modified DHFR, ion peaks for unmodified peptides corresponded well with the mass spectrum of DHFR V10F (cf. Figure 4 and Figure S1 of the Supporting Information).
To strengthen the conclusions concerning the source of regio- and stereoselectivity in the selected ribosomes, we attempted to identify ribosomes having regio- and stereochemical preferences opposite from those discussed above. This involved conducting a separate selection using β2-puromycin (Figure 2). In the initial selection using β3-puromycin, clones exhibiting <40% inhibition of cell growth in the presence of β3-puromycin were not employed for in vitro translation experiments.12 We reasoned that some of those clones might show enhanced sensitivity to a β-puromycin derivative (β2-puromycin) having a configuration opposite from that of β3-puromycin. Therefore, we used that set of clones for a β2-puromycin selection assay and discovered six new clones showing >40% inhibition of cell growth (Figure 5A). The IC50 assay for cells harboring the six clones in the presence of β3-puromycin and β2-puromycin identified three clones (010335, 010374, and 070215) with 1.5–2-fold lower IC50 values for β2-puromycin than for β3-puromycin (Figure 5B). Following plasmid isolation and sequencing, two clones (010335 and 010374) were found to have mutations in both critical regions (nucleotides 2057–2063 and 2502–2507) in the 23S rRNA gene (Table 2). As described previously, region 1 was mutated to confer resistance against erythromycin such that cell growth in the presence of erythromycin would partially block the growth depending on wild-type ribosomes, thus allowing enhanced production of the modified ribosomes.12 Therefore, an IC50 assay using both β-puromycin derivatives in the presence of 2.5 μg/mL erythromycin was conducted. The clones exhibited >2-fold improved sensitivity to β2-puromycin relative to β3-puromycin (Table 2), in the presence of erythromycin. Thus, we succeeded in discovering two clones demonstrating enhanced recognition for a puromycin derivative having the opposite regio- and stereoconfiguration relative to those selected using β3-puromycin. Figure 6 illustrates the opposite preferences for methylated β-alanines exhibited by clones 010335 and 040329.
The variants of the ribosomes tested were modified in two regions of 23S rRNA, nucleotides 2057–2063 (region 1) and nucleotides 2502–2507 or 2496–2501 (region 2). The clones selected using β3-puromycin had the same sequence in region 1, responsible for erythromycin resistance, but different sequences in region 2 (Table 1). The wild-type sequence 2057GAAAGAC2063 in region 1 was changed to 2057AGCGUGA2063 for all the clones. In the second region, clones 0403x4 (2502AGCCAG2507) and 040329 (2502UGGCAG2507) exhibited good homology. Perhaps for this reason, both of these clones yielded similar results for the incorporation of methyl-β-alanine derivatives (Table 1). Clone 040321 (2502AGAUAA2507) had little homology to either of the clones mentioned above and demonstrated the best regio- and stereoselectivity. Clone 040217, which produced maximal levels of full length DHFR in the case of methyl-β-alanine derivatives, was modified in a different region (2496AUAGAA2501) altogether. For the clones selected using β2-puromycin, both regions had sequences different from those of clones selected using β3-puromycin. Clones 010335 and 010374 both had the same sequence of nucleotides 2057UGCGUGG2063, which shared good homology with region 1 (2057AGCGUGA2063) of clones selected using β3-puromycin (Table 2). In region 2, clone 010335 (2502AGAUGA2507) shared reasonable homology with clone 040321 (2502AGAUAA2507), but poor homology with clone 010374 (2502CGCUCG2507).
In an effort to understand the basis for the enhanced recognition of β-amino acids by the ribosomal clones employed in this study, we employed a reference structure for E. coli 23S rRNA (PDB entry 2WWQ)18 to model the placement of the nucleotides in the modified loop residues in regions of nucleotides 2057–2063 and 2496–2507 of clones 010335 and 040329 (Figure 7). While not definitive, it is interesting that all six clones considered in this study contained U2061 (in lieu of G2061 present in the wild type), and that this residue is putatively involved in a H-bonding interaction with residue A2451, which is believed to be important catalytically in peptide bond formation.22−25 Further, all six clones shared the common pentanucleotide sequence 2058GCGUG2062, while the wild type has the corresponding sequence 2058AAAGA2062. An in silico analysis of potential internucleotide H-bonding interactions was conducted for five of the six clones in comparison with the wild type and is presented in Table S1 of the Supporting Information. Strikingly, each of the clones was predicted to have a region of the 23S rRNA in which several of the contiguous H-bonds present in wild type were absent. Plausibly, this may actually reflect a somewhat more flexible structure, amenable to recognition of β-amino acids in addition to the canonical α-amino acids. Additionally, it should be noted that there are numerous reports suggesting that the ribose moiety of the 3′-terminal adenosine on aminoacyl-tRNA may also be an important factor in determining the facility of ribosomally mediated peptide bond formation.52−59 Accordingly, the foregoing analysis, based only on ribosome structure, may not address all of the factors involved.
While there have been numerous reports of the effects of one or more β-amino acids as constituents of polypeptides, virtually all of these have involved substrates prepared by chemical synthesis. In this study, we incorporated a β-amino acid into a protein motif (RRM1 of hnRNP LL) by the use of a modified ribosome. The unmodified protein is capable of binding to an i-motif DNA despite exhibiting a random coil structure. The incorporation of β-mAla in lieu of alanine at position 35 conferred a preferred structure to the smaller α-helix of RRM1 (Figure 9B) without changing the DNA binding ability of the protein (Figure 10C–E). We believe that this represents the first report of a stabilizing effect of a β-amino acid in a ribosomally elaborated protein. We hypothesize that the increased stability of the α-helix as a result of the successful incorporation of β-mAla may be due to the greater flexibility as well as hydrophobicity of β-amino acids, which may provide an advantage for alternative hydrogen bonding with the neighboring amino acids. In the case presented here, the documented stabilization of the α-helix was achieved without any apparent effect of RRM1 on DNA binding. A more detailed analysis would be required to determine whether any change in DNA binding could be noted under a variety of experimental conditions. A previous report has suggested that the alternative hydrogen bonds involving the terminal amino acids of an α-helix contribute to helix capping.60 The presence of alanine at the beginning of the smaller α-helix may play an important role in the helical stability by taking part in such processes. Therefore, this study involving β-mAla may provide a vehicle for better defining the potential effects of β-amino acids on protein structure through further experiments and modeling using different amino acid positions of RRM1.
Conclusions
We have successfully demonstrated the ribosome-mediated incorporation of four new β-amino acids (1–4) into full length DHFR and amino acid 4 into the RRM1 domain of hnRNP LL. We have verified that the modified ribosomes, in general, displayed preferred regio- and stereoselectivity in accordance with the regio- and stereochemistry of the β-puromycin derivatives used for their selection. We conclude that it is possible to select for modified ribosomes whose architectures recognize features intrinsic to the puromycins used for their selection, including specific elements of regio- and stereochemistry. We provide a preliminary analysis of structural features of the modified ribosomes conducive to the additional recognition of β-amino acids and demonstrate the stabilizing effect of 3(S)-methyl-β-alanine on a short α-helix present in RRM1.
Acknowledgments
We thank Dr. Shujian Cun for assistance with the preparation of the construct for expression of the RRM1 domain of hnRNP LL and Dr. Dayn Sommers for assistance with the CD measurements.
Supporting Information Available
Remaining synthetic schemes and experimental and characterization data for the corresponding cyanomethyl and pdCpA esters of β-amino acids 1–4 and for β2-puromycin. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00389.
This study was supported by National Institutes of Health Research Grant GM103861 awarded by the National Institute of General Medical Sciences.
The authors declare no competing financial interest.
In a recent publication, Englander et al. have studied the basis for ribosomal discrimination of amino acid chirality.61
Supplementary Material
References
- Kurland C. G.; Ehrenberg M. (1987) Growth-optimizing accuracy of gene expression. Annu. Rev. Biophys. Biophys. Chem. 16, 291–317. [DOI] [PubMed] [Google Scholar]
- Ling J.; Reynolds N.; Ibba M. (2009) Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 63, 61–78. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Van Deventer J. A.; Tirrell D. A. (2010) Residue-specific incorporation of non-canonical amino acids into proteins: Recent developments and applications. Curr. Opin. Chem. Biol. 14, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter E. A.; Weisblum B.; Gellman S. H. (2002) Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial β-peptides. J. Am. Chem. Soc. 124, 7324–7330. [DOI] [PubMed] [Google Scholar]
- Heckler T. G.; Roesser J. R.; Xu C.; Chang P. I.; Hecht S. M. (1988) Ribosomal binding and dipeptide formation by misacylated tRNAPhe’s. Biochemistry 27, 7254–7262. [DOI] [PubMed] [Google Scholar]
- Roesser J. R.; Xu C.; Payne R. C.; Surratt C. K.; Hecht S. M. (1989) Preparation of misacylated aminoacyl-tRNAPhe’s useful as probes of the ribosomal acceptor site. Biochemistry 28, 5185–5195. [DOI] [PubMed] [Google Scholar]
- Starck S. R.; Qi X.; Olsen B. N.; Roberts R. W. (2003) The puromycin route to assess stereo- and regiochemical constraints on peptide bond formation in eukaryotic ribosomes. J. Am. Chem. Soc. 125, 8090–8091. [DOI] [PubMed] [Google Scholar]
- Bain J. D.; Wacker D. A.; Kuo E. E.; Chamberlin A. R. (1991) Site-specific incorporation of non-natural residues into peptides: Effect of residue structure on suppression and translation efficiencies. Tetrahedron 47, 2389–2400. [Google Scholar]
- Tan Z.; Forster A. C.; Blacklow S. C.; Cornish V. W. (2004) Amino acid backbone specificity of the Escherichia coli translation machinery. J. Am. Chem. Soc. 126, 12752–12753. [DOI] [PubMed] [Google Scholar]
- Nathans D.; Neidle A. (1963) Structural requirements for puromycin inhibition of protein synthesis. Nature 197, 1076–1077. [DOI] [PubMed] [Google Scholar]
- Pestka S. (1971) Inhibitors of ribosome functions. Annu. Rev. Microbiol. 25, 487–562. [DOI] [PubMed] [Google Scholar]
- Dedkova L. M.; Fahmi N. E.; Paul R.; del Rosario M.; Zhang L.; Chen S.; Feder G.; Hecht S. M. (2012) β-Puromycin selection of modified ribosomes for in vitro incorporation of β-amino acids. Biochemistry 51, 401–415. [DOI] [PubMed] [Google Scholar]
- Maini R.; Nguyen D. T.; Chen S.; Dedkova L. M.; Chowdhury S. R.; Alcala-Torano R.; Hecht S. M. (2013) Incorporation of β-amino acids into dihydrofolate reductase by ribosomes having modifications in the peptidyltransferase center. Bioorg. Med. Chem. 21, 1088–1096. [DOI] [PubMed] [Google Scholar]
- Kang H.-J.; Kendrick S.; Hecht S. M.; Hurley L. H. (2014) The transcriptional complex between the BCL2 i-motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 136, 4172–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder M.; Golovine S.; Hecht S. M. (1997) A chemical deprotection strategy for the elaboration of misacylated transfer RNA’s. J. Org. Chem. 62, 778–779. [Google Scholar]
- Lodder M.; Golovine S.; Laikhter A. L.; Karginov V. A.; Hecht S. M. (1998) Misacylated transfer RNAs having a chemically removable protecting group. J. Org. Chem. 63, 794–803. [DOI] [PubMed] [Google Scholar]
- Huynh M. L.; Russell P.; Walsh B. (2009) Tryptic digestion of in-gel proteins for mass spectrometry analysis. Methods Mol. Biol. 519, 507–513. [DOI] [PubMed] [Google Scholar]
- Seidelt B.; Innis C. A.; Wilson D. N.; Gartmann M.; Armache J.; Villa E.; Trabuco L. G.; Becker T.; Mielke T.; Schulten K.; Steitz T. A.; Beckmann R. (2009) Structural insight into nascent polypeptide chain-mediated translational stalling. Science 326, 1412–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacek N.; Mankin A. S. (2005) The ribosome transferase center: Structure, function, evolution, inhibition. Crit. Rev. Biochem. Mol. Biol. 40, 385–311. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech M.; Nierhaus K. H. (2008) Ribosomal peptide-bond formation. Chem. Biol. 15, 417–419. [DOI] [PubMed] [Google Scholar]
- Carrasco N.; Hiller D. A.; Strobel S. A. (2011) Minimal transition state charge stabilization of the oxyanion during peptide bond formation by the ribosome. Biochemistry 50, 10491–10498. [DOI] [PubMed] [Google Scholar]
- Polikanov Y. S.; Steitz T. A.; Innis C. A. (2014) A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 21, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P.; Hansen J.; Ban N.; Moore P. B.; Steitz T. A. (2000) The structural basis of ribosome activity in peptide bond formation. Science 289, 920–930. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M. A.; Stuart A.; Fiser A.; Sánchez R.; Melo F.; Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325. [DOI] [PubMed] [Google Scholar]
- Eswar N., Marti-Renom M. A., Webb B., Madhusudhan M. S., Shen D. E. M., Pieper U., and Sali A. (2006) Current Protocols in Bioinformatics, Unit 15, 1, John Wiley & Sons, Inc., New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov I. B.; Gou Z.; Li R.; Hwang S. (2006) Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Bioinformatics 64, 19–27. [DOI] [PubMed] [Google Scholar]
- Hwang S.; Gou Z.; Kuznetsov I. B. (2007) DP-Bind: A web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics 23, 634–636. [DOI] [PubMed] [Google Scholar]
- Marshall S. A.; Lazar G. A.; Chirino A. J.; Desjarlais J. R. (2003) Rational design and engineering of therapeutic proteins. Drug Discovery Today 8, 212–221. [DOI] [PubMed] [Google Scholar]
- Leblond F.; Davis S. C.; Valdes P. A.; Pogue B. W. (2010) Pre-clinical whole-body fluorescence imaging: Review of instruments, methods and applications. J. Photochem. Photobiol., B 98, 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y. T.; Tsai K. L.; Sawaya M. R.; Asturias F. J.; Yeates T. O. (2013) Structure and flexibility of nanoscale protein cages designed by symmetric self-assembly. J. Am. Chem. Soc. 135, 7738–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon R. A.; van Pelt S. (2013) Enzyme immobilization in biocatalysis: Why, what and how. Chem. Soc. Rev. 42, 6223–6235. [DOI] [PubMed] [Google Scholar]
- Heckler T. G.; Zama Y.; Naka T.; Hecht S. M. (1983) Dipeptide formation with misacylated tRNAPhes. J. Biol. Chem. 258, 4492–4495. [PubMed] [Google Scholar]
- Bain J. D.; Glabe C. G.; Dix T. A.; Chamberlin A. R. (1989) Biosynthetic site-specific incorporation of a non-natural amino acid into a polypeptide. J. Am. Chem. Soc. 111, 8013–8014. [Google Scholar]
- Bain J. D.; Diala E. S.; Glabe C. G.; Wacker D. A.; Lyttle M. H.; Dix T. A.; Chamberlin A. R. (1991) Site-specific incorporation of nonnatural residues during in vitro protein biosynthesis with semi-synthetic aminoacyl-tRNAs. Biochemistry 30, 5411–5421. [DOI] [PubMed] [Google Scholar]
- Noren C. J.; Anthony-Cahill S. J.; Griffith M. C.; Schultz P. G. (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244, 182–188. [DOI] [PubMed] [Google Scholar]
- Nowak M. W.; Kearney P. C.; Sampson J. R.; Saks M. E.; Labarca C. G.; Silverman S. K.; Zhong W.; Thorson J.; Abelson J. N.; Davidson N.; Schultz P. G.; Dougherty D. A.; Lester H. A. (1995) Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science 268, 439–442. [DOI] [PubMed] [Google Scholar]
- Li S.; Millward S.; Roberts R. W. (2002) In vitro selection of mRNA display libraries containing an unnatural amino acid. J. Am. Chem. Soc. 124, 9972–9973. [DOI] [PubMed] [Google Scholar]
- Takahashi T. T.; Austin R. J.; Roberts R. W. (2003) mRNA display: Ligand discovery, interaction analysis and beyond. Trends Biochem. Sci. 28, 159–165. [DOI] [PubMed] [Google Scholar]
- Seebach D.; Beck A. K.; Bierbaum D. J. (2004) The world of β- and γ-peptides comprised of homologated proteinogenic amino acids and other components. Chem. Biodiversity 1, 1111–1239. [DOI] [PubMed] [Google Scholar]
- Appella D. H.; Christianson L. A.; Klein D. A.; Powell D. R.; Huang X.; Barchi J. J. Jr.; Gellman S. H. (1997) Residue-based control of helix shape in β-peptide oligomers. Nature 387, 381–384. [DOI] [PubMed] [Google Scholar]
- Cheng R. P.; DeGrado W. F. (2001) De novo design of a monomeric helical β-peptide stabilized by electrostatic interactions. J. Am. Chem. Soc. 123, 5162–5163. [DOI] [PubMed] [Google Scholar]
- Frackenpohl J.; Arvidsson P. I.; Schreiber J. V.; Seebach D. (2001) The outstanding biological stability of β- and γ-peptides toward proteolytic enzymes: An in vitro investigation with fifteen peptidases. ChemBioChem 2, 445–455. [DOI] [PubMed] [Google Scholar]
- Hart S. A.; Bahadoor A. B.; Matthews E. E.; Qiu X. J.; Schepartz A. (2003) Helix macrodipole control of β3-peptide 14-helix stability in water. J. Am. Chem. Soc. 125, 4022–4023. [DOI] [PubMed] [Google Scholar]
- Cristau M.; Devin C.; Oiry C.; Chaloin O.; Amblard M.; Bernad N.; Heitz A.; Fehrentz J. A.; Martinez J. (2000) Synthesis and biological evaluation of bombesin constrained analogs. J. Med. Chem. 43, 2356–2361. [DOI] [PubMed] [Google Scholar]
- Porter E. A.; Wang X.; Lee H. S.; Weisblum B.; Gellman S. H. (2000) Non-hemolytic β-amino-acid oligomers. Nature 404, 565. [DOI] [PubMed] [Google Scholar]
- Kritzer J. A.; Stephens O. M.; Guarracino D. A.; Reznik S. K.; Schepartz A. (2005) β-Peptides as inhibitors of protein-protein interactions. Bioorg. Med. Chem. 13, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach D.; Gardiner J. (2008) β-Peptidic peptidomimetics. Acc. Chem. Res. 41, 1366–1375. [DOI] [PubMed] [Google Scholar]
- Qiu J. X.; Petersson E. J.; Matthews E. E.; Schepartz A. (2006) Toward β-amino acid proteins: A cooperatively folded β-peptide quaternary structure. J. Am. Chem. Soc. 128, 11338–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. S. P.; Nguyen J. B.; Schepartz A. (2014) Design and high-resolution structure of a β3-peptide bundle catalyst. J. Am. Chem. Soc. 136, 6810–6813. [DOI] [PubMed] [Google Scholar]
- Weinger J. S.; Strobel S. A. (2006) Participation of the tRNA A76 hydroxyl groups throughout translation. Biochemistry 45, 5939–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Zhou J.; Lodder M.; Hecht S. M. (2006) Tandemly Activated Transfer RNAs Participate in Protein Synthesis. J. Biol. Chem. 281, 13865–13868. [DOI] [PubMed] [Google Scholar]
- Koch M.; Huang Y.; Sprinzl M. (2008) Peptide-bond synthesis on the ribosome: No free vicinal hydroxy group required on the terminal ribose residue of peptidyl-tRNA. Angew. Chem., Int. Ed. 47, 7242–7245. [DOI] [PubMed] [Google Scholar]
- Duca M.; Chen S.; Hecht S. M. (2008) Modeling the reactive properties of tandemly activated tRNAs. Org. Biomol. Chem. 6, 3292–3299. [DOI] [PubMed] [Google Scholar]
- Duca M.; Trindle C.; Hecht S. M. (2011) Structural basis for the exceptional stability of bisaminoacylated nucleotides and transfer RNAs. J. Am. Chem. Soc. 133, 11368–11377. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Sprinzl M. (2011) Peptide bond formation on the ribosome: The role of the 2′-OH group on the terminal adenosine of peptidyl-tRNA and of the length of nascent peptide chain. Angew. Chem., Int. Ed. 50, 7287–7289. [DOI] [PubMed] [Google Scholar]
- Zaher H. S.; Shaw J. J.; Strobel S. A.; Green R. (2011) The 2′-OH group of the peptidyl-tRNA stabilizes an active conformation of the ribosomal PTC. EMBO J. 30, 2445–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monajemi H.; Zain S. M.; Abdullah W. A. T. W. (2015) The P-site A76 2′-OH acts as a peptidyl shuttle in a stepwise peptidyl transfer mechanism. RSC Adv. 5, 25489–25503. [Google Scholar]
- Aurora R.; Rose G. D. (1998) Helix capping. Protein Sci. 7, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander M. T.; Avins J. L.; Fleisher R. C.; Liu B.; Effraim P. R.; Wang J.; Schulten K.; Leyh T. S.; Gonzalez R. L. Jr.; Cornish V. W. (2015) The ribosome can discriminate the chirality of amino acids within its peptidltransferase center. Proc. Natl. Acad. Sci. U.S.A. 112, 6038–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.