

Abstract
The importance of the thyroid hormone axis in the regulation of skeletal growth and maintenance has been well established from clinical studies involving patients with mutations in proteins that regulate synthesis and/or actions of thyroid hormone. Data from genetic mouse models involving disruption and overexpression of components of the thyroid hormone axis also provide direct support for a key role for thyroid hormone in the regulation of bone metabolism. Thyroid hormone regulates proliferation and/or differentiated actions of multiple cell types in bone including chondrocytes, osteoblasts and osteoclasts. Thyroid hormone effects on the target cells are mediated via ligand-inducible nuclear receptors/transcription factors, thyroid hormone receptor (TR) α and β, of which TRα seems to be critically important in regulating bone cell functions. In terms of mechanisms for thyroid hormone action, studies suggest that thyroid hormone regulates a number of key growth factor signaling pathways including insulin-like growth factor-I, parathyroid hormone related protein, fibroblast growth factor, Indian hedgehog and Wnt to influence skeletal growth. In this review we describe findings from various genetic mouse models and clinical mutations of thyroid hormone signaling related mutations in humans that pertain to the role and mechanism of action of thyroid hormone in the regulation of skeletal growth and maintenance.
Keywords: thyroid hormone, bone, cartilage, growth factors, bone cells
Introduction
Thyroid hormone (TH) plays an important role in normal endochondral ossification and is essential for skeletal development, linear growth, maintenance of bone mass, and efficient fracture healing (1). Juvenile hypothyroidism causes growth arrest with delayed bone formation and mineralization, and T4 replacement induces rapid catch-up growth (2). By contrast, childhood thyrotoxicosis accelerates bone formation with premature closure of the growth plates and skull sutures, leading to short stature and craniosynostosis (3). Although there is considerable evidence regarding the importance of TH in skeletal development, the molecular mechanisms of TH action in bone are poorly understood. In this chapter, we discuss regulation and mechanisms of action of TH during skeletal development with particular emphasis on areas in which recent advances have been made.
Physiology of TH: Regulation, metabolism and TH receptor
Regulation
Systemic TH levels are maintained by the classical negative feedback loop involving the hypothalamus-pituitary-thyroid (HPT) axis (Figure 1). Thyrotropin releasing hormone (TRH) is synthesized in the paraventricular nucleus (PVN) of the hypothalamus and stimulates synthesis and secretion of thyroid stimulating hormone (TSH) from thyrotroph cells in the anterior pituitary gland. TSH subsequently acts via the TSH receptor (TSHR) on thyroid follicular cells to stimulate synthesis and release of 3,5,3′,5′-L-tetraiodothyronine (thyroxine, T4) and 3,5,3′-L-triiodothyronine (T3). The circulating T4 and T3 are predominantly bound to carrier proteins including thyroxine binding globulin, transthyretin (previously known as thyroxine binding pre-albumin) and albumin, with only approximately 0.2% of the total T3 and 0.02% of the total T4 available as free unbound hormones (fT3, fT4) in plasma.
Figure 1.
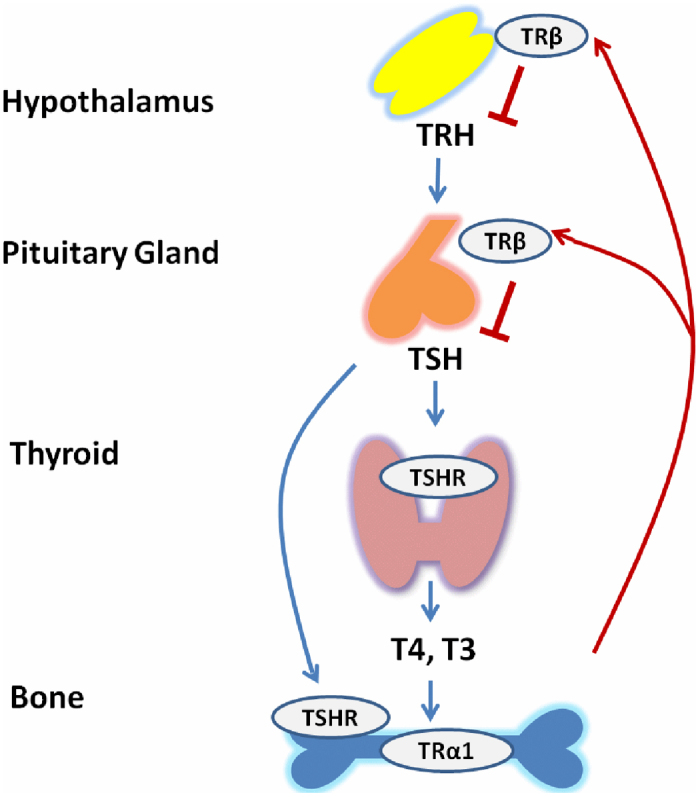
TRH-TSH-T3 feedback loop. The hypothalamic neurons secrete thyrotropin releasing hormone (TRH) which is carried down to the adenohypophysis of the pituitary by the hypothalamic portal vein where it releases thyroid stimulating hormone (TSH). The released TSH reaches thyroid glands via blood stream to bind to TSH receptor (TSHR) to stimulate production and release of thyroxin (T4) and T3. T3 exerts its actions on bone mainly by binding to TRa. TSH can also act directly on bone cells by binding to TSHR. Increased levels of T3 can act by negative feedback loop via TRβ to inhibit release of TRH and TSH, thereby preventing hyperparathyroidism.
TH is known to act via the nuclear TH receptor β (TRβ) in the hypothalamus and pituitary to inhibit TRH and TSH production and secretion (4,5) and thus complete a negative feedback loop that maintains systemic thyroid status within a normal reference range. This negative feedback loop maintains a physiological inverse relationship between TSH and circulating T3 and T4 levels that defines the HPT axis set-point (6,7).
Metabolism
The predominant circulating TH is the pro-hormone T4, which can be converted to the biologically more potent hormone, T3. TH metabolism is mediated by three iodothyronine deiodinases. The type 1 and type 2 enzymes (D1 and D2) convert T4 to T3 by catalyzing removal of a 5′-iodine atom. By contrast, the type 3 enzyme (D3) irreversibly removes a 5-iodine atom from either T4 or T3 to generate the inactive metabolites 3,3′,5′-L-triiodo-thyronine (reverse T3, rT3) and 3,3′-diiodothyronine (T2), respectively (8,9). D1 is not expressed in skeletal cells (10,11), indicating D1 does not influence T3 action on bone directly. D2 is restricted to mature primary osteoblasts but is undetectable in chondrocytes and osteoclasts (12).
The cellular influx as well as efflux of iodothyronines is known to be mediated by several specific membrane transporter proteins including the monocarboxylate transporters 8 and 10 (MCT8 and MCT10), sodiumdependent organic anion co-transporting polypeptide 1 (OATP1), the sodium taurocholate co-transporting polypeptide (NTCP) and the L-type amino acid transporter 1 (LAT1) and LAT2 (13–15). A study by Capelo et al revealed that MCT8, LAT1 and LAT2 are expressed in the skeletal tissues of mice as well as in osteoblastic MC3T3-E1 cells (16). Thus, the intra-cellular levels of the active hormone, T3, and its availability to nuclear TH receptors (TRs) are determined by the relative activities of D2 and D3 as well as expression levels of TH transport proteins.
TH receptor/ TH action
The major action of TH is exerted through nuclear TH receptors (TRs), which are ligand-inducible transcription factors. Based on chromosomal localization and amino acid homology, two classes of TRs, a and β, have been identified. Due to differential splicing of these two genes, multiple TRs are generated as α1, α2, α3, β1, β2, and β3, as well as three truncated forms, Δa1, Δa2, Δβ (17,18). The α2 and α3 isoforms and all of the truncated receptors are non-T3 binding proteins that function as antagonists of TH signaling (18–20). TRa1 and TRβ1 are expressed in virtually all tissues, but their abundance and roles differ, depending on the developmental stage of the organism and on the particular tissue type (21). TRa1 is more abundantly expressed in heart, brain, and bone, while TRβ1 is more highly expressed in liver and pituitary (22). By contrast, expression of TRβ2 is restricted to the hypothalamus and pituitary where it mediates inhibition of TRH and TSH expression and the cochlea and retina where it regulates sensory organ development (23,24) and TRβ3 is expressed in kidney, liver, and lung (25). Thus, TH action in target tissues is determined in part by the types and abundance of TH receptors present.
In the nucleus, TRs form homodimers with another TR or heterodimers with retinoid X receptors (RXR) and bind to specific TH response element sequences (TREs) located in promoter regions of T3-target genes and regulate their expression in a ligand-dependent manner. Unliganded TRs bind TREs in T3 target genes and mediate transcriptional repression. Co-repressor proteins such as nuclear receptor corepressor protein/silencing mediator of retinoid and TH receptors are recruited to the RXR-TR heterodimer in the absence of T3 and inhibit target gene expression. T3 binding displaces the co-repressor, allowing co-activator proteins such as CBP/p300, pCAF, and SRC-1 to interact with the RXR-TR heterodimer and activate gene transcription in a hormone-dependent manner (26–28).
Besides the genomic actions of T3, nongenomic mechanism of TH analogues are increasingly recognized to have downstream consequences at the level of specific gene transcription (26,29). The nongenomic mechanisms of TH are known to be initiated at the plasma membrane, in the cytoplasm or in the intracellular organelles, such as mitochondria. At the membrane level, TH may interact with integrin aV/β3 to activate ERK1/2 which culminates in regulation of ion transport systems or cell proliferation (30). The relative contribution of nongenomic mechanisms in mediating TH effects on skeletal development is yet to be determined.
Skeletal development
The skeleton in different parts of the body develops through two distinct processes, intramembranous ossification and endochondral ossification. Intramembranous ossification, which occurs in the flat bones of the skull, involves direct differentiation of embryonic mesenchymal cells into bone-forming osteoblasts without an intermediate cartilage model (31). By contrast, endochondral ossification, which occurs in the remainder of the skeleton, involves the replacement of a cartilage model by bone tissue. Mesenchymal precursor cells condense and differentiate into chondrocytes, which secrete matrix proteins to form a cartilage template. The model expands through chondrocyte proliferation. Ossification of the cartilage model is preceded by hypertrophy of the chondrocytes in the prospective mid-shaft of the bone. Subsequently, blood vessels, osteoclasts (cartilage- and bone-resorbing cells), as well as bone marrow and osteoblast precursors then invade the model from the bone collar and proceed to form the primary ossification center. The primary center expands towards the ends of the cartilage model, as the osteoclasts remove cartilage extracelluar matrix (ECM) and osteoblasts deposit bone on cartilage remnants. In long bones, secondary ossification centers form within cartilage at the ends of long bones and remain separated from the primary ossification center by the epiphyseal growth plates. Skeletal maturity occurs when the expanding primary ossification center meets the secondary ossification center, thus obliterating the growth plate. The ordered process of growth plate chondrocyte proliferation, hypertrophic differentiation, apoptosis and subsequent new bone formation mediates linear growth until adulthood (32).
The process of endochondral ossification and the rate of linear growth are tightly regulated by multiple systemic hormones (including THs, growth hormone (GH), glucocorticoids and sex steroids) and various cytokines and growth factors (including insulin-like growth factor 1 (IGF-1), parathyroid hormone-related peptide (PTHrP), Indian hedgehog (Ihh), bone morphogenetic protein (BMP)s, fibroblast growth factor (FGF)s and vascular endothelial growth factor (VEGF)s that act in a paracrine and autocrine manner (33). The interaction between systemic hormones and local factors play a critical role in regulating linear growth, bone mass accumulation and mineralization processes until peak bone mass (34) is achieved in early adulthood. Through-out adult life there is a gradual loss of bone mass, which in women is accelerated at the menopause. Excessive TH as in the case of hyperthyroidism in adults is known to induce increased bone turnover and fracture incidence (35,36). While TH is known to be involved in regulating development of peak bone mass during early childhood and in the maintenance of bone mass in adults, this review will focus on the mechanism of TH action in regulating skeletal development.
Effects of TH on skeletal development
A number of genetic mouse models (37,38) have been generated to date to elucidate the role of the TH axis on growth and development of a number of tissues including bone. The skeletal phenotypes of these mutant mouse models will be described below.
Skeletal phenotype of TH signaling related mutant mice
Mouse mutants with altered TSH or TH levels
Pax8−/− mice lack the thyroid specific transcription factor Pax8 required for thyroid follicular cell formation (39,40) and hyt/hyt mice have a loss-of-function mutation in the TSH receptor (TSHRP556L) (41–43). Both mutants have a 2000-fold elevation of TSH and undetectable THs, but the TSHR is functional in Pax8−/− mice whereas it is non-functional in hyt/hyt mice. Thus, the reciprocal relationship between THs and TSH remains intact in Pax8−/− mice but is disrupted in hyt/hyt mice (44). Both mutants exhibited a similar skeletal phenotype of impaired linear growth, delayed endochondral ossification, impaired chondrocyte differentiation, reduced cortical bone, impaired trabecular bone remodeling and reduced bone mineralization (44,45). These data indicate that any action of TSH in bone is likely to be minor when compared to the effects of T3.
Table 1. Skeletal phenotype of TH signaling related mutant mice.
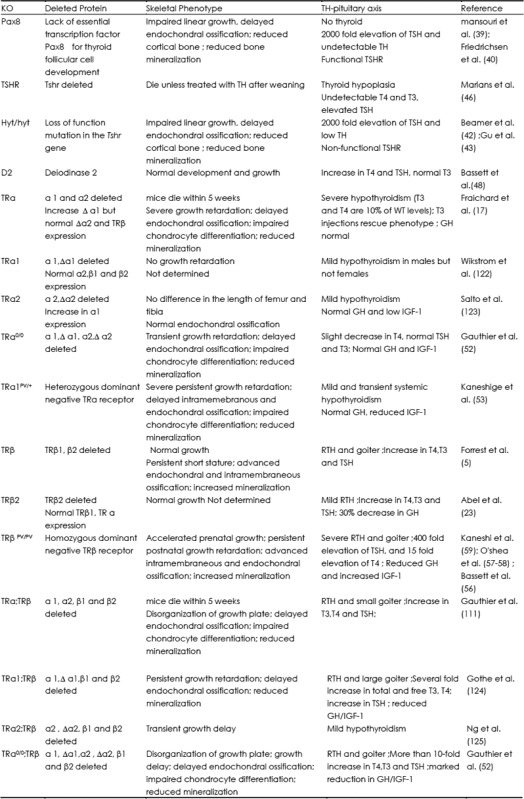
TSHR knockout mice (TSHR−/−) present with developmental and growth delays and severe osteoporosis at all sites and profound hypothyroidism, with no detectable T4 and T3 and elevated TSH. The TSHR knockout mice die within 1 week of weaning unless treated with TH supplementation (46). TH replacement after weaning did not normalize the low bone mineral density in TSHR−/− mice and these mice showed a dramatic increase in bone turnover. These findings confirmed that the bone loss was independent of T3 and T4 levels. Thus, an independent role for TSH in negative regulation of bone remodeling was proposed (47).
Deiodinase D2 knockout mice were generated to investigate the effects of osteoblast-specific T3 deficiency (48) because D2 expression was detected only in mature osteoblasts (12). The D2 knockout mice exhibit pituitary resistance to feedback regulation by T4 characterized by an increase in TSH and T4, but normal T3. Overall growth as well as skeletal development was found to be normal in D2 knockout mice indicating an insignificant role for D2 during endochondral and intramembranous ossification in vivo. However, adult D2 knockout mice had brittle bones due to increased mineralization and reduced bone formation because of restricted cellular hypothyroidism in osteoblasts (48). Further studies are needed to determine the mechanism for reduced bone formation in the D2 conditional knockout mice.
Dual oxidase generates the hydrogen peroxide required by thyroid peroxidase for the incorporation of iodine into thyroglobulin. Dual oxidase 2 (Duox2) mutant mice, genetic model for congenital hypothyroidism, exhibit low T4 and high TSH caused by lack of T4 feedback to the pituitary gland and is associated with dwarfing and hearing impairment. Concomitant with small size and low aBMD, circulating levels of IGF-1 are lower in the mutants. These phenotypes of Duox2 mutant mice are consistent with lack of TH support, a key role for TH in skeletal development (49). Further studies are needed to investigate the specific functions of Duox2 in other tissues.
TRa mutants
Several TRa knockout mice have been generated and this has led to the identification of additional TRa isoforms expressed from a promoter within intron 7 of the THRA gene (50). As a result only TRa0/0 mice lack all TRa isoforms whereas other TRa mutants retain truncated isoforms with dominant-negative activity. TRa0/0 mice lack all TRa isoforms and are systemically euthyroid. During development TRa0/0 mice display features of skeletal hypothyroidism with transient growth retardation and with delayed endochondral ossification characterized by impaired chondrocyte differentiation and reduced mineralization (51,52).
Mice harboring dominant-negative mutations of TRa1 in different genetic backgrounds have mild and transient systemic hypothyroidism but they also exhibit a more severe phenotype of delayed skeletal development than TRa0/0 mice. TRa1PV/+ mice are most severely affected, displaying persistent post-natal growth retardation and markedly delayed endochondral ossification and decreased mineralization (38,53).
Deletion or mutation of TRa does not affect systemic thyroid status but caused local skeletal hypothyroidism while the presence of a dominant-negative TRa leads to a more severe skeletal phenotype than receptor deficiency alone. These findings demonstrate an important role for unliganded TRa1 acting as a potent dominant-negative antagonist (54). The repressive actions of the unliganded receptor, therefore, have a greater physiologic effect than having no receptor at all. Consistent with this phenotype, skeletal expression of the T3 target genes, fibroblast growth factor receptors (FGFR) 1 and 3, were reduced (51,55,56).
TRβ mutants
Deletion of TRβ results in elevated T4, T3 and TSH levels consistent with resistance to TH (RTH). In contrast to TRa mutants, during development TRβ−/− mice display features of skeletal hyperthyroidism with advanced endochondral and intramembranous ossification, accelerated chondrocyte differentiation, increased mineralization and persistent short stature due to premature growth plate quiescence (5,23,51).
Mice harboring a dominant-negative mutation in THRB (TRβPV/PV) have severe RTH and exhibit a more severe phenotype than TRβ−/− with accelerated prenatal growth characterized by advanced endochondral and intramembraneous ossification (57–59). Consistent with phenotype, increased skeletal expression of the T3 target genes, FGFR1 and 3, (51,55,56) revealed the presence of enhanced T3 action resulting from supraphysiological stimulation of TRα in bone. Thus, deletion or mutation of TRβ disrupts the HPT axis resulting in skeletal thyrotoxicosis. The presence of a dominant negative TRβ leads to a more severe skeletal phenotype than receptor deficiency alone. In summary, during development reduced T3 action in TRa mutant mice results in delayed ossification and reduced mineralization whereas increased T3 action in TRβ mutant mice leads to advanced ossification and increased mineralization.
Clinical manifestation of TH signaling related mutations in humans
The developing skeleton is sensitive to thyroid status and childhood hypothyroidism is characterized by growth retardation, delayed bone age and short stature, whereas juvenile thyrotoxicosis accelerates growth and advances bone age but results in persistent short stature due to premature fusion of the epiphyses (2,60,61). A loss of function mutation of the TSH β-subunit results in TSH deficiency and congenital hypothyroidism. Two affected siblings received TH replacement from birth but despite the lifelong absence of TSH, their skeletal development and bone mineral density were normal (62). These findings suggest that TSH is not required for normal skeletal development and growth. TSHR mutations result in wide spectrum of clinical manifestations ranging from mild to severe hypothyroidism and hyperthyroidism (63). Up to this date more than 40 kinds of loss of function mutations in the TSHR gene have been reported as the causative defect in congenital hypothyroidism (64). By contrast, gain of function mutations in the TSHR gene were identified in familial non-autoimmune hyperthyroidism or sporadic non-autoimmune hyperthyroidism (65).
Table 2. Clinical manifestation of TH signaling related mutations in humans.
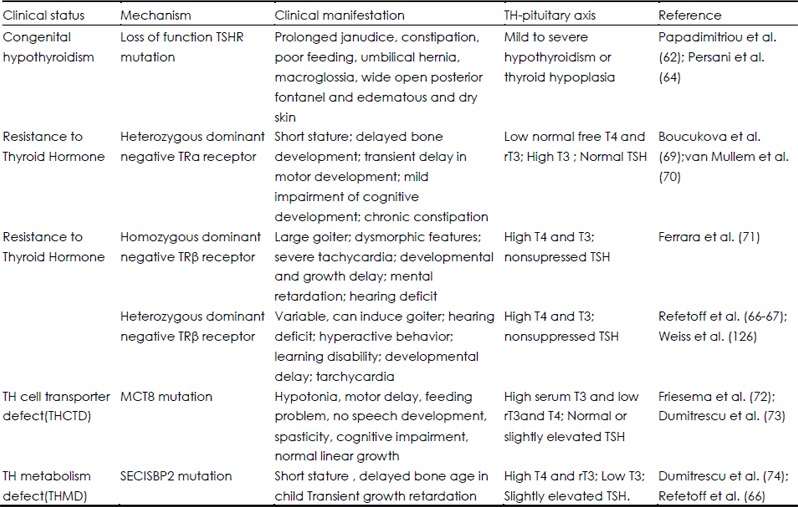
The syndrome of resistance to TH (RTH) is caused by decreased tissue responsiveness to TH and was first described in 1967 (66,67). Clinical features include goiter, elevated circulating TH levels, nonsuppressed serum TSH level, clinical euthyroidism, and tachycardia; some individuals also demonstrate attention deficit disorder and deficits of linear growth, hearing, and bone formation (66). The RTH genetic defect is caused mostly by mutations of TH receptor β gene. A dominant-negative mutation of TRβ blunts negative feedback in the HPT axis and is characterized by elevated T4 and T3 and an inappropriately normal or elevated TSH. RTH patients display variable skeletal phenotypes that are difficult to interpret due to confounding effects of treatment and the heterogeneity of TRβ mutations (66,68). Nevertheless, reported features of increased bone turnover, osteoporosis, fracture and craniosynostosis in RTH suggest a predominant role for T3 in bone development (68). Last year, two families with a heterozygous mutation of THRA, resulting in expression of a dominant negative TRa protein, have been the first to be reported (69,70). Levels of free T4 and rT3 in these patients were in the low-normal range and T3 in the high-normal range, with normal TSH. Despite near normal TH levels, patients displayed a phenotype consistent with the characteristic features of hypothyroidism that included short stature, delayed bone development, skeletal dysplasia and chronic constipation, thus suggesting that TRa1 plays a major role in human skeletal development. Recently, several patients with homozygous TRβ mutations were reported (71). The clinical manifestations are a combination of those found in individuals heterozygous for a mutation only in TRα or in TRβ. Patients have a more severe RTH phenotype, goiter, hearing loss, and much greater elevations of serum T3, T3, and TSH, than heterozygous individuals and also have neurological impairment and growth retardation, more characteristic of a deficiency in the action of TRα (69,70). These clinical phenotypes suggest the possible interference of the mutant TRβ with function of TRa1.
A sex-linked form of mental retardation with motor abnormalities, named the Allan-Herdon Dudley syndrome, was described in 1944. When MCT8, a specific transporter of TH, was sequenced in these patients, inactivating mutations were identified in some individuals (72,73). Patients manifest with truncal hypotonia, poor head control, and later spasticity and were found to have abnormal thyroid function (elevated serum T4 and rT3 and low T3).
Deiodinases are selenoproteins that catalyze iodothyronine deiodination and are important in TH activation and inactivation. Selenium is an essential trace element required for the biosynthesis of selenoproteins and selenocysteine insertion sequence binding protein 2 (SECISBP2), a key trans-acting factor. Patients with mutations in the SECISBP2 gene presented with transient growth retardation associated with abnormal thyroid function, low T3, high T4 and rT3, and slightly elevated TSH (74). Thus, there are a number of clinical genetic studies with mutations in genes related to the TH signaling pathway that attest the importance of TH in the regulation of skeletal metabolism.
Effects of T3 in bone cells in vitro
Chondrocytes
TRa1 and TRβ1 are expressed in resting and proliferating chondrocytes in the growth plate, suggesting these cells are direct targets for T3 actions (75). T3 stimulates clonal expansion of resting chondrocyte progenitor cells but inhibits subsequent chondrocyte proliferation, while stimulating hypertrophic differentiation (75–79). Accordingly, T3 induces markers of hypertrophic chondrocyte differentiation, including alkaline phosphatase and collagen X expression in primary growth-plate chondrocyte cultures, and enhances cartilage matrix mineralization (75). T3 also stimulates the expression of proteoglycan and collagen-degrading enzymes including aggrecanase-2 (a disintegrin and metalloproteinase with thrombospondin motifs1, ADAMTS5) and matrix metalloproteinase 13 (MMP13) (78,80,81). T3 regulation of growth plate chondrocyte proliferation and differentiation in vitro has been shown to involve a number of growth factor signaling pathways including IGF-1, Wnt, the Ihh/PTHrP feedback loop and FGFR3 (Figure 2) (55,82,83). In conclusion, TH stimulates maturation of chondrocytes and the progression of endochondral ossification and is essential for linear growth.
Figure 2.

Mechanisms for T3 regulation of chondrocyte differentiation. T3 can modulate local actions of growth factors such as IGF-I, FGFs, Wnts, Ihh and PTHrp to regulate proliferation and differentiation of cells of chondrocytic lineage. The differentiating promoting actions of T3 on chondrocytes are known to be mediated via inhibition of Sox9 and stimulation of Runx2 expression.
Osteoblasts
T3 has been found to stimulate, inhibit, or exert no effect on osteoblastic cell proliferation, but a consensus suggests that T3 stimulates osteoblast activity (1). T3 has been shown to increase expression of the osteoblast differentiation markers collagen I, osteocalcin, osteopontin, alkaline phosphatase, MMP9 and MMP13 in osteoblasts (81,84–87). In addition, T3 modulates key pathways involved in osteoblast proliferation and differentiation. T3 stimulates osteoblast responses to IGF1, PTH and FGFs both in cell cultures and in vivo (88–90). These observations demonstrate that T3 stimulates osteoblast activity by complex direct and indirect mechanisms involving many growth factors and cytokines (Figure 3).
Figure 3.

Mechanisms for T3 regulation of osteoblast differentiation. T3 increases local IGF-I actions by modulating production of IGF-I and/or its binding proteins and thereby stimulate differentiation of mesenchymal cells into osteoblast lineage. T3 can also stimulate local FGF actions by increasing FGF receptor expression in osteoblasts. T3 can also exert direct effects via TRE to regulate transcription of bone formation genes.
Moreover, the nuclear factors constituting the TH receptor coactivator complex and the molecular pathways by which TH mediates its effects on target gene expression in osteoblasts remain poorly understood. Recently, we have shown that diabetes- and obesity-related protein (DOR), a key modulator of TH function in muscle cells, is differentially regulated by TH and acts as a stimulatory mediator of TH effects in osteoblasts (91).
Osteoclasts
TH directly stimulates bone resorption in organ cultures of mouse calvaria (92) and fetal rat limb bones (93,94). Earlier reports showed that T3 stimulated osteoclastic bone resorption in the presence of osteoblasts, but not in their absence (95,96). These findings imply that TH indirectly stimulates osteoclasts via increased expression of RANKL and other cytokines involved in osteoclasto-genesis including interleukin 6 (IL-6), IL-8 and prosta-glandin E2 (PGE2) in osteoblasts (92,97,98). Several studies have demonstrated apparent expression of TR proteins in all bone cell lineages, however, it is not clear whether osteoclasts express TRa1 and TRβ1 mRNAs because currently available TR antibodies are of low affinity, thus compromising the detection of endogenous TRs (75,99). Thus, it remains unclear whether the effects of T3 to promote bone resorption result from direct actions in osteoclasts or indirect effects mediated by osteoblasts.
Mechanism of TH action in the developing skeleton
Interaction with the Growth hormone /IGF-1 signaling pathway
The importance of IGF-1 in skeletal development is well established because peak bone mass is considerably reduced as a consequence of deficiency of IGF-1 action in both humans and experimental animals (100–102). IGF-1 is the major determinant of post-natal growth, it mediates both GH-dependent and −independent effects and is involved in chondrocyte recruitment, proliferation and hypertrophic differentiation (103).
IGF-1 signaling is also required to maintain the Ihh-PTHrP loop during skeletogenesis. In fetal IGF1−/− mice, expression of Ihh was reduced in long bones, whereas expression of PTHrP was increased (104). Furthermore, Wang et al (105) have demonstrated that IGF-1/IGF1R induces Wnt4 expression and β-catenin activation. IGF-1/IGF1R actions on growth plate chondrocytes are neutralized by the Wnt antagonists sFRP3 and Dkk1, confirming that the Wnt/ β -catenin signaling pathway is downstream of IGF-1 signaling (Figure 2).
It is now known that IGF-1 expression in bone is regulated by GH as well as many systemic and local regulators of bone growth. In this regard, our previous studies showed that days 7 to 14 of the prepubertal period and days 23 to 31 of the pubertal period represent critical time points for skeletal growth as well as increases in serum IGF-I levels in mice (106). It was also determined that there is an important critical period during prepubertal growth when the effects of GH are small and IGF-I remains an important regulator (101). Recently, we demonstrated that TH is a major regulator of IGF-I expression, independent of GH, during the prepubertal growth period (45). When we measured the postnatal changes in serum levels of total T3 and IGF-1 in C57BL/6J mice, as seen in Figure 4, serum levels of IGF-1 increased almost three fold in mice during the prepuberal growth period. This increase in IGF-1 was preceded by changes in serum T3 levels, which increased two-fold between day 7 and 14. Studies using genetic mouse models deficient in TH found that serum levels of IGF-1 were reduced by more than 50% at day 21 compared to wild-type mice as a consequence of a decrease in IGF-1 expression in liver and bone. Daily administration of T3/T4 during the prepubertal growth period (days 5 to 14) increased IGF-1 expression in both liver and bone and normalized the serum IGF-1 levels. Furthermore, in vitro studies in osteoblasts revealed that TH, in the presence of TRa1, bound to a TH response element in intron 1 of the IGF-1 gene to stimulate transcription (45). Thus, TH is a key regulator of both local and endocrine IGF-1 action during the prepubertal growth period.
Figure 4.
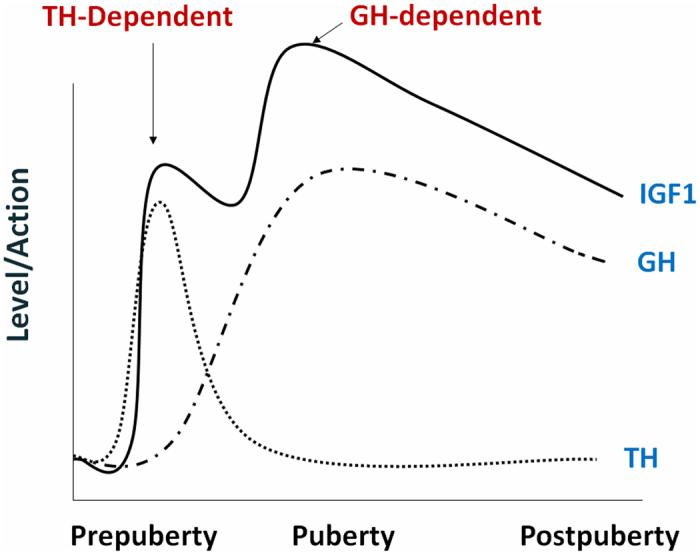
Model of TH regulation of skeletal growth in mice. It is proposed that the increase in IGF-I expression and bone accretion during prepubertal and pubertal growth periods are mediated via TH and GH-dependent mechanisms, respectively. Some of the effects of TH on bone growth are direct and occur independent of GH.
In terms of the target cell types for TH effects on IGF-1 expression, we and others have found that TH treatment increased IGF-1 expression in bone cells and chondrocytes (45,58,107). There is also evidence that TH is able to act directly on growth plate chondrocytes through GH-independent mechanisms (108). Lewinson et al demonstrated that TH is able to stimulate longitudinal bone growth in animals in which GH secretion has been ablated by hypophysectomy (109). TH also stimulated chondrocyte differentiation in both thyroidectomized and hypophysectomized rats, a role for which growth hormone cannot substitute (110). Thus, there is ample evidence that TH is essential for skeletal growth during the prepubertal growth period.
The relationship between TH signaling and GH/IGF-1 signaling is potentially complex due to multiple possible points of interaction at both the systemic and local levels. Through studies using a variety of transgenic mice in which TR function has been altered, our understanding of this complexity has been improved. TRa0/0 and TRa1PV/+ mice showed normal pituitary GH production, but diminished expression of GH receptor, IGF-1, and IGF-1 receptor in growth plate chondrocytes (52,58). By contrast, pituitary GH production was reduced and GH receptor and IGF-1 expression in the growth plate was increased in TRβ−/− and TRβPV/PV mice (5,58,111). The different phenotypes between the TRapv/+ mice and TRβPV/PV mice can be explained by the known finding that the pituitary gland is a TRβ-responsive tissue, while the growth plate is primarily a TRa-responsive tissue. Thus, GH/IGF1 signaling is also a local downstream mediator of T3 action in the growth plate.
GH therapy normalized the decreased serum IGF-1 level of hypothyroid pups. However, growth failure, despite normal serum IGF-1 concentrations in GH-treated hypothyroid rats, has been described earlier (112). Consistent with this, we found that inhibition of IGF-1 action only partially reduced TH effects on osteoblast differentiation and the rate of metatarsal bone mineralization (45). These results provide indirect evidence that TH acts via other pathways, in addition to IGF-1. It is known that TH influences fibroblast growth factor (FGF) receptor signaling in bone and interacts with the Wnt/β-catenin and Ihh/PTHrP signaling pathways to regulate endochondral ossification (52–54). The extent to which IGF-1 interaction with other growth factor signaling pathways is involved in TH regulation of skeletal metabolism remains to be determined.
Interaction with the Ihh/PTHrP feedback loop
The pace of chondrocyte differentiation is precisely regulated by the Ihh/PTHrP paracrine negative feedback loop. Prehypertrophic chondrocytes secrete Ihh which diffuses to periarticular cells to induce synthesis of PTHrP. PTHrP, acting via its receptor PTHR1, then completes the loop by stimulating chondrocyte proliferation and inhibiting further hypertrophic differentiation (113,114). The effect of TH on the Ihh/PTHrP feedback loop has been studied in thyroid manipulated rats (83). In hypothyroid rats, growth plates were grossly disorganized and the expression of PTHrP was increased and extended throughout the growth plate, while the PTHrP receptor was expressed in the same location as in euthyroid animals. These changes in PTHrP receptor expression can lead to inhibition of hypertrophic chondrocyte differentiation and result in arrest of linear growth (Figure 2). In thyrotoxic growth plates, histology essentially was normal but PTHrP receptor (PTHrP-R) mRNA was undetectable, while PTHrP mRNA expression was unchanged. An absence of the PTHrP receptor results in negative PTHrP signaling and progression of hypertrophic chondrocyte differentiation to accelerate linear growth. Furthermore, recent studies in chicken tibia explants have shown that Ihh stimulates degradation of the type 2 deiodinase enzyme resulting in an induction of PTHrP expression (114). Together, these findings suggest that TH regulates the set point of the Ihh/PTHrP feedback loop to modulate the pace of chondrocyte differentiation and endochondral bone formation during postnatal growth.
Interaction with Fibroblast Growth Factor signaling
Signaling through the FGF pathway has been demonstrated to negatively regulate proliferation of growth plate chondrocytes. During endochondral ossification, FGFR2 is expressed in condensing mesenchyme and perichondrium, FGFR1 is present in prehypertrophic and hypertrophic chondrocytes, and proliferating chondrocytes express FGFR3. During linear growth, FGFR2 is not expressed in the growth plate, but FGFR1 expression persists in prehypertrophic and hypertrophic cells, and proliferating chondrocytes express FGFR3 (33,115). The role of FGFR1 and FGFR2 in the developing growth plate is poorly understood. However, FGFR3 has been predicted to play an important role. Activating mutations in FGFR-3 results in achondroplasia, the most common form of dwarfism in humans (115,116), whereas Fgfr3 knockout mice display limb overgrowth (117,118), indicating that FGFR3 is a negative regulator of linear growth. T3 stimulates expression of FGFR-1 and FGFR-2 mRNA in the ATDC5 chondrocytic cell line undergoing chondrogenesis, but stimulation of FGFR3 by T3 was greater and persisted longer, coinciding with the period in which T3 inhibited chondrocyte proliferation and stimulated hypertrophic differentiation (55). Investigation of the FGF/FGFR signaling pathway in TR mutant mice revealed that FGFR 3 was reduced in growth plates of TRa0/0 and TRa1PV/+ mice and increased in TRβ−/− and TRβPV/PV mice (55–58). Taken together, these findings suggest that FGFR3 may play a role in mediating effects of T3 on chondrogenesis. Some of the growth inhibitory actions of FGFR3 are mediated via reduced activity of the Ihh/PTHrP feedback loop (119). The interactions between FGFR3 and other growth factor signaling pathways in regulating chondrocytes still need to be worked out.
Interaction with Wnt/β-catenin signaling
Wnt/β-Catenin signaling also has been recognized as an important signal-transduction pathway in regulating terminal differentiation of growth plate chondrocytes. Inhibition of β-catenin signaling in Col2a1-ICAT transgenic mice results in reduced chondrocyte proliferation and differentiation, delayed formation of the secondary ossification center, and reduced skeletal growth (120). Wang et al have shown that T3 promotes growth plate chondrocyte terminal differentiation by increasing Wnt-4 expression, β-catenin accumulation, TCF/LEF transcriptional activity, and expression of the Wnt/β-catenin target gene Runx2/cbfa1 (82). Furthermore, they also have shown that T3 treatment stimulates PI3K/Akt/GSK-3 β signaling (105). PI3K and Akt are important signal transducers of IGF-1 signaling. Akt can inactivate GSK-3β, a negative regulator of the canonical Wnt/β-catenin pathway (121). The inhibition of PI3K/Akt activity by LY294002 prevents T3-induced Wnt4 expression and β-catenin activation. These results indicate that TH promotes growth plate cell differentiation and longitudinal bone growth by activating β-catenin signaling via modulation of IGF-1/IGF1R signaling through the Wnt and PI3K/Akt pathways. While a number of growth factor signaling pathways have been implicated to play an important role in the TH regulation of skeletal growth (Figure 3), the extent to which these growth factor signaling pathways contribute to TH action in vivo remains to be determined.
Conclusions
There is now considerable data in the literature both from mouse genetic studies and human clinical studies involving mutations in genes related to the TH signaling pathway that demonstrate a key role for TH in the regulation of skeletal growth. In terms of target cell types for TH action, while much focus is on chondrocytes, there is also recent evidence that other bone cell types including osteoblasts and osteoclasts are also regulated by TH signaling. In terms of the mechanism for TH action, studies suggest that TH regulates a number of key growth factor signaling pathways including IGF-1, Wnt, PTHrP and FGF to regulate skeletal growth. However, the relative contribution of these growth factor signaling pathways in mediating TH effects on bone in vivo remain to be determined. The issue of how the various growth factor signaling pathways interact to mediate TH effects in various target cell types needs to be investigated. Also, studies are needed to address the issue of whether all of the TH effects on bone cells are mediated via genomic actions of TH or some of the TH effects are mediated via non-genomic actions. Future development and application of mice with conditional disruption of genes involved in TH signaling pathway in time and space are required to identify the cell-specific mechanisms of TH action and interaction with various pathways in vivo.
Acknowledgments
Financial support was received from funding agencies in the United States (NIH grant AR048139 and VA merit review grant). The authors thank Dr. Donna Strong for proof reading the article.
References
- Harvey CB, O'Shea PJ, Scott AJ, Robson H, Siebler T, Shalet SM, Samarut J, Chassande O, Williams GR. Molecular mechanisms of thyroid hormone effects on bone growth and function. Mol Genet Metab. 2002;75:17–30. doi: 10.1006/mgme.2001.3268. [DOI] [PubMed] [Google Scholar]
- Rivkees SA, Bode HH, Crawford JD. Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N Engl J Med. 1988;318:599–602. doi: 10.1056/NEJM198803103181003. [DOI] [PubMed] [Google Scholar]
- Segni M, Leonardi E, Mazzoncini B, Pucarelli I, Pasquino AM. Special features of Graves' disease in early childhood. Thyroid. 1999;9:871–877. doi: 10.1089/thy.1999.9.871. [DOI] [PubMed] [Google Scholar]
- Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE. Critical role for thyroid hormone receptor beta2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107:1017–1023. doi: 10.1172/JCI10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, Curran T. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: evidence for tissue-specific modulation of receptor function. EMBO J. 1996;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- Bassett JH, Williams GR. Critical role of the hypothalamic-pituitary-thyroid axis in bone. Bone. 2008;43:418–426. doi: 10.1016/j.bone.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Andersen S, Bruun NH, Pedersen KM, Laurberg P. Biologic variation is important for interpretation of thyroid function tests. Thyroid. 2003;13:1069–1078. doi: 10.1089/105072503770867237. [DOI] [PubMed] [Google Scholar]
- Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Germain DL, Galton VA, Hernandez A. Minireview: Defining the roles of the iodothyronine deiodinases: current concepts and challenges. Endocrinology. 2009;150:1097–1107. doi: 10.1210/en.2008-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia CH, Christoffolete MA, Zaitune CR, Dora JM, Harney JW, Maia AL, Bianco AC. Type 2 iodothyronine selenodeiodinase is expressed throughout the mouse skeleton and in the MC3T3-E1 mouse osteoblastic cell line during differentiation. Endocrinology. 2005;146:195–200. doi: 10.1210/en.2004-1043. [DOI] [PubMed] [Google Scholar]
- LeBron BA, Pekary AE, Mirell C, Hahn TJ, Hershman JM. Thyroid hormone 5′-deiodinase activity, nuclear binding, and effects on mitogenesis in UMR-106 osteoblastic osteosarcoma cells. J Bone Miner Res. 1989;4:173–178. doi: 10.1002/jbmr.5650040207. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Robson H, Kester MH, van Leeuwen JP, Shalet SM, Visser TJ, Williams GR. Iodothyronine deiodinase enzyme activities in bone. Bone. 2008;43:126–134. doi: 10.1016/j.bone.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser WE, Friesema EC, Visser TJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol. 2011;25:1–14. doi: 10.1210/me.2010-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Jansen J, Milici C, Visser TJ. Thyroid hormone transporters. Vitam Horm. 2005;70:137–167. doi: 10.1016/S0083-6729(05)70005-4. [DOI] [PubMed] [Google Scholar]
- Heuer H, Visser TJ. Minireview: Pathophysiological importance of thyroid hormone transporters. Endocrinology. 2009;150:1078–1083. doi: 10.1210/en.2008-1518. [DOI] [PubMed] [Google Scholar]
- Capelo LP, Beber EH, Huang SA, Zorn TM, Bianco AC, Gouveia CH. Deiodinase-mediated thyroid hormone inactivation minimizes thyroid hormone signaling in the early development of fetal skeleton. Bone. 2008;43:921–930. doi: 10.1016/j.bone.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraichard A, Chassande O, Plateroti M, Roux JP, Trouillas J, Dehay C, Legrand C, Gauthier K, Kedinger M, Malaval L, Rousset B, Samarut J. The T3R alpha gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J. 1997;16:4412–4420. doi: 10.1093/emboj/16.14.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GR. Cloning and characterization of two novel thyroid hormone receptor beta isoforms. Mol Cell Biol. 2000;20:8329–8342. doi: 10.1128/mcb.20.22.8329-8342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig RJ, Lazar MA, Hodin RA, Brent GA, Larsen PR, Chin WW, Moore DD. Inhibition of thyroid hormone action by a nonhormone binding c-erbA protein generated by alternative mRNA splicing. Nature. 1989;337:659–661. doi: 10.1038/337659a0. [DOI] [PubMed] [Google Scholar]
- Plateroti M, Gauthier K, Domon-Dell C, Freund JN, Samarut J, Chassande O. Functional interference between thyroid hormone receptor alpha (TRalpha) and natural truncated TRDeltaalpha isoforms in the control of intestine development. Mol Cell Biol. 2001;21:4761–4772. doi: 10.1128/MCB.21.14.4761-4772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Sjoberg M, Vennstrom B. Contrasting developmental and tissue-specific expression of alpha and beta thyroid hormone receptor genes. EMBO J. 1990;9:1519–1528. doi: 10.1002/j.1460-2075.1990.tb08270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SY. Isoform-dependent actions of thyroid hormone nuclear receptors: lessons from knockin mutant mice. Steroids. 2005;70:450–454. doi: 10.1016/j.steroids.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Abel ED, Boers ME, Pazos-Moura C, Moura E, Kaulbach H, Zakaria M, Lowell B, Radovick S, Liberman MC, Wondisford F. Divergent roles for thyroid hormone receptor beta isoforms in the endocrine axis and auditory system. J Clin Invest. 1999;104:291–300. doi: 10.1172/JCI6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Reh TA, Rusch A. Neurodevelopmental control by thyroid hormone receptors. Curr Opin Neurobiol. 2002;12:49–56. doi: 10.1016/s0959-4388(02)00289-1. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett JH, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol. 2003;213:1–11. doi: 10.1016/j.mce.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid. 2002;12:441–446. doi: 10.1089/105072502760143791. [DOI] [PubMed] [Google Scholar]
- Kim SW, Ho SC, Hong SJ, Kim KM, So EC, Christoffolete M, Harney JW. A novel mechanism of thyroid hormone-dependent negative regulation by thyroid hormone receptor, nuclear receptor corepressor (NCoR), and GAGA-binding factor on the rat cD44 promoter. J Biol Chem. 2005;280:14545–14555. doi: 10.1074/jbc.M411517200. [DOI] [PubMed] [Google Scholar]
- Farach-Carson MC, Davis PJ. Steroid hormone interactions with target cells: cross talk between membrane and nuclear pathways. J Pharmacol Exp Ther. 2003;307:839–845. doi: 10.1124/jpet.103.055038. [DOI] [PubMed] [Google Scholar]
- Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005;146:2864–2871. doi: 10.1210/en.2005-0102. [DOI] [PubMed] [Google Scholar]
- Rice DP, Rice R. Locate, condense, differentiate, grow and confront: developmental mechanisms controlling intramembranous bone and suture formation and function. Front Oral Biol. 2008;12:22–40. doi: 10.1159/000115030. [DOI] [PubMed] [Google Scholar]
- Ballock RT, O'Keefe RJ. The biology of the growth plate. J Bone Joint Surg Am. 2003;85-A:715–726. [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Bonjour JP, Theintz G, Law F, Slosman D, Rizzoli R. Peak bone mass. Osteoporos Int. 1994;4 Suppl 1:7–13. doi: 10.1007/BF01623429. [DOI] [PubMed] [Google Scholar]
- Vestergaard P, Mosekilde L. Fractures in patients with hyperthyroidism and hypothyroidism: a nationwide follow-up study in 16,249 patients. Thyroid. 2002;12:411–419. doi: 10.1089/105072502760043503. [DOI] [PubMed] [Google Scholar]
- Vestergaard P, Rejnmark L, Mosekilde L. Influence of hyper- and hypothyroidism, and the effects of treatment with antithyroid drugs and levothyroxine on fracture risk. Calcif Tissue Int. 2005;77:139–144. doi: 10.1007/s00223-005-0068-x. [DOI] [PubMed] [Google Scholar]
- O'Shea PJ, Williams GR. Insight into the physiological actions of thyroid hormone receptors from genetically modified mice. J Endocrinol. 2002;175:553–570. doi: 10.1677/joe.0.1750553. [DOI] [PubMed] [Google Scholar]
- Bassett JH, Williams GR. The skeletal phenotypes of TRalpha and TRbeta mutant mice. J Mol Endocrinol. 2009;42:269–282. doi: 10.1677/JME-08-0142. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. 1998;19:87–90. doi: 10.1038/ng0598-87. [DOI] [PubMed] [Google Scholar]
- Friedrichsen S, Christ S, Heuer H, Schafer MK, Mansouri A, Bauer K, Visser TJ. Regulation of iodothyronine deiodinases in the Pax8-/-mouse model of congenital hypothyroidism. Endocrinology. 2003;144:777–784. doi: 10.1210/en.2002-220715. [DOI] [PubMed] [Google Scholar]
- Beamer WJ, Eicher EM, Maltais LJ, Southard JL. Inherited primary hypothyroidism in mice. Science. 1981;212:61–63. doi: 10.1126/science.7209519. [DOI] [PubMed] [Google Scholar]
- Beamer WG, Cresswell LA. Defective thyroid ontogenesis in fetal hypothyroid (hyt/hyt) mice. Anat Rec. 1982;202:387–393. doi: 10.1002/ar.1092020311. [DOI] [PubMed] [Google Scholar]
- Gu WX, Du GG, Kopp P, Rentoumis A, Albanese C, Kohn LD, Madison LD, Jameson JL. The thyrotropin (TSH) receptor transmembrane domain mutation (Pro556-Leu) in the hypothyroid hyt/hyt mouse results in plasma membrane targeting but defective TSH binding. Endocrinology. 1995;136:3146–3153. doi: 10.1210/endo.136.7.7789342. [DOI] [PubMed] [Google Scholar]
- Bassett JH, Williams AJ, Murphy E, Boyde A, Howell PG, Swinhoe R, Archanco M, Flamant F, Samarut J, Costagliola S, Vassart G, Weiss RE, Refetoff S, Williams GR. A lack of thyroid hormones rather than excess thyrotropin causes abnormal skeletal development in hypothyroidism. Mol Endocrinol. 2008;22:501–512. doi: 10.1210/me.2007-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W, Govoni KE, Donahue LR, Kesavan C, Wergedal J, Long C, Bassett JH, Gogakos A, Wojcicka A, Williams GR, Mohan S. Genetic evidence that thyroid hormone is indispensable for prepubertal insulin-like growth factor-I expression and bone acquisition in mice. J Bone Miner Res. 2012;27:1067–1079. doi: 10.1002/jbmr.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A. 2002;99:15776–15781. doi: 10.1073/pnas.242322099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe E, Marians RC, Yu W, Wu XB, Ando T, Li Y, Iqbal J, Eldeiry L, Rajendren G, Blair HC, Davies TF, Zaidi M. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115:151–162. doi: 10.1016/s0092-8674(03)00771-2. [DOI] [PubMed] [Google Scholar]
- Bassett JH, Boyde A, Howell PG, Bassett RH, Galliford TM, Archanco M, Evans H, Lawson MA, Croucher P, St Germain DL, Galton VA, Williams GR. Optimal bone strength and mineralization requires the type 2 iodothyronine deiodinase in osteoblasts. Proc Natl Acad Sci U S A. 2010;107:7604–7609. doi: 10.1073/pnas.0911346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Marden CC, Ward-Bailey P, Gagnon LH, Bronson RT, Donahue LR. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol Endocrinol. 2007;21:1593–1602. doi: 10.1210/me.2007-0085. [DOI] [PubMed] [Google Scholar]
- Chassande O, Fraichard A, Gauthier K, Flamant F, Legrand C, Savatier P, Laudet V, Samarut J. Identification of transcripts initiated from an internal promoter in the c-erbA alpha locus that encode inhibitors of retinoic acid receptor-alpha and triiodo-thyronine receptor activities. Mol Endocrinol. 1997;11:1278–1290. doi: 10.1210/mend.11.9.9972. [DOI] [PubMed] [Google Scholar]
- Bassett JH, O'Shea PJ, Sriskantharajah S, Rabier B, Boyde A, Howell PG, Weiss RE, Roux JP, Malaval L, Clement-Lacroix P, Samarut J, Chassande O, Williams GR. Thyroid hormone excess rather than thyrotropin deficiency induces osteoporosis in hyperthyroidism. Mol Endocrinol. 2007;21:1095–1107. doi: 10.1210/me.2007-0033. [DOI] [PubMed] [Google Scholar]
- Gauthier K, Plateroti M, Harvey CB, Williams GR, Weiss RE, Refetoff S, Willott JF, Sundin V, Roux JP, Malaval L, Hara M, Samarut J, Chassande O. Genetic analysis reveals different functions for the products of the thyroid hormone receptor alpha locus. Mol Cell Biol. 2001;21:4748–4760. doi: 10.1128/MCB.21.14.4748-4760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneshige M, Suzuki H, Kaneshige K, Cheng J, Wimbrow H, Barlow C, Willingham MC, Cheng S. A targeted dominant negative mutation of the thyroid hormone alpha 1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci U S A. 2001;98:15095–15100. doi: 10.1073/pnas.261565798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassande O. Do unliganded thyroid hormone receptors have physiological functions? J Mol Endocrinol. 2003;31:9–20. doi: 10.1677/jme.0.0310009. [DOI] [PubMed] [Google Scholar]
- Barnard JC, Williams AJ, Rabier B, Chassande O, Samarut J, Cheng SY, Bassett JH, Williams GR. Thyroid hormones regulate fibroblast growth factor receptor signaling during chondrogenesis. Endocrinology. 2005;146:5568–5580. doi: 10.1210/en.2005-0762. [DOI] [PubMed] [Google Scholar]
- Bassett JH, Nordstrom K, Boyde A, Howell PG, Kelly S, Vennstrom B, Williams GR. Thyroid status during skeletal development determines adult bone structure and mineralization. Mol Endocrinol. 2007;21:1893–1904. doi: 10.1210/me.2007-0157. [DOI] [PubMed] [Google Scholar]
- O'Shea PJ, Harvey CB, Suzuki H, Kaneshige M, Kaneshige K, Cheng SY, Williams GR. A thyrotoxic skeletal phenotype of advanced bone formation in mice with resistance to thyroid hormone. Mol Endocrinol. 2003;17:1410–1424. doi: 10.1210/me.2002-0296. [DOI] [PubMed] [Google Scholar]
- O'Shea PJ, Bassett JH, Sriskantharajah S, Ying H, Cheng SY, Williams GR. Contrasting skeletal phenotypes in mice with an identical mutation targeted to thyroid hormone receptor alpha1 or beta. Mol Endocrinol. 2005;19:3045–3059. doi: 10.1210/me.2005-0224. [DOI] [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci U S A. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma B, Otten BJ, Stoelinga GB, Wit JM. Catch-up growth after prolonged hypothyroidism. Eur J Pediatr. 1996;155:362–367. doi: 10.1007/BF01955262. [DOI] [PubMed] [Google Scholar]
- Segni M, Gorman CA. The aftermath of childhood hyperthyroidism. J Pediatr Endocrinol Metab. 2001;14 Suppl 5:1277–1282. 1297–1298. [PubMed] [Google Scholar]
- Papadimitriou A, Papadimitriou DT, Papadopoulou A, Nicolai-dou P, Fretzayas A. Low TSH levels are not associated with osteoporosis in childhood. Eur J Endocrinol. 2007;157:221–223. doi: 10.1530/EJE-07-0247. [DOI] [PubMed] [Google Scholar]
- Refetoff S. Resistance to thyrotropin. J Endocrinol Invest. 2003;26:770–779. doi: 10.1007/BF03347364. [DOI] [PubMed] [Google Scholar]
- Persani L, Calebiro D, Cordella D, Weber G, Gelmini G, Libri D, De Filippis T, Bonomi M. Genetics and phenomics of hypothyroidism due to TSH resistance. Mol Cell Endocrinol. 2010;322:72–82. doi: 10.1016/j.mce.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Gozu HI, Lublinghoff J, Bircan R, Paschke R. Genetics and phenomics of inherited and sporadic non-autoimmune hyperthyroidism. Mol Cell Endocrinol. 2010;322:125–134. doi: 10.1016/j.mce.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Refetoff S, Dumitrescu AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab. 2007;21:277–305. doi: 10.1016/j.beem.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab. 1967;27:279–294. doi: 10.1210/jcem-27-2-279. [DOI] [PubMed] [Google Scholar]
- Weiss RE, Refetoff S. Treatment of resistance to thyroid hormone—primum non nocere. J Clin Endocrinol Metab. 1999;84:401–404. doi: 10.1210/jcem.84.2.5534. [DOI] [PubMed] [Google Scholar]
- Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, Henning E, Reinemund J, Gevers E, Sarri M, Downes K, Offiah A, Albanese A, Halsall D, Schwabe JW, Bain M, Lindley K, Muntoni F, Vargha-Khadem F, Dattani M, Farooqi IS, Gurnell M, Chatterjee K. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med. 2012;366:243–249. doi: 10.1056/NEJMoa1110296. [DOI] [PubMed] [Google Scholar]
- van Mullem A, van Heerebeek R, Chrysis D, Visser E, Medici M, Andrikoula M, Tsatsoulis A, Peeters R, Visser TJ. Clinical phenotype and mutant TRalpha1. N Engl J Med. 2012;366:1451–1453. doi: 10.1056/NEJMc1113940. [DOI] [PubMed] [Google Scholar]
- Ferrara AM, Onigata K, Ercan O, Woodhead H, Weiss RE, Refetoff S. Homozygous thyroid hormone receptor beta-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab. 2012;97:1328–1336. doi: 10.1210/jc.2011-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175. doi: 10.1086/380999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Abdullah MS, Lado-Abeal J, Majed FA, Moeller LC, Boran G, Schomburg L, Weiss RE, Refetoff S. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37:1247–1252. doi: 10.1038/ng1654. [DOI] [PubMed] [Google Scholar]
- Robson H, Siebler T, Stevens DA, Shalet SM, Williams GR. Thyroid hormone acts directly on growth plate chondrocytes to promote hypertrophic differentiation and inhibit clonal expansion and cell proliferation. Endocrinology. 2000;141:3887–3897. doi: 10.1210/endo.141.10.7733. [DOI] [PubMed] [Google Scholar]
- Miura M, Tanaka K, Komatsu Y, Suda M, Yasoda A, Sakuma Y, Ozasa A, Nakao K. Thyroid hormones promote chondrocyte differentiation in mouse ATDC5 cells and stimulate endochondral ossification in fetal mouse tibias through iodothyronine deiodinases in the growth plate. J Bone Miner Res. 2002;17:443–454. doi: 10.1359/jbmr.2002.17.3.443. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Genge BR, Wuthier RE, Wu LN. Thyroid hormone inhibits growth and stimulates terminal differentiation of epiphyseal growth plate chondrocytes. J Bone Miner Res. 1998;13:1398–1411. doi: 10.1359/jbmr.1998.13.9.1398. [DOI] [PubMed] [Google Scholar]
- Himeno M, Enomoto H, Liu W, Ishizeki K, Nomura S, Kitamura Y, Komori T. Impaired vascular invasion of Cbfa1-deficient cartilage engrafted in the spleen. J Bone Miner Res. 2002;17:1297–1305. doi: 10.1359/jbmr.2002.17.7.1297. [DOI] [PubMed] [Google Scholar]
- Mello MA, Tuan RS. Effects of TGF-beta1 and triiodothyronine on cartilage maturation: in vitro analysis using long-term high-density micromass cultures of chick embryonic limb mesenchy-mal cells. J Orthop Res. 2006;24:2095–2105. doi: 10.1002/jor.20233. [DOI] [PubMed] [Google Scholar]
- Makihira S, Yan W, Murakami H, Furukawa M, Kawai T, Nikawa H, Yoshida E, Hamada T, Okada Y, Kato Y. Thyroid hormone enhances aggrecanase-2/ADAM-TS5 expression and proteo-glycan degradation in growth plate cartilage. Endocrinology. 2003;144:2480–2488. doi: 10.1210/en.2002-220746. [DOI] [PubMed] [Google Scholar]
- Pereira RC, Jorgetti V, Canalis E. Triiodothyronine induces colla-genase-3 and gelatinase B expression in murine osteoblasts. Am J Physiol. 1999;277:E496–E504. doi: 10.1152/ajpendo.1999.277.3.E496. [DOI] [PubMed] [Google Scholar]
- Wang L, Shao YY, Ballock RT. Thyroid hormone interacts with the Wnt/beta-catenin signaling pathway in the terminal differentiation of growth plate chondrocytes. J Bone Miner Res. 2007;22:1988–1995. doi: 10.1359/jbmr.070806. [DOI] [PubMed] [Google Scholar]
- Stevens DA, Hasserjian RP, Robson H, Siebler T, Shalet SM, Williams GR. Thyroid hormones regulate hypertrophic chondrocyte differentiation and expression of parathyroid hormone-related peptide and its receptor during endochondral bone formation. J Bone Miner Res. 2000;15:2431–2442. doi: 10.1359/jbmr.2000.15.12.2431. [DOI] [PubMed] [Google Scholar]
- Gouveia CH, Schultz JJ, Bianco AC, Brent GA. Thyroid hormone stimulation of osteocalcin gene expression in ROS 17/2.8 cells is mediated by transcriptional and post-transcriptional mechanisms. J Endocrinol. 2001;170:667–675. doi: 10.1677/joe.0.1700667. [DOI] [PubMed] [Google Scholar]
- Varga F, Rumpler M, Zoehrer R, Turecek C, Spitzer S, Thaler R, Paschalis EP, Klaushofer K. T3 affects expression of collagen I and collagen cross-linking in bone cell cultures. Biochem Biophys Res Commun. 2010;402:180–185. doi: 10.1016/j.bbrc.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga F, Rumpler M, Luegmayr E, Fratzl-Zelman N, Glantschnig H, Klaushofer K. Triiodothyronine, a regulator of osteoblastic differentiation: depression of histone H4, attenuation of c-fos/c-jun, and induction of osteocalcin expression. Calcif Tissue Int. 1997;61:404–411. doi: 10.1007/s002239900356. [DOI] [PubMed] [Google Scholar]
- Banovac K, Koren E. Triiodothyronine stimulates the release of membrane-bound alkaline phosphatase in osteoblastic cells. Calcif Tissue Int. 2000;67:460–465. doi: 10.1007/s002230001171. [DOI] [PubMed] [Google Scholar]
- Stevens DA, Harvey CB, Scott AJ, O'Shea PJ, Barnard JC, Williams AJ, Brady G, Samarut J, Chassande O, Williams GR. Thyroid hormone activates fibroblast growth factor receptor-1 in bone. Mol Endocrinol. 2003;17:1751–1766. doi: 10.1210/me.2003-0137. [DOI] [PubMed] [Google Scholar]
- Gu WX, Stern PH, Madison LD, Du GG. Mutual up-regulation of thyroid hormone and parathyroid hormone receptors in rat osteoblastic osteosarcoma 17/2.8 cells. Endocrinology. 2001;142:157–164. doi: 10.1210/endo.142.1.7905. [DOI] [PubMed] [Google Scholar]
- Huang BK, Golden LA, Tarjan G, Madison LD, Stern PH. Insulin-like growth factor I production is essential for anabolic effects of thyroid hormone in osteoblasts. J Bone Miner Res. 2000;15:188–197. doi: 10.1359/jbmr.2000.15.2.188. [DOI] [PubMed] [Google Scholar]
- Linares GR, Xing W, Burghardt H, Baumgartner B, Chen ST, Ricart W, Fernandez-Real JM, Zorzano A, Mohan S. Role of diabetes- and obesity-related protein in the regulation of osteoblast differentiation. Am J Physiol Endocrinol Metab. 2011;301:E40–E48. doi: 10.1152/ajpendo.00065.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaushofer K, Hoffmann O, Gleispach H, Leis HJ, Czerwenka E, Koller K, Peterlik M. Bone-resorbing activity of thyroid hormones is related to prostaglandin production in cultured neonatal mouse calvaria. J Bone Miner Res. 1989;4:305–312. doi: 10.1002/jbmr.5650040304. [DOI] [PubMed] [Google Scholar]
- Mundy GR, Shapiro JL, Bandelin JG, Canalis EM, Raisz LG. Direct stimulation of bone resorption by thyroid hormones. J Clin Invest. 1976;58:529–534. doi: 10.1172/JCI108497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann O, Klaushofer K, Koller K, Peterlik M, Mavreas T, Stern P. Indomethacin inhibits thrombin-, but not thyroxin-stimulated resorption of fetal rat limb bones. Prostaglandins. 1986;31:601–608. doi: 10.1016/0090-6980(86)90168-1. [DOI] [PubMed] [Google Scholar]
- Allain TJ, Chambers TJ, Flanagan AM, McGregor AM. Tri-iodothyronine stimulates rat osteoclastic bone resorption by an indirect effect. J Endocrinol. 1992;133:327–331. doi: 10.1677/joe.0.1330327. [DOI] [PubMed] [Google Scholar]
- Britto JM, Fenton AJ, Holloway WR, Nicholson GC. Osteoblasts mediate thyroid hormone stimulation of osteoclastic bone resorption. Endocrinology. 1994;134:169–176. doi: 10.1210/endo.134.1.8275930. [DOI] [PubMed] [Google Scholar]
- Siddiqi A, Burrin JM, Wood DF, Monson JP. Tri-iodothyronine regulates the production of interleukin-6 and interleukin-8 in human bone marrow stromal and osteoblast-like cells. J Endocrinol. 1998;157:453–461. doi: 10.1677/joe.0.1570453. [DOI] [PubMed] [Google Scholar]
- Miura M, Tanaka K, Komatsu Y, Suda M, Yasoda A, Sakuma Y, Ozasa A, Nakao K. A novel interaction between thyroid hormones and 1,25(OH)(2)D(3) in osteoclast formation. Biochem Biophys Res Commun. 2002;291:987–994. doi: 10.1006/bbrc.2002.6561. [DOI] [PubMed] [Google Scholar]
- Abu EO, Bord S, Horner A, Chatterjee VK, Compston JE. The expression of thyroid hormone receptors in human bone. Bone. 1997;21:137–142. doi: 10.1016/s8756-3282(97)00097-5. [DOI] [PubMed] [Google Scholar]
- Govoni KE, Wergedal JE, Florin L, Angel P, Baylink DJ, Mohan S. Conditional deletion of insulin-like growth factor-I in collagen type 1alpha2-expressing cells results in postnatal lethality and a dramatic reduction in bone accretion. Endocrinology. 2007;148:5706–5715. doi: 10.1210/en.2007-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan S, Richman C, Guo R, Amaar Y, Donahue LR, Wergedal J, Baylink DJ. Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology. 2003;144:929–936. doi: 10.1210/en.2002-220948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, Clemens TL. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24:782–801. doi: 10.1210/er.2002-0033. [DOI] [PubMed] [Google Scholar]
- Wang Y, Nishida S, Sakata T, Elalieh HZ, Chang W, Halloran BP, Doty SB, Bikle DD. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology. 2006;147:4753–4761. doi: 10.1210/en.2006-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Shao YY, Ballock RT. Thyroid hormone-mediated growth and differentiation of growth plate chondrocytes involves IGF-1 modulation of beta-catenin signaling. J Bone Miner Res. 2010;25:1138–1146. doi: 10.1002/jbmr.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman C, Kutilek S, Miyakoshi N, Srivastava AK, Beamer WG, Donahue LR, Rosen CJ, Wergedal JE, Baylink DJ, Mohan S. Postnatal and pubertal skeletal changes contribute predominantly to the differences in peak bone density between C3H/HeJ and C57BL/6J mice. J Bone Miner Res. 2001;16:386–397. doi: 10.1359/jbmr.2001.16.2.386. [DOI] [PubMed] [Google Scholar]
- Lakatos P, Caplice MD, Khanna V, Stern PH. Thyroid hormones increase insulin-like growth factor I content in the medium of rat bone tissue. J Bone Miner Res. 1993;8:1475–1481. doi: 10.1002/jbmr.5650081210. [DOI] [PubMed] [Google Scholar]
- Thorngren KG, Hansson LI. Effect of thyroxine and growth hormone on longitudinal bone growth in the hypophysectomized rat. Acta Endocrinol (Copenh) 1973;74:24–40. doi: 10.1530/acta.0.0740024. [DOI] [PubMed] [Google Scholar]
- Lewinson D, Bialik GM, Hochberg Z. Differential effects of hypothyroidism on the cartilage and the osteogenic process in the mandibular condyle: recovery by growth hormone and thyroxine. Endocrinology. 1994;135:1504–1510. doi: 10.1210/endo.135.4.7925111. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Isgaard J, Tornell J, Nilsson A, Isaksson OG, Lindahl A. Endocrine regulation of longitudinal bone growth. Acta Paediatr Suppl. 1993;82 Suppl 391:33–40. doi: 10.1111/j.1651-2227.1993.tb12925.x. [DOI] [PubMed] [Google Scholar]
- Gauthier K, Chassande O, Plateroti M, Roux JP, Legrand C, Pain B, Rousset B, Weiss R, Trouillas J, Samarut J. Different functions for the thyroid hormone receptors TRalpha and TRbeta in the control of thyroid hormone production and post-natal development. EMBO J. 1999;18:623–631. doi: 10.1093/emboj/18.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RA, Smith RM, Meller DJ, Dahlenburg GW, Lineham JD. Effect of growth hormone on growth and myelination in the neonatal hypothyroid rat. J Endocrinol. 1988;119:117–125. doi: 10.1677/joe.0.1190117. [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Dentice M, Bandyopadhyay A, Gereben B, Callebaut I, Christoffolete MA, Kim BW, Nissim S, Mornon JP, Zavacki AM, Zeold A, Capelo LP, Curcio-Morelli C, Ribeiro R, Harney JW, Tabin CJ, Bianco AC. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7:698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Coumoul X, Deng CX. Roles of FGF receptors in mammalian development and congenital diseases. Birth Defects Res C Embryo Today. 2003;69:286–304. doi: 10.1002/bdrc.10025. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–449. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- Chen M, Zhu M, Awad H, Li TF, Sheu TJ, Boyce BF, Chen D, O'Keefe RJ. Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J Cell Sci. 2008;121:1455–1465. doi: 10.1242/jcs.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, Forrest D, Thoren P, Vennstrom B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J. 1998;17:455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salto C, Kindblom JM, Johansson C, Wang Z, Gullberg H, Nordstrom K, Mansen A, Ohlsson C, Thoren P, Forrest D, Vennstrom B. Ablation of TRalpha2 and a concomitant overexpression of alpha1 yields a mixed hypo- and hyperthyroid phenotype in mice. Mol Endocrinol. 2001;15:2115–2128. doi: 10.1210/mend.15.12.0750. [DOI] [PubMed] [Google Scholar]
- Gothe S, Wang Z, Ng L, Kindblom JM, Barros AC, Ohlsson C, Vennstrom B, Forrest D. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev. 1999;13:1329–1341. doi: 10.1101/gad.13.10.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L, Hurley JB, Dierks B, Srinivas M, Salto C, Vennstrom B, Reh TA, Forrest D. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27:94–98. doi: 10.1038/83829. [DOI] [PubMed] [Google Scholar]
- Weiss RE. “They have ears but do not hear” (Psalms 135:17): non-thyroid hormone receptor beta (non-TRbeta) resistance to thyroid hormone. Thyroid. 2008;18:3–5. doi: 10.1089/thy.2007.0373. [DOI] [PubMed] [Google Scholar]