

Abstract
Fibroblast growth factor (FGF)/fibroblast growth factor receptor (FGFR) signaling plays essential roles in bone development and diseases. Missense mutations in FGFs and FGFRs in humans can cause various congenital bone diseases, including chondrodysplasia syndromes, craniosynostosis syndromes and syndromes with dysregulated phosphate metabolism. FGF/FGFR signaling is also an important pathway involved in the maintenance of adult bone homeostasis. Multiple kinds of mouse models, mimicking human skeleton diseases caused by missense mutations in FGFs and FGFRs, have been established by knock-in/out and transgenic technologies. These genetically modified mice provide good models for studying the role of FGF/FGFR signaling in skeleton development and homeostasis. In this review, we summarize the mouse models of FGF signaling-related skeleton diseases and recent progresses regarding the molecular mechanisms, underlying the role of FGFs/FGFRs in the regulation of bone development and homeostasis. This review also provides a perspective view on future works to explore the roles of FGF signaling in skeletal development and homeostasis.
Introduction
Skeletons are formed through two distinct developmental modes, namely intramembranous ossification and endochondral ossification. The former is directly accomplished by osteoblast differentiation from mesenchymal cells; the latter involves initial differentiation of mesenchymal cells into chondrocytes to form a cartilage template and subsequent replacement by bone.1 The cranium and medial clavicles are formed through intramembranous ossification. Long bones, including the appendicular skeleton, facial bones and vertebrae, are formed through endochondral ossification.2,3
Various signaling molecules control the process of skeleton development, such as fibroblast growth factor (FGF), wingless-type MMTV integration site family members (Wnt) and bone morphogenetic protein (BMP) signaling pathways. Among these signaling pathways, FGF/fibroblast growth factor receptor (FGFRs) signaling is very essential. The 22 members of the FGF family mediate their cellular responses by binding to FGFRs. There are four distinct FGF receptors with differential FGF-binding properties.4,5 A typical FGFRs contains an extracellular ligand-binding domain, a transmembrane region and an intracellular divided tyrosine kinase domain. FGFs bind to the extracellular domain of FGFRs and induce the phosphorylation of tyrosine residues in the intracellular domain of FGFRs. The activated FGFRs recruits target proteins to its cytoplasmic tail and modifies these proteins by phosphorylation,6 leading to the activation of intracellular downstream signaling pathways, such as mitogen-activated protein kinase (Ras/MAPK), phosphoinositide 3-kinase/Akt (also known as protein kinase B), phospholipase C and protein kinase C pathways. Furthermore, FGF signaling can also stimulate the signal transducers and activators of transcription (STAT) 1/p21 pathway2,7 (Figure 1). Multiple kinds of mouse models with genetic modifications of FGF/FGFRs have been generated. In our review, we summarize the use of these mouse models in the research of the role of FGF/FGFRs signaling in skeleton development and homeostasis.
Figure 1.

Signaling pathways activated by FGF/FGFR. FGFs induce dimerization, kinase activation and transphosphorylation of tyrosine residues of FGFRs, leading to activation of downstream signaling pathways. Multiple pathways are stimulated by FGF/FGFR signaling such as Ras-MAP kinase, PI-3 kinase/AKT and PLC-γ pathways. Furthermore, FGF signaling can also stimulate STAT1/p21 pathway. FGF/FGFR signaling also phosphorylates the Shc and Src protein. FGF/FGFR play crucial roles in the regulation of proliferation, differentiation and apoptosis of chondrocytes via downstream signaling pathways.
Role of FGFRs in bone genetic diseases and homeostasis
FGFR1
FGFR1 is first expressed in the early limb bud.8–10 At the epiphyseal growth plate, FGFR1 is expressed in perichondrium, prehypertrophic and hypertrophic chondrocytes.9,11,12 FGFR1 is also expressed in osteoblasts and osteocytes (Table 1).13–16
Table 1. The expression patterns of FGFs/FGFRs during skeleton development.2,7,11,31,46,60,161,162,204,262,263.
| FGFs/FGFRs | Limb bud | Osteoblast lineage | Cartilage | Cranial bone | Receptor specificity |
|---|---|---|---|---|---|
| FGF2 |
Developing condensation |
Periosteal cells, Osteoblasts in trabecular bone |
Perichondrium, Chondrocytes |
Mesenchymal cells in the suture |
FGFR1, FGFR2, FGFR3c, FGFR4 |
| FGF4 |
Posterior AER at E10.5-11.0 |
|
|
Sutural mesenchyme in early craniofacial skeletogenesis |
FGFR1c, FGFR2c, FGFR3c |
| FGF7 |
Loose mesenchyme |
|
Perichondrium |
|
FGFR2b |
| FGF8 |
AER |
Cortical bone at embryonic stage |
Perichondrium, Chondrocytes |
Osteoblasts |
FGFR2c, FGFR3c, FGFR4 |
| FGF9 |
AER, Developing condensation |
Periosteum, Primary spongiosa |
Perichondrium, Chondrocyte primordia |
Mesenchyme of suture in early craniofacial development stages |
FGFR2c, FGFR3, FGFR4 |
| FGF10 |
Lateral plate mesoderm |
|
|
|
FGFR2b |
| FGF18 |
Perichondrium and presumptive joint positions |
|
Chondrocytes, |
Mesenchymal cells in the suture separating the two osteogenic fronts |
FGFR2c, FGFR3c |
| FGF21 |
|
|
Chondrocytes |
|
FGFR1-4 |
| FGF23 |
|
Osteoblasts, Osteocytes |
Resting and hypertrophic zone |
|
FGFR1, FGFR3c, FGFR4 |
| FGFR1 |
Mesenchyme (IIIc) |
Osteoblasts in trabecular bone, Osteocytes |
Prehypertrophic and hypertrophic chondrocytes of growth plate, Perichondrium, Cartilage of the cranial base |
Dura mater and periosteum, Calvarial mesenchyme and later in osteoblasts |
|
| FGFR2 |
AER (IIIb), Early limb bud mesenchyme (IIIc) |
Periosteum, Trabecular bone (IIIc), Osteocytes |
Prechondrogenic condensation, Resting zone of growth plate, Perichondrium, Cartilage of the cranial base |
Proliferating osteoprogenitor cells and differentiating osteoblasts |
|
| FGFR3 |
Center of the mesenchyme condensation |
Osteoblasts, Osteocytes |
Resting zone and proliferating chondrocytes of growth plate, Cartilage of the cranial base |
Low levels in sutural osteogenic fronts at late stages of development |
|
| FGFR4 | Strictly in osteoblasts between the periosteal and endosteal layers | Resting and proliferative zones of growth plate |
A series of mouse models of Fgfr1 have been generated to genetically dissect the functions of Fgfr1 during gastrulation and later developmental processes. Fgfr1-deficient (Fgfr1−/−) embryos display severe growth retardation, and died prior to or during gastrulation because of intrinsic blocks in mesodermal differentiation.17,18 Deletion of the Ig domain IIIc of Fgfr1 (Fgfr1IIIc) leads to gastrulation defects resembling the Fgfr1−/− alleles. However, mice with Fgfr1IIIb ablation are viable and fertile, suggesting that IIIc is the dominant isoform for the majority of FGFR1 functions in embryogenesis.19 Chimeras were generated by injecting Fgfr1−/− embryonic stem cells into wild-type blastocysts to circumvent the gastrulation defect. The milder mutant chimeras exhibit deformed limb buds and varying degrees of reduction in limb skeletal elements.19–21
Mice with targeted deletion of FGFR1 in all limb bud mesenchymal cells (via T (brachyury)-cre),22 or posterior limb bud mesenchyme (via Shh-cre)23 were used to further study the role of FGFR1 in limb development. T-cre; Fgfr1 mice die at birth and show reduced limb skeleton, misshapen forelimb/hindlimb bud and missing digits, whereas Shh-cre Fgfr1 mice display normal limb bud size, but missed a digit.10 Li et al.24 assessed the roles of FGFR1 signaling in forelimb and hindlimb development by disrupting this gene, using AP2-Cre and Hoxb6-Cre transgenic mice that express Cre recombinase in complementary temporal and spatial patterns during limb bud formation. The results indicate that disruption of Fgfr1 at an earlier stage, prior to thickening of limb mesenchyme, results in more severe defects, characterized by malformation of the apical ectodermal ridge (AER).
FGF receptor-specific substrates (Frs) act as the principal mediators for FGFR1 signal transduction. Mice that lack the Frs-binding site on FGFR1 (Fgfr1ΔFrs/ΔFrs) die during late embryogenesis, and exhibit defects in neural tube closure, and in the development of the tail bud and pharyngeal arches. However, mutant FGFR1 still has functions during gastrulation and somitogenesis, indicating that distinct signal transduction mechanisms of FGFR1 signaling in different developmental contexts.25
Osteoglophonic dysplasia (OD) patients, resulting from activating mutations of FGFR1, exhibit rhizomelic dwarfism,26 indicating that FGFR1 is a negative regulator of long bone growth. Embryos with conditional deletion of Fgfr1 in osteochondro-progenitor cell lineages show increased height of the hypertrophic zone due to delayed degradation, or maturation of hypertrophic chondrocytes, or decreased osteoclastogenesis.15
Studies in humans and mice also reveal that FGFR1 play crucial role in bone formation. A gain-of-function mutation in FGFR1 (P252R) leads to Pfeiffer syndrome (PS), one type of craniosynostoses, characterized by premature fusion of one or several calvarial sutures.27 Several activating mutations of FGFR1 in OD patients also lead to craniosynostosis in addition to rhizomelic dwarfism.26 Mice carrying a P250R mutation in FGFR1 were generated to mimic human PS. Studies using these mutant mice uncovered that FGFs/FGFR1 signals may regulate intramembranous bone formation.28
Jacob et al.15 found that adult mice, with deletion of Fgfr1, exhibited increased bone mass. Deletion of Fgfr1, in osteochondro-progenitor cells in mice (via Col2-cre), leads to increased proliferation and delayed differentiation, and matrix mineralization of osteoblasts, while inactivation of Fgfr1 in differentiated osteoblasts (via Col1-cre) causes accelerated osteoblast mineralization differentiation.15 It has been proposed that FGFR1 promotes the differentiation of mesenchymal progenitors into preosteoblasts, but inhibits the proliferation of mesenchymal progenitor cells, as well as the maturation and mineralization of osteoblasts.15 Impaired osteoclast activity is another reason for increased bone mass in mice with Fgfr1-deficient in differentiated osteoblasts. To explore the direct effect of FGFR1 on osteoclasts, Lu et al.29 generated mice with targeted deletion of Fgfr1 in bone marrow monocytes and osteoclasts using LysM-cre. The mutant mice exhibit increased bone mass, impaired osteoclast formation and activity indicating the positive regulation of FGFR1 on osteoclasts. The role of FGFR1 in osteocytes is still not clarified and should be studied by deletion of Fgfr1 in osteocytes using dentin matrix protein-1(Dmp1)-Cre (Figure 2).
Figure 2.
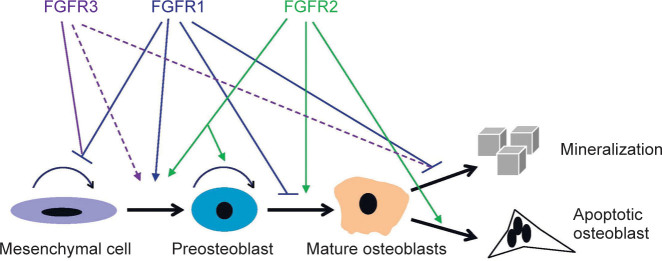
The regulation of osteogenesis by FGFR1-3. FGFRs play distinct roles during the differentiation of osteoblast. There are some conflicting results about the effect of FGFR3 on osteogenesis, which is marked by dotted lines.
In addition to the effect of FGFR1 on limb development and bone formation or remodeling, FGFR1 also participates in phosphorus metabolism. Osteoglophonic dysplasia patients have non-ossifying bone lesions, hypophosphatemia and increased serum level of FGF23, a member of the FGF family, which is a circulating phosphaturic hormone produced mainly by osteoblasts and osteocytes.30,31 Pharmacological inhibition of FGFR1 inhibits FGF23 transcription in bone of animal models.32 Integrative nuclear FGFR1 can activate the transcription factor cyclic AMP response element-binding protein (CREB),33 which also binds the proximal Fgf23 promoter; thus, it is hypothesized that FGFR1 may regulate FGF23 by binding CREB.
FGFR2
FGFR2 is expressed in condensing mesenchyme of early limb bud,9,34,35 and later appears as the marker of prechondrogenic condensations. In developing bone, FGFR2 is predominantly localized to perichondrial and periosteal tissue, and weakly to endosteal tissue and trabecular bone.36 FGFR2 is intensely expressed in the cartilage of the cranial base and growth plate.11,37–40 In cranial sutures, FGFR2 is mainly expressed in osteoprogenitor cells13 and differentiating osteoblasts.41,42 The expression pattern of FGFR2 indicates its important role in skeleton development (Table 1).
Mice with deletion of transmembrane domain and part of kinase I domain of Fgfr2 (Fgfr2−/−) die at E4.5–5.5 due to stopped inner cell mass growth.43 Targeted deletion of the Ig domain III of FGFR2 results in embryonic lethality at E10–11 because of failures in the formation of functional placenta. Mutant embryos also fail to form limb buds completely, indicating that FGFR2 Ig domain III is essential for limb initiation.24,44,45
Activating FGFR2 mutations have variable effects on cranial cell replication, or differentiation in mice and humans.40,46 More than 10 gain-of-function mutations in FGFR2 cause multiple types of craniosynostoses, such as Apert syndrome (AS), Crouzon syndrome (CS) and PS, as well as Beare–Stevenson cutis gyrata syndrome (BSS).40,47 Among them, AS is one of the most severe craniosynostoses. S252W and P253R mutations in FGFR2 are responsible for nearly all known cases of AS.2,47
Several gain-of-function mutant mouse models, mimicking human craniosynostoses, have been generated to study the mechanism of FGFR2 for regulating the suture development. Fgfr2+/S252W mutant mice mimicking human AS have smaller body size, midline sutural defect and craniosynostosis with abnormal osteoblastic proliferation and differentiation.48 Fgfr2+/S252W mice also show ectopic cartilage at the midline sagittal suture, increased cartilage in the basicranium, nasal turbinates and trachea. These mutant mice display long bone abnormalities, as evidenced by the disorganized growth plates and more prominent cartilage mineralization.48 Fgfr2+/P253R mice have growth retardation of the synchondroses of cranial base and growth plates of the long bones with decreased proliferation of chondrocytes, which may be responsible for the smaller body size and shortened cranial base in Fgfr2+/P253R mice.39 Furthermore, Fgfr2+/P253R mice also show ectopic cartilages in the sagittal sutures39,49 consistent with the skull phenotypes in Fgfr2+/S252W mice and the symptom in AS patients.48,50 However, Chen et al.51 found that the growth plates of Fgfr2+/S250W mice showed slightly shorter columns of proliferating chondrocytes, but no abnormal hypertrophic zone; and premature closure of cranial base synchondrosis have been detected in Fgfr2+/S250W mice.
In Fgfr2+/P253R mice or Fgfr2+/Y394C mice mimicking human BSS (also characterized by skull abnormalities), the premature fusion of coronal suture is associated with enhanced osteoblast differentiation similar to Fgfr2+/S252Wmice.39,49,52 In another mouse model with a C342Y mutation in FGFR2IIIc (Fgfr2c+/C342Y) (equivalent to mutation in human causes CS/PS), premature fusion of cranial sutures is accompanied by enhanced osteogenesis and increased proliferation of osteoprogenitor cells in the coronal sutures.53 Chen et al.51 also found decreased bone formation and premature closure of the coronal suture in Fgfr2+/S250W mice similar to phenotypes in human AS.51 However, increased apoptosis is responsible for premature fusion in Fgfr2+/S250W coronal suture.51 These results suggest that different activating mutations in FGFR2 result in craniosynostosis through distinct mechanisms.
Fgfr2IIIc−/− mice also show delayed differentiation and mineralization of the skull vault, and premature coronal suture due to decreased cell proliferation.54 The retarded ossification in Fgfr2IIIc−/− mice is correlated with the decreased osteoblast markers OP and Cbfa1, which is emphasized by increased osteogenesis of Crouzon-like mutant Fgfr2c+/C342Y mice with upregulated OP and Cbfa1 levels.55 These results suggest that FGFR2IIIc is a positive regulator of intramembranous ossification. Conditional deletion of Fgfr2 in mesenchymal condensations of mice via Dermol-Cre results in skeletal dwarfism and decreased bone density. The proliferation of osteoprogenitors and the function of mature osteoblasts are impaired in mutant mice. However, the differentiation of osteoblast lineage in mutant mice is not disturbed,36 which is distinct from the delayed differentiation in Fgfr2IIIc–/– mice.54 This finding may reflect the differences in the timing of Fgfr2 inactivation.36 These differences can be further explored by conditional deletion of FGFR2 in osteoblastic cells.
Fgfr2IIIc–/– mice also exhibit dwarfisms, reduced growth of the skull base and axial, as well as appendicular skeletons, which is associated with decreased proliferating chondrocytes and hypertrophic zone in these endochondral bones. This leads to premature loss of skull base sutures and smaller-than-normal long bones and vertebrae.54 The expressions of chondrocyte markers Ihh and PTH-related peptide (PTHrP) are also diminished in Fgfr2IIIc–/– mice.54 These results suggest that Fgfr2IIIc regulates chondrocyte lineages. Fgfr2; Dermol-Cre mice have decreased bone length without apparent defects in chondrocyte proliferation, but show shortened hypertrophic zone, which is similar to that in Fgfr2IIIc–/– mice.54 Increased osteoclast activity may account for decreased hypertrophic zone by increasing the removal of calcified hypertrophic chondrocyte matrix.36
Genetic mouse models are also used to find therapeutic strategy. Activated ERK1/2 and p38 signaling pathways may participate in the regulation of coronal suture by FGFR2.39,49,52 The premature fusion of cranial sutures can be partially rescued by blocking ERK1/2 or p38 activation, respectively in vitro and in vivo.39,49,52,56 Shukla et al.56,57 showed that RNA interference targeting the mutant form of FGFR2 S252W completely rescued Apert-like syndrome in mice, and local treatment of the Apert mice with U0126, an inhibitor of MEK1/2, significantly alleviated craniosynostosis. Using genetically modified mice simultaneously carrying C342Y, L424A and R424A mutations in the same FGFR2c (Fgfr2cCLR), researchers found that the activated FGFR2 signaling caused by C342Y mutation, cannot recruit and stimulate tyrosine phosphorylation of FRS2.53 Thus, premature fusion of sutures, mediated by activated FGFR2IIIc, is prevented by attenuation of the signaling pathways through selective uncoupling between the docking protein FRS2α and activated FGFR2IIIc.53 These studies provide opportunities for developing novel therapeutic strategies for craniosynostoses.
FGFR3
FGFR3 is first expressed in chondrocytes, differentiated initially from the core of the mesenchyme condensation.58 FGFR3 is expressed in reserve and proliferating chondrocytes as the epiphyseal growth plate is formed.12,58,59 Immunohistochemistry results have indicated that FGFR3 is also expressed in mature osteoblasts and in osteocytes.14 During calvarial bone development FGFR3 is expressed at low levels in sutural osteogenic fronts at the late stages (Table 1).34,38
Gain-of-function point mutations in FGFR3 cause several types of the human skeletal dysplasias, including achondroplasia (ACH), hypochondroplasia (HCH), thanatophoric dysplasia (TD) and severe achondroplasia, with developmental delay and acanthosis nigricans (SADDAN).60 Among these diseases, ACH is the most common type of human dwarfism characterized by short stature, especially in the proximal upper and lower limbs, central facial dysplasia, macrocephaly and spine protrusion.61–63 The phenotype of HCH is similar to ACH, but much milder than ACH, whereas TD is the most common form of lethal skeletal dysplasia characterized by macrocephaly, narrow bell-shaped thorax, severe shortening of the limbs and lethality in the neonatal period. TD has been classified into TDI and TDII. TDI patients have curved, short femurs, with or without cloverleaf skull, and TDII patients have relatively longer femurs with severe cloverleaf skull.64 Patients with SADDAN exhibit acanthosis nigricans and anomalies in the central nervous system, in addition to severe skeletal dysplasia.65,66
Currently, multiple FGFR3-related mouse models have been generated using genetic approach to study the role of FGFR3 in skeleton development and diseases. Mice carrying activating mutations of FGFR3 mimicking human ACH exhibit smaller body size, dome-shaped skull and shortened long bones with disorganized chondrocyte columns in growth plates.60,67–70 Mice carrying FGFR3 K644E mutation mimicking human TDII die within few hours after birth, whereas mice carrying FGFR3 S365C mutation, which corresponds to FGFR3 S371C mutation in human TDI, exhibit skeletal dysplasia more severe than ACH.71,72 FGFR3 negatively regulates chondrogenesis of long bones by affecting the proliferative activity and differentiation of chondrocytes. A number of reports have demonstrated that FGFR3 signaling inhibits chondrocyte proliferation through STAT1 signaling by inducing the expression of cell cycle suppressor genes such as the CDK inhibitor p21.73–76 Loss of Stat1 restored the reduced chondrocyte proliferation in ACH mice, but did not rescue the reduced hypertrophic zone or the delayed formation of secondary ossification centers in ACH mice. The expression of a constitutively active mutant of MEK1 in chondrocytes of Fgfr3-deficient mice inhibits skeletal overgrowth, strongly suggesting that FGFR3 inhibits chondrocyte differentiation through the ERK/MAPK pathway.76 In contrast, evidence suggests that FGFR3 promote chondrocyte terminal hypertrophic differentiation.77,78 Conversely, mice carrying targeted deletion of FGFR3 exhibit overgrowth of long bone, wider hypertrophic zone, proliferative zone and enhanced proliferative activity of chondrocytes.59,79
Moreover, the activity and the signaling outcomes of the FGFR3 pathway during chondrogenesis are also influenced by many intracellular and extracellular signals. Activated FGFR3 inhibits BMP4 expression in post-natal mouse growth plates,80 while BMP treatment rescues the retarded growth of long bone in ACH mouse model.77 These studies emphasize the antagonistic interaction between FGFR3 and BMP signaling in the control of chondrogenesis. Moreover, IHH expression is reduced in mice carrying activating FGFR3.80 PTHrP partially reverses the inhibition of long bone growth caused by FGFR3 activation.72 It was suspected that FGFR3 signaling may act upstream of the IHH/PTHrP system in regulating the onset of hypertrophic differentiation.77 In addition, it was reported that IGF1 prevents the apoptosis, induced by FGFR3 mutation, through the phosphoinositide 3-kinase pathway and MAPK pathways.81
FGFR3 signaling is also an important regulator of osteogenesis. Chondrocyte-specific activation of FGFR3 in mice causes premature synchondrosis closure and enhanced osteoblast differentiation around synchondroses. Premature synchondrosis closure is also observed in the spine and cranial base in human cases of homozygous ACH and TD, as well as in mouse models of ACH, with increased bone formation.70,72,82 Activated FGFR3 leads to decreased bone mass by regulating both osteoblast and osteoclast activities.83,84 Mice lacking FGFR3 also have decreased bone mineral density and osteopenia.14,85 FGFR3 can inhibit proliferation of BMSCs in vitro.83,85 However, both deletion and activation of FGFR3 can lead to increased differentiation, but impaired mineralization of osteoblasts (Figure 2).83,85 The reasons for these seemingly inconsistent results need to be explored.
Given its causal role in some skeletal disorders, including ACH, FGFR3 and/or its downstream pathways, are attractive targets for therapy. C-type natriuretic peptide is a newly identified potential therapeutic antagonist of FGFR3 signaling that alleviates the dwarfism phenotype of mice mimicking human ACH through its inhibition on FGFR3/MAPK pathway.86,87 It was reported that parathyroid hormone (PTH) (1–34) stimulates the longitudinal bone growth in rats and improves the growth of the cultured femurs from mice carrying a gain-of-function mutation (G380R) of FGFR3.88,89 In addition, we have found previously that PTHrP partially reversed the shortening of cultured bone rudiments from ACH mice.72 Recently, we found that systemic intermittent injection of PTH (1–34) can rescue the lethal phenotype of TDII mice and significantly alleviate the retarded skeleton development of ACH mice.90 We also have identified a novel inhibitory peptide for FGFR3 signaling, which alleviated the bone growth retardation in bone rudiments from mice mimicking human TDII and reversed the neonatal lethality of TDII mice.91
FGFR4
In addition to its expression in the resting and proliferative zones of growth plates,11 FGFR4 is also highly expressed in rudimentary membranous bone and strictly localized in osteoblasts between the periosteal and endosteal layers (Table 1).92 Interestingly, Fgfr4-deficient mice are developmentally normal, but the Fgfr3/Fgfr4 double null mice grow more slowly.93 However, the effect of FGFR4 on bone development remains unclear and needs further studies.
FGFs participate in skeleton development and bone metabolism
FGF2
FGF2 is one of the earliest members identified in the FGF polypeptide family, and is expressed in majority of cells and tissues including limb bud, chondrocytes and osteoblasts. FGF2 is stored in the extracellular matrix.11,94–96
FGF2 contributes to the growth and patterning of the limb.96 Overexpression of human FGF2 in mice (TgFGF2) results in dwarfism, with shortening and flattening of long bones and moderate macrocephaly.97 Deletion of Stat1 leads to a significant correction of the chondrodysplasic phenotype of TgFGF2 mice.98 These results indicate the essential role of STAT1 in FGF-mediated regulation of epiphyseal growth plates. Fgf2-knockout (Fgf2−/−) mice have normal limbs. The normal skeleton in Fgf2−/− mice indicates that the function of FGF2 may be replaced by FGF8 and FGF4,99 which is also expressed in the limb bud. FGF2 also plays important roles in bone homeostasis. Deletion of Fgf2 in mice leads to decreased bone mass, bone formation and mineralization.95,100 Endogenous FGF2 promotes the differentiation of bone marrow stromal cells (BMSCs) into osteoblasts, since FGF2 deficiency results in adipogenesis and reduced osteogenesis of BMSCs.95,101 Similar to Fgf2−/− mice, TgFGF2 mice also have reduced bone mass, which may result from impaired endochondrol ossification, or continuous exposure to high levels of FGF2 in vivo.14,102 Targeted overexpression of FGF2 in chondrocytes and osteoblasts should provide important information about the role of FGF2 in dwarfism and bone formation.102
Other important factors for bone homeostasis also exert their effects through FGF2. PTH and BMP2-induced bone formation in Fgf2−/− mice are greatly impaired, and osteoclast formation stimulated by PTH and BMP2 are also disrupted in Fgf2−/− bone marrow stromal cultures.103–105 The impaired bone anabolic effect of PTH in Fgf2−/− mice is associated with reduced expression of activating transcription factor 4, a critical regulator for osteoblast differentiation and function.106 Furthermore, prostaglandin F2α also induces osteoblast proliferation through endogenous FGF2.107
FGF2 has three isoforms: a low molecular weight isoform (lmw, 18 kDa) and two high molecular weight isoforms (hmw, 21 and 22 kDa). FGF2lmw is secreted and activates FGFRs, whereas FGF2hmw remains intranuclear. Their roles in bone formation are largely unknown. Transgenic mice with targeted overexpression of FGF2lmw and FGF2hmw in immature and mature osteoblast lineage (via Col3.6-cre) are used to elucidate the differential functions of FGF2 isoforms in bone formation.108,109 Col3.6-FGF2lmw mice have increased bone mineral density (BMD), bone mass and enhanced mineralization of BMSCs, which is related to the reduced expression of the Wnt antagonist secreted frizzled receptor 1.110 In contrast to TgFGF2lmw mice, Fgf2lmw−/− mice show significantly reduced BMD and impaired mineralization.108
Col3.6-FGF2hmw mice display dwarfism, decreased BMD, increased FGF23 level, hypophosphatemia and rickets/osteomalacia, which is similar to X-linked hypophosphatemia (XLH).109,110 A potential mechanism is that FGF2 enhances FGF23/FGFR1/KLOTHO signaling, and then downregulates renal Na+/Pi cotransporter NPT2a, causing Pi wasting, osteomalacia and decreased BMD.109 The upregulation of FGF23 level by FGF2hwm depends on FGFR1/MAPK pathway.110 These studies indicate that FGF2 isoforms have important effects on bone homeostasis and different FGF2 isoforms perform distinct roles.
FGF4
Vertebrate limb development largely depends on signals from the AER. During limb development, FGF4 is first expressed in the developing murine forelimb bud at E10.0. Its expression is strongest in the posterior AER at E10.5–11.0 and is undetectable at E12.0.111 FGF4 provides mitogenic and morphogenic signals to regulate normal limb development.111,112 Fgf4 knockout (Fgf4−/−) mice die on E4.5 (early embryonic stages),113 preventing the direct evaluation of FGF4 function in the developing limb. Mice with targeted deletion of Fgf4 in limbs (via Rarb-Cre) are viable and have normal skeletal patterns.111 The expression pattern of Sonic hedgehog (Shh), another key signaling molecule in AER maintenance, is normal in the limb buds, suggesting that FGF4–Shh feedback loop is not essential for limb development.
In addition to its essential roles in the AER of normal embryo, FGF4 can also promote intramembranous ossification and participate in the development of calvarial bone. FGF4 is expressed in sutural mesenchyme during early craniofacial skeletogenesis.60 Treatment with FGF4 on developing mouse coronal suture leads to synostotic coronal sutures accompanied by the induction of apoptosis and accelerated mineralization.114 FGF4 can also cause premature suture fusion with increased cell proliferation, both in cultured calvaria and in mice.115 Furthermore, systemic administration of FGF4 and its 134 amino-acid residues leads to increased bone formation in rats and mice in vivo.116 FGF4 can also promote BMSC proliferation in vitro,117,118 and strongly stimulate Runx2 expression in osteoblast-like MC3T3-E1 and murine premyoblast C2C12 cells.119 However, studies especially genetic studies on the role of FGF4 in bone formation, are still lacking.
FGF8
FGF8 is expressed throughout the AER, indicating its important role in limb development.120–122 Mice with deleted Fgf8 show early embryonic lethality before limb development.123,124 Lewandoski et al. generated mice with targeted deletion of Fgf8124 (via Msx2-cre) in limb bud.112 These mice display failed limb development with substantial reduction in limb-bud size, and hypoplasia or aplasia of specific skeletal elements.112 However, the Msx2 promoter drived cre is not expressed sufficiently early to completely ablate Fgf8 function during forelimb formation, which results in a complex forelimb phenotype. Using Rarb-Cre mice, Fgf8 is conditionally deleted in the developing forelimb AER. These mice have severe forelimb deformity, including the absence of radius and first digit.125,126
In addition to its important role in limb development, FGF8 also regulates osteoblast and chondrocyte differentiation. FGF8 is expressed in chondrocytes and perichondrium of dorsal costal bone, as well as in the osteoblast compartment of calvarial bone in cortical bone and the growth plate of developing bones.60,127 FGF8 can effectively predetermine mouse BMSCs and C2C12 cell line to differentiate to osteoblasts and increase bone formation in vitro.128,129 However, Lin et al.130 found that FGF8 stimulated the proliferation of MC3T3E1 or primary rat osteogenic cells, but inhibited osteogenic differentiation and mineralization. These controversial results may be attributed to the different cells used in in vitro experiments. As to cartilage, FGF8 can promote the degradation of cartilage and exacerbation of osteoarthritis.131 However, the influence of FGF8 on bone and cartilage remains unclear.
FGF9
FGF9 has the highest affinity to FGFR3, and can also bind FGFR2 with a lower affinity (Table 1).132 FGF9 is broadly expressed in different tissues including in AER, perichondrium/periosteum, chondrocytes of growth plate, as well as primary spongiosa.133–135
Colvin et al.136 generated Fgf9 knockout (Fgf9−/−) mice and showed that deletion of Fgf9 alleles led to lethality at the neonatal stage mainly due to malformations of the lung, and causing male-to-female sex reversal.136,137 Fgf9−/− mice display disproportionate shortening of the proximal skeletal elements (rhizomelia), but the limb bud development and mesenchymal condensations are normal.135 These results indicate that loss of Fgf9 in AER does not lead to limb patterning defects that primarily affected mesenchymal condensation. The rhizomelia results from the loss of Fgf9 function after mesenchymal condensation. Similarly, transgenic mice, with overexpression of Fgf9 in chondrocytes (Col2a1–Fgf9), also show dwarfism, short limb and vertebral defect because of the reduced proliferation and terminal differentiation of chondrocytes. These results are similar to bone phenotypes, caused by activated FGFR3.133 These seemingly inconsistent results between Fgf9 null and transgenic mice may result from distinct effect of FGF9 on different stages of skeletogenesis.
In addition, Fgf9−/− mice also show impaired osteogenesis, which may be secondary to the earlier defective chondrogenesis and vascularization,135 or FGF9 may directly regulate osteogenesis, as demonstrated by in vitro calvarial bone cell culture studies.138 Furthermore, the loss of Fgf9 results in a deficiency of osteoclasts in the perichondrium and primary spongiosa of developing bone.135 These findings suggest that FGF9 can positively regulate osteogenesis and osteoclastogenesis in endochondral ossification.
FGF9 is also expressed in the mesenchyme of suture in the early craniofacial development stages.115 By contrast to its promoting effects on osteogenesis in endochondral ossification, targeted overexpression of FGF9 in cranial mesenchymal cells leads to a switch from intramembranous to endochondral ossification in mouse parietal bones, indicating that FGF9 may regulate bone development by affecting the direction of mesenchyme differentiation.139
Recently, missense mutations in FGF9 have been identified to result in elbow-knee synostosis, premature fusion of cranial sutures in mice140 and multiple synostosis syndrome in humans.141 These data further suggest the important effect of FGF9 on bone development.
However, the different impacts of FGF9 on different stages of limb development and the direct effect of FGF9 on adult bone homeostasis are still unclear. Targeted deletion of Fgf9 in different stages and cells using Fgf9 CKO mice142 are necessary to answer these questions in the future.
FGF10
FGF10 is expressed in the lateral plate mesoderm and serves as a mesenchymally expressed limb bud initiator,44,143,144 and the expression persists in the mesenchyme under AER after initial limb bud formation. FGF10 acts epistatically at the upstream of FGF8.145 Positive feedback exists between FGF8 and FGF10, which is essential for limb development.44 To define the role of FGF10, Fgf10 knockout (Fgf10−/−) mouse strain was generated. These mice show complete absence of fore- and hindlimbs, and die after birth associated with complete absence of lungs.145,146 The limb bud formation in Fgf10−/− embryos is initiated but outgrowth of the limb buds is impaired, while the clavicle formation is normal.146 However, the impact of FGF10 on postnatal bone development and modeling remains unclear.
FGF18
FGF18 is expressed in osteogenic mesenchymal cells and differentiating osteoblasts of developing calvaria, in the perichondrium and joints, as well as growth plates of developing long bones.11,147,148
Fgf18 knockout (Fgf18−/−) mice die shortly after birth, and display expanded zones of proliferating and hypertrophic chondrocytes with increased chondrocyte proliferation and differentiation, similar to that observed in mice lacking Fgfr3.147,148 Bone cultures of fetal mouse tibias treated with FGF18 show decreased bone length and hypertrophic differentiation of chondrocytes.87,149 These studies demonstrate the inhibitory effect of FGF18 in chondrogenesis. In contrast to the negative role of FGF18 in chondrogenesis found in Fgf18−/− mice or FGF18-treated cultured bone, the proliferation and differentiation of primary chondrocytes and prechondrocytic ATDC5 cells are stimulated by FGF18 treatment in vitro.150 FGF18 also enhances BMP function and stimulate chondrogenesis in earlier stages of cartilage formation by suppressing noggin expression.151 These seemingly contradictory data suggest that the in vivo role of FGF18 in chondrogenesis need to be further studied. In addition, FGF18 regulates bone development by inducing skeletal vascularization and subsequent recruitment and formation of osteoclasts in developing long bone.152
Fgf18−/− mice also show delayed suture closure with decreased proliferation of calvarial osteogenic mesenchymal cells and delayed osteogenic differentiation. The calvarial bone mineralization in Fgf18−/− mice is also decreased.148,152 The delayed osteogenic differentiation is also observed in the developing long bones of Fgf18−/− mice.152 In vitro studies show that FGF18 treatment results in enhanced proliferation of MC3T3-E1 cells and perichondrial cells in cultured metatarsals,150 supporting the promoting effect of FGF18 on osteogenesis. These data indicate that FGF18 may be an important modulator for both endochondral and intramembranous bone formation in adult mice.
Although FGF18 is a key regulator for chondrogenesis, osteogenesis and vascularization of early skeleton development, the mechanism and the direct effect of FGF18 on the three critical stages in skeleton developmental or bone homeostasis at adult period need to be further studied.
FGF21
FGF21 is a member of the FGF19/21/23 subfamily that functions as an endocrine hormone.153,154 FGF21 is a powerful regulator of glucose and lipid metabolism.155–158 Recently, FGF21 has also been found to participate in bone homeostasis. The overexpression of Fgf21 in liver driven by Apoe promoter in transgenic mice show decreased bone mass, impaired bone formation and increased osteoclast function, which is consistent with the phenotypes of mice with pharmacological FGF21 treatment. In contrast, Fgf21−/− mice have increased bone mass with improved osteogenesis and decreased osteoclast function. The possible mechanism is that FGF21 stimulates adipogenesis from bone marrow mesenchymal stem cells by potentiating the activity of peroxisome proliferator-activated receptor γ, but inhibits osteoblastogenesis.159 These results indicate that FGF21 is a negative regulator of bone turnover and a key integrator of bone and energy metabolism, and underscores the importance of the whole body energy metabolism in bone physiology.159
Furthermore, FGF21 is expressed in the growth plate,160,161 and is associated with reduced skeletal growth and growth hormone (GH) insensitivity caused by undernutrition. After food restriction, FGF21 expression is increased in the tibial growth plates of mice. Fgf21−/− mice exhibit greater body and tibia growth than their wild-type controls after food restriction because of reduced GH binding and GH receptor expression in the liver and in the growth plates of wild-type mice, but not in that of Fgf21−/− mice.161 FGF21 also has direct effect on chondrocytes. Higher concentrations of FGF21 inhibit chondrocyte proliferation and differentiation by reducing GH binding in cultured chondrocytes.160 FGFR1 may participate as receptors of FGF21 in the regulation of chondrocytes by FGF21.160,162
Owen et al.163 found that physiological levels of FGF21 regulate the HPA axis and glucocorticoid levels, as well as the kisspeptin pathway in female fertility, which may also have effect on bone homeostasis.
FGF23
FGF23 is an approximately 32-kDa protein with an N-terminal FGF homology domain and a novel 72-amino-acid C-terminus, which permits interaction with FGF receptor-α–Klotho coreceptor complexes in cell membranes of target tissues.31,164 FGF23 is mainly secreted by osteoblasts and osteocytes,165–167 and as a hormone to regulate systemic phosphate homeostasis and vitamin D metabolism.
FGF23 downregulates serum phosphate. Mutations in an RXXR site in FGF23 prevents its cleavage resulting in autosomal-dominant hypophosphatemic rickets (ADHR), characterized by low serum phosphorus concentrations, rickets, osteomalacia, lower extremity deformities, short stature, bone pain and dental abscesses.168–172 The overproduction of FGF23 by tumors173 and osteogenic cells in fibrous dysplastic lesions174 may be responsible for the hypophosphatemia in tumor-induced osteomalacia and fibrous dysplasia, respectively. In addition to its role in hypophosphatemic diseases, FGF23 is involved in hyperphosphatemic diseases. Hyperphosphatemic familial tumoral calcinosis is a relatively rare genetic disease characterized by enhanced renal tubular phosphate reabsorption and elevated serum phosphorus, as well as paraarticular calcific tumors.175 Multiple mutations in FGF23 gene that lead to decreased FGF23 activity have been identified in patients with hyperphosphatemic familial tumoral calcinosis.176–178 These human studies help to define the critical role of FGF23 in regulating phosphate metabolism.
The transgenic mice, ubiquitously expressing human FGF23, reproduce the common clinical features of hypophosphatemia, including decreased serum phosphorus concentration, increased renal phosphate wasting, inappropriately low serum 1,25-dihydroxyvitamin D [1,25(OH)2D] level, and rachitic bone.179 Overexpression of human FGF23 in osteoblastic lineage or FGF23R176Q (a mutant form that fails to be degraded by furin proteases) in liver results in phenotypic changes similar to those of patients with ADHR or transgenic mice expressing FGF23 ubiquitously.180,181 Serum phosphate level is regulated by renal NaPi-2a in the brush border membrane of proximal tubules.182 The renal phosphate wasting in the transgenic mice is accompanied by the reduced expression of NaPi-2a.179 The reduction of serum 1,25(OH)2D levels may result from a significant decrease in renal mRNA level for 25-hydroxyvitamin D-1a-hydroxylase (1a-OHase) and a simultaneous elevation of 24-hydroxylase mRNA, induced by increased serum level of FGF23 (Figure 3).183
Figure 3.
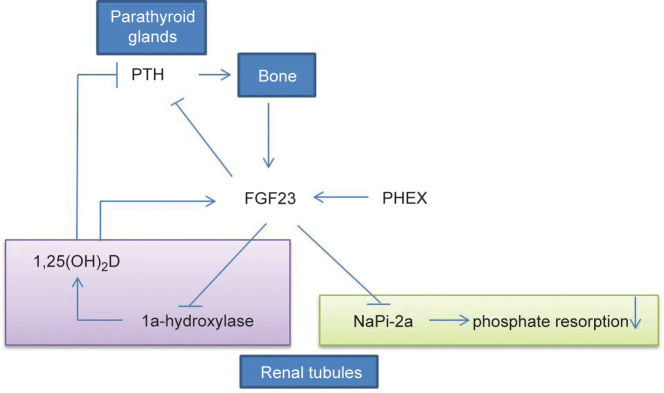
FGF23 regulates systemic phosphate homeostasis and vitamin D metabolism. FGF23 can reduce expression of NaPi-2a in kidney tubules and lead to renal phosphate wasting. FGF23 downregulates activity of 25-hydroxyvitamin D 1a-hydroxylase in kidney tubules and reduces 1,25(OH)2D level. Furthermore, FGF23 also have relationship with PTH and PHEX.
Consistently, Fgf23 knockout (Fgf23−/−) mice have opposite features including significantly increased serum levels of phosphate, calcium and 1,25(OH)2D because of the upregulated renal phosphate reabsorption and enhanced expression of renal 1a-OHase, respectively.184 The Fgf23−/− mice also exhibit premature aging-like phenotypes including reduced lifespan, infertility, osteoporosis and renal dysfunction.184 The elimination or reduction of vitamin D activity from Fgf23−/− mice can rescue the premature aging-like features and ectopic calcifications. These in vivo experimental data strongly support the very essential roles of FGF23 in the regulation of phosphate homeostasis, vitamin D activity and in the pathogenesis of premature aging.185
Recent studies have indicated the regulation of iron on FGF23. Reduced serum iron concentrations are strongly correlated with increased serum FGF23 in ADHR patients,186 and C-terminal FGF23 is negatively correlated with ferritin.187 To investigate the effect of iron on the development of the ADHR phenotype, R176Q-Fgf23 knock-in mice mimicking human ADHR are generated and placed on control or low-iron diets.188,189 R176Q-Fgf23 knock-in mice on low-iron diet have elevated intact C-terminal Fgf23 with hypophosphatemic osteomalacia and low serum 1,25(OH)2D. Iron chelation in vitro results in a significantly increased Fgf23 mRNA level that depends on MAPK signaling.189 However, the mechanism for the regulation of FGF23 by iron is still unclear.
Increased FGF23 level is also found in patients with hypophosphatemic diseases including XLH and autosomal dominant hypophosphatemic rickets (ARHR). XLH is caused by inactivating mutations in phosphate regulating gene with homologies to endopeptidases on the X chromosome (PHEX).190,191 Mice with ablation of Phex gene (Hyp mice) have increased FGF23 expression and hypophosphatemia.192 Both the serum phosphate levels and skeletal changes in Hyp mice can be reversed by introducing Fgf23 null mutation into Hyp mice,166,193,194 indicating that enhanced FGF23 level is responsible for the hypophosphatemia in XLH patients and Hyp mice. The increased FGF23 level is due to the improved Fgf23 expression, but not decreased degradation.165,194,195 ARHR results from missense mutations in DMP-1. Dmp1 knockout mice exhibit hypophosphatemic rickets and osteomalacia similar to ARHR patients.196,197 Both Dmp1 null mice and patients with ARHR show elevated serum FGF23 levels. Considering the role of FGF23 in ADHR and other hypophosphatemic diseases, ARHR has been proposed to be associated with excessive actions of FGF23.
FGF23 also participates in some clinical pathological processes, in addition to its role in genetic diseases. In patients with chronic kidney disease (CKD), FGF23 level is elevated due to increased serum calcium and phosphate concentrations and PTH,31,198 and is associated with increased FGF23 transcription in bone.199 Some researchers proposed that FGF23 might be an early biomarker for earlier interventions in CKD.200 However, the reason for the high serum levels of FGF23 in CKD is unclear. Furthermore, elevated level of FGF23 in CKD patients have been linked to greater risks of left ventricular hypertrophy (LVH).201,202 Using animal models, Faul et al.203 found that increased level of FGF23 in mice resulted in pathological hypertrophy of cardiomyocytes and LVH. To avoid redundancy and give full attention of the exciting results from FGF23 studies, we encourage you to read the recently published review by Quarles and Bhattacharyya.31,204
Other FGFs
In addition to the FGFs mentioned above, the roles of majority of these 22 FGFs are not defined in skeleton development and homeostasis. Researchers have generated knockout or CKO mouse models of these FGFs (Table 2). Fgf3 knockout mice show a short, dorsally curled tail, caudal vertebrae and smaller body.205 Some mouse models show normal skeleton phenotypes, such as mice lacking FGF1, FGF5, FGF6, FGF7, FGF17 or FGF22.206–212 The skeleton phenotypes of knockout mice lacking FGF11–FGF16, or FGF20, are still not analyzed.213–219 The effect of these FGFs on bone development or homeostasis need be further studied.
Table 2. Mouse models with genetically modified FGF/FGFR signaling in skeletal development and homeostasis.
| Gene | Model | Exon | Cre line (tissue) | Survival | Phenotype | Reference | Related human skeleton disease |
|---|---|---|---|---|---|---|---|
| FGFR1 |
KO |
Exon 4 |
Germline |
Die at E7.5–9.5 |
Severe growth retardation, defect of mesodermal differentiation |
[17] |
NA |
| KO |
Exons 8–14 |
Germline |
Die at E7.5–9.5 |
Early growth defects, aberrant mesodermal patterning |
[18] |
NA |
|
| FGFR1-deficient ES chimeras |
Exon 4; Exons 8–14 |
Germline |
Die during gastrulation |
Defective cell migration through primitive streak, malformation of chimeric limb buds |
[21,228] |
NA |
|
| KO |
Exon 3 (α-isoforms) |
Germline |
Die at E9.5–12.5 |
Distal truncation of limb bud, lethal at E9.5–12.5 due to posterior embryonic defects |
[20] |
NA |
|
| KO |
Exon 8 (IIIb) |
Germline |
Viable |
No obvious phenotype |
[19] |
NA |
|
| KO |
Exon 9 (IIIc) |
Germline |
Lethal |
Gastrulation defects |
[19] |
NA |
|
| KO |
Exons 8–17 (Frs2/3-binding site) |
Germline |
Die during late embryogenesis |
Defects in neural tube closure and in the development of the tail bud and pharyngeal arches |
[25] |
NA |
|
| CKO |
Exons 8–14 |
T (brachyury)-cre (all LMB cells) |
Die at birth |
Later reduction of limb skeleton, misshapen forelimb/hindlimb bud, missing digits |
[10,229] |
NA |
|
|
Shh-cre (posterior LBM cells) |
Normal limb bud size, missing digit 3 |
[10,229] |
NA |
||||
|
Ap2-Cre (progress zone of the mouse limb at E10.5) |
Abnormal development of the anterior digits |
[24] |
NA |
||||
|
Hoxb6-Cre (lateral plate mesoderm of E8.5) |
Severe abnormalities in autopod formation in hindlimbs |
[24] |
NA |
||||
| CKO |
Exons 8–15 |
Col2a1-cre (osteo-chondrocyte lineage |
Viable |
Increased bone mass, delayed osteoblast differentiation, increased proliferation of osteochondro-progenitor cells, increased height of the hypertrophic chondrocyte zone at E16.5 |
[15,230] |
NA |
|
|
Col1-cre (differentiated osteoblasts) |
Viable |
Increased bone mass, accelerated osteoblast differentiation and mineralization, impaired osteoclast activity |
[15,230] |
NA |
|||
|
LysM-cre |
Viable |
Increased bone mass, impaired osteoclast formation and activity |
[29] |
NA |
|||
|
OC-cre |
Viable |
Increased bone mass |
Su et al. unpublished data |
NA |
|||
| CKO |
Exon 4 |
Meox2-Cre (various) |
Die at E11.5 |
Developmental delay, mesodermal migration and patterning defects, craniorachischisis and posterior truncations |
[25] |
NA |
|
| DN |
Transgene (Tyrp1-FGFR1*IIIc) |
Retinal pigment epithelium |
|
No skeleton phenotype was reported |
[231] |
NA |
|
| GOF (KI) |
Exon 7 (P250R) |
Germline |
Viable |
PS including decreased body size, premature suture closure, increased bone formation at suture |
[28] |
PS (P250R) |
|
| OE |
Transgene (BAC-FGFR1P252R) |
Various |
Viable |
Premature suture closure |
[232] |
PS (P250R) |
|
| FGFR2 |
KO |
Exons 10,11 and part of exon 12 (transmembrane domain and part of its kinase I domain) |
Germline |
Die at E4.5–5.5 |
The growth of the inner cell mass stopped, no visceral endoderm formed, trophoblast defects |
[43] |
NA |
| KO |
Exons 7–9 (Entire Ig III) |
Germline |
Die at E10–11 |
Failure of limb bud initiation and placenta formation |
[44,45] |
NA |
|
| KO-LacZ |
Exon 8 (IIIb) |
Germline |
Die at birth |
Impaired limb outgrowth, severe dysgenesis of multiple organs |
[233] |
NA |
|
| KO |
Exon 9 (IIIc) (A translational stop codon inserted into exon 9) |
Germline |
Viable |
Delayed ossification in the sphenoid region of the skull base, dwarfism in the long bones and axial skeleton |
[54] |
NA |
|
| CKO |
Exon 8 (IIIb) |
CMV-Cre (germline) |
Die at birth |
Defects of limb outgrowth and branching morphogenesis |
[234] |
NA |
|
| CKO |
Exon 9 (IIIc) (Resulting in a GOF mutation associated with exon switching within the Fgfr2 gene) |
ZP3-Cre (germline) |
Die within 9 days |
Coronal synostosis, ocular proptosis, precocious sternal fusion, and abnormalities in secondary branching in several organs |
[235] |
CS/PS |
|
| CKO |
Exons 8–10 |
Dermo1-Cre (mesenchymal condensations) |
Viable |
Skeletal dwarfism and decreased bone density, impaired proliferation of osteoprogenitors and function of mature osteoblasts |
[36] |
NA |
|
| KD (RNAi) |
Transgene (U6-ploxPneo-Fgfr2) |
EIIa-Cre (germline) |
Lethal |
Displayed limb defects |
[57] |
NA |
|
| GOF (KI) |
Exon 7 (S250W) |
Germline |
Viable |
Several features similar to AS including smaller body size, brachycephaly, and midface hypoplasia |
[51] |
AS |
|
| GOF (KI) |
Exon 7 (S252W) |
Germline |
Neonatal lethality |
Smaller size, midline sutural defect and craniosynostoses, increased cartilage in the basicranium, nasal turbinates and long bone |
[48] |
AS(S252W) |
|
| GOF (KI) |
Exon 7 (P253R) |
Germline |
Viable |
Smaller body size, brachycephalyand syndactyly, premature of cranial sutures |
[39] |
AS (P253R) |
|
| GOF (KI) |
Exon 7 (P253R) |
Germline |
Die at P1–3w |
Smaller body size, brachycephaly and syndactyly, premature of cranial sutures |
[49] |
AS (P253R) |
|
| GOF (ENU-induced) |
Exon 7 (W290R) |
Germline |
Neonatal lethality |
Features resembling those found in patients with CS |
[236] |
CS |
|
| GOF (KI) |
Exon 9 (IIIc) ( C342Y ) |
Germline |
Viable |
Shortened face, protruding eyes, premature fusion of cranial sutures, and enhanced Spp1 expression in the calvaria, just like human Crouzon syndrome/Pfeiffer syndrome |
[55] |
CS/PS (C342Y) |
|
| GOF (KI) |
Exon 9 (IIIc) and Exon 10 (transmembrane domain) (C342Y; L424A; R424A, CLR) |
Germline |
Viable |
Normal skull development |
[53] |
NA |
|
| GOF (KI) |
Exon 10 (transmembrane domain) (Y394C) |
Germline |
Postnatal lethality |
Epidermal hyperplasia and premature closure of cranial sutures (craniosynostosis) due to abnormal cell proliferation and differentiation |
[52] |
BSS |
|
| FGFR3 |
KO |
Exon 5 |
Germline |
Viable |
Bone overgrowth, decreased bone mass |
[79] |
CATSHL syndrome |
| KO |
From Ig-like domain II to the transmembrane domain |
Germline |
Viable |
Bone overgrowth, defective bone mineralization and osteopenia, early arthritis, deafness |
[59,85,237] |
CATSHL syndrome |
|
| KO (a stop codon inserted) |
Exon 8 (IIIb) |
Germline |
Viable |
No obvious phenotype |
[238] |
NA |
|
| KO (a stop codon inserted) |
Exon 9 (IIIc) |
Germline |
Viable |
Skeletal overgrowth, decreased bone mineral density |
[238] |
NA |
|
| CKO |
Exons 9–10 |
EIIa-Cre |
Viable |
Increased length of long bone and decreased bone mineral density |
[239] |
NA |
|
| GOF (KI) |
Exon 7 (P244R) |
Germline |
Viable |
Abnormal craniofacial morphology |
[240] |
MS (P250R) |
|
| GOF ( KI) |
Exon 9 (Y367C) |
Germline |
Viable (die at 6–8 weeks after birth) |
Skeletal dysplasia more severe than ACH |
[241] |
TD I (Y373C) |
|
| GOF ( KI) |
Exon 10 (S365C) |
Germline |
Viable |
Skeletal dysplasia more severe than ACH |
[72] |
TD I |
|
| GOF ( KI) |
Exon 10 (G369C) |
Germline |
Viable |
Macrocephaly and shortened limbs due to retarded endochondral bone growth and premature closure of cranial base synchondroses |
[70] |
ACH (G375R) |
|
| GOF ( KI) |
Exon 10 (G374R) |
Germline |
Viable |
Small size, short tail, macrocephaly and dome-shaped heads, the narrower epiphyseal growth plates and decreased hypertrophic chondrocyte zone |
[67,68] |
ACH (G380R) |
|
| GOF ( KI) |
K644E cDNA knock-in |
Germline |
Viable |
Retardation of bone growth, macrocephaly and shortening of the long bones resembling ACH patients |
[73] |
ACH |
|
| GOF ( KI) |
Exon 15 (K644E) |
Germline |
Neonatal lethality |
Die within few hours after birth, skeletal dysplasia more severe than ACH |
[71] |
TD II (K650E) |
|
| GOF ( KI) |
Exon15 (K644M) |
Germline |
Viable |
Acanthosis nigricans and anomalies in central nervous system in addition to severe skeletal dysplasia |
[242] |
SADDAN |
|
| OE |
Transgene (Col2- G374R) |
Chondrocyte |
Viable |
Mice are dwarfed, with axial, appendicular and craniofacial, skeletal hypoplasia |
[80] |
ACH |
|
| OE |
Transgene (FGFR3- hG380R) |
Germline |
Viable |
Disproportionate dwarfism similar to those of human achondroplasia |
[243] |
ACH |
|
| FGFR4 |
KO |
Exon 6 (Ig II) |
Germline |
Viable |
Morphologically normal, no obvious defects in skeleton |
[93] |
NA |
| KI |
Exon 8 (G385R) |
Germline |
Viable |
Skeleton phenotype not reported |
[244] |
NA |
|
| FGFR3/FGFR4 |
Double KO |
|
Germline |
Viable |
Neonatal growth retardation, lung abnormalities |
[93] |
NA |
| FGF1 |
KO |
Exon1 |
Germline |
Viable |
No obvious phenotype |
[206] |
NA |
| FGF2 |
KO |
Exon1 (all three isoforms) |
Germline |
Viable |
Impaired cerebral cortex development, blood pressure regulation |
[245] |
NA |
| KO |
Exon1 (all three isoforms) |
Germline |
Viable |
Decreased bone mass. decreased vascular smooth muscle contractility, low blood pressure and thrombocytosis |
[95,100] |
NA |
|
| KO |
Exon1 (All three isoforms) |
Germline |
Viable |
Delayed wound healing and neuronal defects and impaired development of the cerebral cortex |
[99] |
NA |
|
| KO |
Exon 1 (CTGCAG replacing the wild-type CCATGC) (Lmw) |
Germline |
Viable |
Decreased bone mineral content, bone, BMD and impaired mineralization of BMSCs |
[108,246] |
NA |
|
| KO |
Exon 1 (the 14-bp oligo was designed to introduce stop codons in all three reading frames) (hmw) |
Germline |
Viable |
Skeleton phenotype was not reported |
[247] |
NA |
|
| Heterozygous (Fgf2+/−) |
Exon1 |
Germline |
Viable |
Decreased bone mass and bone formation |
[104] |
NA |
|
| OE |
Transgene (PGK-hFGF2) |
Various |
Viable |
Dwarf mouse with premature closure of the growth plate and shortening of bone length, defective bone mineralization and osteopenia |
[97,102] |
NA |
|
| OE |
Transgene (3.6 kb) Col1a-18-kDa FGF2-IRES-GFPsaph) |
Immature and mature osteoblast lineage |
Viable |
Increased BMD, bone volume, trabecular thickness, and cortical bone thickness |
[108] |
NA |
|
| OE |
Transgene (3.6 kb) Col1a-HMW FGF2-IRES-GFPsaph |
Immature and mature osteoblast lineage |
Viable |
Dwarfism, decreased BMD, osteomalacia, increased FGF23 level and hypophosphatemia |
[109,110] |
similar to XLH |
|
| FGF3 |
KO |
Exon1b (leaky expression of the mutant Fgf3) |
Germline |
Die in the early postnatal period |
A short, dorsally curled tai and caudal vertebrae, smaller body, Inner ear defects |
[205] |
NA |
| CKO |
Exons1b-3 |
EIIa-Cre (germline) |
Viable |
Shortened, thickened and curved tail, normal inner ears |
[248,249] |
NA |
|
| CKO |
Exon 2 |
CMV-cre (germline) |
Viable |
Short, curly tails, abnormal otic morphologies |
[250] |
NA |
|
| CKO |
Exon 2 |
|
|
Not used for skeleton research |
[251] |
NA |
|
| FGF3/4 |
OE |
Upregulation of FGF3/4 caused by retroviral insertion |
Cranial sutures |
Viable |
Facial shortening with increased interorbital distance and precocious closure of several cranial sutures (craniosynostosis) |
[252] |
Craniosynostosis |
| FGF4 |
KO |
Exon 1 |
Disrupted prior to limb bud initiation |
Died at E5.0 |
Severely impaired proliferation of the inner cell mass |
[113] |
NA |
| CKO-AP |
Exons 1–3 |
Rarb/Cre (developing forelimb region) |
Viable |
Normal forelimbs and hindlimbs |
[111,126] |
NA |
|
| CKO |
Exons 2–3 |
MSX2-cre |
Viable |
Normal forelimbs and hindlimbs |
[112] |
NA |
|
| FGF5 |
KO |
Exon 1 |
Germline |
|
Abnormally long hair, impaired skeletal muscle |
[207] |
NA |
| FGF6 |
KO-Lac Z |
Exon 1 |
Germline |
Viable |
No abnormal phenotype of skeleton detected |
[208] |
NA |
| FGF7 |
KO |
Exon 1 |
Germline |
Viable |
No abnormal phenotype of skeleton detected |
[209] |
NA |
| FGF8 |
KO |
Exons 2−3+neo |
Germline |
Lethal |
Early embryonic lethality before limb development |
[123,124] |
NA |
| CKO |
Exons 2–3 |
β-actin-cre (early embryo) |
Lethal |
Early embryonic lethality |
[123,124] |
NA |
|
|
MSX2-cre (functions initiated after FGF8 expression in forelimb, but before FGF8 expression in hindlimb) |
Not mentioned |
Substantial reduction in limb-bud size, and hypoplasia or aplasia of specific skeletal elements |
[112] |
NA |
|||
| CKO-AP |
Exon 5 |
Lefty2-Cre (mesoderm) |
Not mentioned |
Limb bud development proceeded normally |
[125,126] |
NA |
|
|
Rarb-Cre (developing forelimb region) |
Not mentioned |
Severe forelimb deformity including absence of radius and first digit |
[125,126] |
NA |
|||
|
AP2-Cre (limb bud ectoderm of E9.5 embryos) |
Not mentioned |
Absence of both forelimbs and hindlimbs |
[126] |
NA |
|||
| FGF9 |
KO |
Exon 1 |
Germline |
Die at birth |
Lung hypoplasia, male-to-female sex reversal, inner ear morphogenesis defect, slightly smaller body, short proximal skeletal |
[135–137,253] |
NA |
| Heterozygous (Fgf9+/−) |
Exon 1 |
Germline |
Viable |
Reduced bone regeneration, impaired neovascularization and decreased cell proliferation |
[254] |
NA |
|
| CKO |
Exon 1 |
Nestin Cre (germline) |
Die at birth |
Lung hypoplasia, skeleton phenotype not mentioned |
[142] |
NA |
|
| OE |
Transgene (αA-crystallin-FGF9) |
Cranial mesenchymal cells |
Viable |
Parietal bones show a switch from intramembranous to endochondral ossification |
[139] |
NA |
|
| OE |
Transgene (Col2a1-FGF9) |
Chondrocyte |
Viable |
Short limb, vertebral defect, reduced proliferation and terminal differentiation of chondrocytes |
[133] |
NA |
|
| GOF (Spontaneous mutation) |
N143T |
Germline |
Lethal |
EKS with radiohumeral and tibiofemoral synostosis, craniosynostosis; lung hypoplasia |
[140,255] |
EKS (158) |
|
| LOF (ENU screen) |
Y162C |
Germline |
Viable |
Normal skeleton phenotype, no male-to-female sexual reversal, decreased vision and retarded lens growth |
[256] |
NA |
|
| FGF10 |
KO |
Exon 1 |
Germline |
Perinatal lethality |
Complete absence of both fore- and hindlimbs, pulmonary branching morphogenesis was completely disrupted |
[145] |
NA |
| KO |
Exon encoding the ATG translational start site |
Germline |
Die after birth |
Complete truncation of the fore- and hindlimbs, normal clavicles, lung defect |
[146] |
NA |
|
| CKO |
Exon 2 |
|
|
Skeleton phenotype was not analyzed |
[251] |
NA |
|
| FGF11 |
KO |
Insertion of Velocigene cassette ZEN-Ub1 |
Germline |
Viable |
Skeleton phenotype was not analyzed |
[219] |
NA |
| FGF12 |
KO |
Exon 2 |
Germline |
Viable |
Skeleton phenotype was not analyzed |
[213] |
NA |
| FGF13 |
CKO |
Exons 2–3 |
|
Viable |
Skeleton phenotype was not analyzed |
[214] |
NA |
| FGF14 |
KO-LacZ |
Exon 2 |
Germline |
viable |
Skeleton phenotype was not analyzed, developed ataxia and a paroxysmal hyperkinetic movement disorder; reduced responses to dopamine agonists |
[215] |
NA |
| FGF15 |
KO |
Exon 3 |
Germline |
Die at E13.5– P21 |
Skeleton phenotype was not analyzed, enhanced bile acid synthesis and contracted gallbladder |
[216] |
NA |
| FGF16 |
KO-LacZ |
Exons 2–3 |
Germline |
Viable |
No bone phenotype was analyzed, decreased proliferation of embryonic cardiomyocytes |
[217] |
NA |
| FGF17 |
KO |
Exons 1a–1b |
Germline |
Viable |
Normal skeletal patterns, abnormal cerebellar development and social behaviors |
[210,211] |
NA |
| FGF18 |
KO |
Exon 3 |
Germline |
Die just before or at birth |
Impaired ossification and increased chondrocyte proliferation, decreased alveolar spaces in the lung |
[148,257] |
NA |
| KO-LacZ |
Exon 1 |
Germline |
Die after birth |
Impaired ossification and increased chondrocyte proliferation, respiratory failure |
[147,152] |
NA |
|
| Heterozygous |
Exon 3 |
Germline |
Viable |
Reduced bone regeneration |
[258] |
NA |
|
| CKO |
Exon1 |
Germline |
|
Even not be used in bone development |
[259] |
NA |
|
| FGF20 |
KO-LacZ |
Exon1 |
Germline |
Viable |
No bone phenotype was analyzed, deafness |
[218] |
NA |
| FGF21 |
KO |
Exons 2, part of exon1 and exon3 |
Germline |
Viable |
Greater body and tibia growth after food restriction |
[157,161] |
NA |
| KO (LacZ) |
Exons1–3 |
Germline |
Viable |
Skeleton phenotype was not reported |
[156] |
NA |
|
| CKO |
Exons1–3 |
Meox-cre (Germline) |
Viable |
Increased bone mass, metabolic defects including decreased circulating glucose level and oxygen consumption |
[155,159] |
NA |
|
| OE |
Transgene (Apoe-FGF21) |
Liver |
Viable |
Decreased bone mass, increased osteoblast and bone resorption |
[158,159] |
NA |
|
| OE |
Transgene (Apoe-hFGF21) |
Liver |
Viable |
Skeleton phenotype was not reported |
[260] |
NA |
|
| FGF22 |
KO |
Exon1 and part of Exon2 |
Germline |
Viable |
Normal skeletal patterns; decreased susceptibility to pharmacologically induced seizures |
[212] |
NA |
| KO |
Exons1–3 |
Germline |
Viable |
Normal skeletal patterns; decreased incidence of tumors by chemical induction |
[261] |
NA |
|
| FGF23 | KO |
Exon 1 |
Germline |
Viable |
Increased serum levels of phosphate, calcium and 1,25(OH)2D, severe growth retardation with abnormal bone phenotype |
[184] |
NA |
| KO-Lacz |
Exons 1–3 |
Germline |
Viable |
Hyperphosphatemia and impaired skeletogenesis |
[193] |
NA |
|
| KO-eGFP |
Exon 1 |
Germline |
Viable |
Hyperphosphatemia and impaired skeletogenesis |
[166] |
NA |
|
| OE |
Transgene (CAG-hFGF23) |
Various |
Viable |
Hypophosphatemia, low serum 1,25(OH)2D level, and rachitic bone,growth retardation |
[179] |
ADHR |
|
| OE |
Transgene (Col1a-hFGF23) |
Osteoblastic lineage |
Viable |
Smaller body, decreased serum phosphate concentrations, low serum 1,25(OH)2D level |
[180] |
ADHR |
|
| OE |
Transgene (Apoe3-hFGF23*R176Q) |
Liver |
Viable |
Hypophosphatemia, low serum 1,25(OH)2D level and rachitic bone |
[181] |
ADHR |
|
| GOF (KI) | Knock in (R176Q-hFGF23) | Germline | Viable | Increased serum level of FGF23, hypophosphatemi and low serum 1,25(OH)2 vitamin D after receiving low-iron diets | [188,189] | ADHR |
Abbreviations: ACH, achondroplasia; ADHR, autosomal dominant hypophosphatemic rickets; AS, Apert syndrome; BSS, Beare–Stevenson cutis gyrata syndrome; CATSHL, camptodactyly, tall stature and hearing loss; CKO, conditional knockout; CS, Crouzon syndrome; EKS, elbow knee synostosis; GOF, gain of function; KD, knockdown; KI, knock-in; KO, knockout; LMB, limb bud mesenchyme; LOF, loss of function; MS, Muenke syndrome; NA, not applicable; OE, overexpression; PS, Pfeiffer syndrome; SADDAN, severe achondroplasia with developmental delay and acanthosis nigricans; TD, thanatophoric dysplasia; XLH, X-linked hypophosphatemia.
Conclusions
Studies in human patients and mouse models with FGFs/FGFRs mutations have shown important roles of FGF signaling in skeletal development, genetic skeletal diseases and bone homeostasis. So, FGF/FGFR signalings will be attractive targets for treating bone related diseases. FGF/FGFR signals control the balance among skeletal cell growth, differentiation and apoptosis during development and adult homeostasis, as well as regulate systemic phosphate homeostasis. However, many unresolved issues still need to be explored.
Many studies have investigated the role of FGFRs in endochondral and intramembranous bone formation during development, but the effects of FGFRs on osteoclasts, especially on osteocytes, have not be clarified. Osteocytes are the most abundant and longest-living cells in the adult skeleton and have essential roles in bone homeostasis.220,221 Thus, uncovering the impact of FGFRs on osteocytes using osteocyte-specific Cre mice is critical.
Compensation, or crosstalk, may occur between different FGFRs during skeleton development. For example, conditional knock out of Fgfr1 in mature osteoblasts leads to increased FGFR3 expression,15 whereas both cultured bone marrow stromal cells from Fgfr3 null mice, or mice carrying gain-of-function mutation in FGFR3, have increased expression of FGFR1.83,85 Crossing between mouse strains harboring various FGFRs mutations is extremely important to elucidate the interactions between different FGFRs.
So far, only part of the 22 known FGF ligands have been shown to be essential for skeletal development, such as FGF8, FGF9 and FGF10. However, the mechanisms remain unclear because most of the knockout mice die before or after birth. Conditional deletion of these FGFs using bone cell-specific Cre mice is necessary to study their roles during bone development. The function of other unexplored FGFs in skeletogenesis remains to be discovered. Furthermore, which FGFRs are the relatively specific receptors of these unexplored FGFs during bone development and metabolism are unknown. Crossing mouse strains harboring different FGFs mutations with FGFRs mutant mouse models is necessary to discover the interactions between FGFs and FGFRs in skeleton development and homeostasis.
Recently, studies have indicated that the bone is closely related with whole-organism physiology.222 For example, bone can regulate energy metabolism, male reproduction and hematopoiesis.223,224 Some hormone secreted from other organs or tissues also have effect on bone, such as Leptin secreted by adipocyte.222 In addition, systemic disease also influence skeleton such as CDK225 and inflammatory disease.226,227 The roles of FGF signaling in the effect of systemic diseases on bone or bone on whole-organism physiology remain unclear and need further exploration.
Acknowledgments
The work is supported by grants from the National Natural Science Foundation of China (81030036, 81270012, 81170809), the Special Funds for Major State Basic Research Program of China (973 Program) (2014CB942904) and the Committee of Science and Technology of Chongqing (CSTC 2011jjA1468), the foundation from national key laboratory (SKLZZ201017). In addition, we apologized to those whose work could not be cited due to space constraints.
The authors declare no conflict of interest.
References
- Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- Su N, Du X, Chen L. FGF signaling: its role in bone development and human skeleton diseases. Front Biosci. 2008;13:2842–2865. doi: 10.2741/2890. [DOI] [PubMed] [Google Scholar]
- Chen L, Deng CX. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 2005;10:1961–1976. doi: 10.2741/1671. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Williams LT. Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocrine Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi TP, Conlon RA, Rossant J. Expression of the fibroblast growth factor receptor FGFR-1/flg during gastrulation and segmentation in the mouse embryo. Dev Biol. 1992;152:75–88. doi: 10.1016/0012-1606(92)90157-c. [DOI] [PubMed] [Google Scholar]
- Peters KG, Werner S, Chen G, Williams LT. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development. 1992;114:233–243. doi: 10.1242/dev.114.1.233. [DOI] [PubMed] [Google Scholar]
- Verheyden JM, Lewandoski M, Deng C, Harfe BD, Sun X. Conditional inactivation of Fgfr1 in mouse defines its role in limb bud establishment, outgrowth and digit patterning. Development. 2005;132:4235–4245. doi: 10.1242/dev.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus JE, Hegde A, Andrade AC, Nilsson O, Baron J. Fibroblast growth factor expression in the postnatal growth plate. Bone. 2007;40:577–586. doi: 10.1016/j.bone.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Savage MP, Olwin BB, Fallon JF. Changes in the expression of fibroblast growth factor receptors mark distinct stages of chondrogenesis in vitro and during chick limb skeletal patterning. Dev Dyn. 1995;204:446–456. doi: 10.1002/aja.1002040410. [DOI] [PubMed] [Google Scholar]
- Iseki S, Wilkie AO, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development. 1999;126:5611–5620. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- Xiao L, Naganawa T, Obugunde E. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J Biol Chem. 2004;279:27743–27752. doi: 10.1074/jbc.M314323200. [DOI] [PubMed] [Google Scholar]
- Jacob AL, Smith C, Partanen J, Ornitz DM. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol. 2006;296:315–328. doi: 10.1016/j.ydbio.2006.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyono A, Avishai N, Ouyang Z, Landreth GE, Murakami S. FGF and ERK signaling coordinately regulate mineralization-related genes and play essential roles in osteocyte differentiation. J Bone Miner Metab. 2012;30:19–30. doi: 10.1007/s00774-011-0288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994;8:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 1998;12:2332–2344. doi: 10.1101/gad.12.15.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Li C, Takahashi K, Slavkin HC, Shum L, Deng CX. Murine fibroblast growth factor receptor 1alpha isoforms mediate node regression and are essential for posterior mesoderm development. Dev Biol. 1999;208:293–306. doi: 10.1006/dbio.1999.9227. [DOI] [PubMed] [Google Scholar]
- Deng C, Bedford M, Li C. Fibroblast growth factor receptor-1 (FGFR-1) is essential for normal neural tube and limb development. Dev Biol. 1997;185:42–54. doi: 10.1006/dbio.1997.8553. [DOI] [PubMed] [Google Scholar]
- Perantoni AO, Timofeeva O, Naillat F. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development. 2005;132:3859–3871. doi: 10.1242/dev.01945. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Li C, Xu X, Nelson DK, Williams T, Kuehn MR, Deng CX. FGFR1 function at the earliest stages of mouse limb development plays an indispensable role in subsequent autopod morphogenesis. Development. 2005;132:4755–4764. doi: 10.1242/dev.02065. [DOI] [PubMed] [Google Scholar]
- Hoch RV, Soriano P. Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development. 2006;133:663–673. doi: 10.1242/dev.02242. [DOI] [PubMed] [Google Scholar]
- White KE, Cabral JM, Davis SI. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76:361–367. doi: 10.1086/427956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioli T, Flanagan S, Kumar P. Clinical findings in a patient with FGFR1 P252R mutation and comparison with the literature. Am J Med Genet. 2000;93:22–28. doi: 10.1002/1096-8628(20000703)93:1<22::aid-ajmg5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Zhou YX, Xu X, Chen L, Li C, Brodie SG, Deng CX. A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet. 2000;9:2001–2008. doi: 10.1093/hmg/9.13.2001. [DOI] [PubMed] [Google Scholar]
- Lu X, Su N, Yang J. Fibroblast growth factor receptor 1 regulates the differentiation and activation of osteoclasts through Erk1/2 pathway. Biochem Biophys Res Commun. 2009;390:494–499. doi: 10.1016/j.bbrc.2009.09.123. [DOI] [PubMed] [Google Scholar]
- Yu X, White KE. Fibroblast growth factor 23 and its receptors. Ther Apher Dial. 2005;9:308–312. doi: 10.1111/j.1744-9987.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. 2012;8:276–286. doi: 10.1038/nrendo.2011.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohrle S, Bonny O, Beluch N. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res. 2011;26:2486–2497. doi: 10.1002/jbmr.478. [DOI] [PubMed] [Google Scholar]
- Peng H, Myers J, Fang X. Integrative nuclear FGFR1 signaling (INFS) pathway mediates activation of the tyrosine hydroxylase gene by angiotensin II, depolarization and protein kinase C. J Neurochem. 2002;81:506–524. doi: 10.1046/j.1471-4159.2002.00833.x. [DOI] [PubMed] [Google Scholar]
- Delezoide AL, Benoist-Lasselin C, Legeai-Mallet L. Spatio-temporal expression of FGFR 1, 2 and 3 genes during human embryo-fetal ossification. Mech Dev. 1998;77:19–30. doi: 10.1016/s0925-4773(98)00133-6. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Burakova T. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Rice DP, Rice R, Thesleff I. Fgfr mRNA isoforms in craniofacial bone development. Bone. 2003;33:14–27. doi: 10.1016/s8756-3282(03)00163-7. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Yin L, Du X, Li C. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42:631–643. doi: 10.1016/j.bone.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Britto JA, Chan JC, Evans RD, Hayward RD, Thorogood P, Jones BM. Fibroblast growth factor receptors are expressed in craniosynostotic sutures. Plast Reconstr Surg. 1998;101:540–543. [PubMed] [Google Scholar]
- Britto JA, Evans RD, Hayward RD, Jones BM. From genotype to phenotype: the differential expression of FGF, FGFR, and TGFbeta genes characterizes human cranioskeletal development and reflects clinical presentation in FGFR syndromes. Plast Reconstr Surg. 2001;108:2026–2039; discussion 2040–2026. doi: 10.1097/00006534-200112000-00030. [DOI] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci USA. 1998;95:5082–5087. doi: 10.1073/pnas.95.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Weinstein M, Li C. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125:753–765. doi: 10.1242/dev.125.4.753. [DOI] [PubMed] [Google Scholar]
- Li X, Chen Y, Scheele S. Fibroblast growth factor signaling and basement membrane assembly are connected during epithelial morphogenesis of the embryoid body. J Cell Biol. 2001;153:811–822. doi: 10.1083/jcb.153.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ, Coffin JD, Hurley MM. FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J Cell Biochem. 2005;96:888–896. doi: 10.1002/jcb.20582. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res. 2007;10:67–81. doi: 10.1111/j.1601-6343.2007.00389.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xiao R, Yang F. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development. 2005;132:3537–3548. doi: 10.1242/dev.01914. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sun M, Uhlhorn VL. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev Biol. 2010;10:22. doi: 10.1186/1471-213X-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiborg S, Aduss H, Cohen MM., Jr Cephalometric study of the Apert syndrome in adolescence and adulthood. J Craniofac Genet Dev Biol. 1999;19:1–11. [PubMed] [Google Scholar]
- Chen L, Li D, Li C, Engel A, Deng CX. A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003;33:169–178. doi: 10.1016/s8756-3282(03)00222-9. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhou X, Oberoi K. p38 Inhibition ameliorates skin and skull abnormalities in Fgfr2 Beare–Stevenson mice. J Clin Invest. 2012;122:2153–2164. doi: 10.1172/JCI62644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Ozcan F, Lew ED. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci USA. 2006;103:18603–18608. doi: 10.1073/pnas.0609157103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129:3783–3793. doi: 10.1242/dev.129.16.3783. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci USA. 2004;101:12555–12560. doi: 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK–ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39:1145–1150. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- Coumoul X, Shukla V, Li C, Wang RH, Deng CX. Conditional knockdown of Fgfr2 in mice using Cre-LoxP induced RNA interference. Nucleic Acids Res. 2005;33:e102. doi: 10.1093/nar/gni100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Hefferon TW, Ortiz de Luna RI. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368–373. [PMC free article] [PubMed] [Google Scholar]
- Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Rousseau F, el Ghouzzi V, Delezoide AL, Legeai-Mallet L, Le Merrer M, Munnich A, Bonaventure J. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1). Hum Mol Genet. 1996;5:509–512. doi: 10.1093/hmg/5.4.509. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Bamshad MJ, Przylepa KA. Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. Am J Med Genet. 1999;85:53–65. [PubMed] [Google Scholar]
- Tavormina PL, Bellus GA, Webster MK. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. Am J Hum Genet. 1999;64:722–731. doi: 10.1086/302275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Du XL, Li CL. Gly374Arg mutation in Fgfr3 causes achondroplasia in mice. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21:537–541. [PubMed] [Google Scholar]
- Wang Y, Spatz MK, Kannan K. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc Natl Acad Sci USA. 1999;96:4455–4460. doi: 10.1073/pnas.96.8.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naski MC, Colvin JS, Coffin JD, Ornitz DM. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–4988. doi: 10.1242/dev.125.24.4977. [DOI] [PubMed] [Google Scholar]
- Chen L, Adar R, Yang X. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517–1525. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata T, Chen L, Li C. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet. 2000;9:1603–1613. doi: 10.1093/hmg/9.11.1603. [DOI] [PubMed] [Google Scholar]
- Chen L, Li C, Qiao W, Xu X, Deng C. A Ser(365)-->Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet. 2001;10:457–465. doi: 10.1093/hmg/10.5.457. [DOI] [PubMed] [Google Scholar]
- Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35–44. doi: 10.1093/hmg/8.1.35. [DOI] [PubMed] [Google Scholar]
- Su WC, Kitagawa M, Xue N. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature. 1997;386:288–292. doi: 10.1038/386288a0. [DOI] [PubMed] [Google Scholar]
- Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D, Basilico C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1361–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–449. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol. 2003;161:1053–1066. doi: 10.1083/jcb.200302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Naski MC, Ornitz DM. FGF signaling in skeletal development. Front Biosci. 1998;3:781–794. doi: 10.2741/a321. [DOI] [PubMed] [Google Scholar]
- Koike M, Yamanaka Y, Inoue M, Tanaka H, Nishimura R, Seino Y. Insulin-like growth factor-1 rescues the mutated FGF receptor 3 (G380R) expressing ATDC5 cells from apoptosis through phosphatidylinositol 3-kinase and MAPK. J Bone Miner Res. 2003;18:2043–2051. doi: 10.1359/jbmr.2003.18.11.2043. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Wilcox WR, Chan YY. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet. 2009;18:227–240. doi: 10.1093/hmg/ddn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Sun Q, Li C. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Hum Mol Genet. 2010;19:1199–1210. doi: 10.1093/hmg/ddp590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ, Miraoui H, Severe N. FGF/FGFR signaling in bone formation: progress and perspectives. Growth Factors. 2012;30:117–123. doi: 10.3109/08977194.2012.656761. [DOI] [PubMed] [Google Scholar]
- Valverde-Franco G, Liu H, Davidson D. Defective bone mineralization and osteopenia in young adult FGFR3−/− mice. Hum Mol Genet. 2004;13:271–284. doi: 10.1093/hmg/ddh034. [DOI] [PubMed] [Google Scholar]
- Yasoda A, Komatsu Y, Chusho H. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10:80–86. doi: 10.1038/nm971. [DOI] [PubMed] [Google Scholar]
- Ozasa A, Komatsu Y, Yasoda A. Complementary antagonistic actions between C-type natriuretic peptide and the MAPK pathway through FGFR-3 in ATDC5 cells. Bone. 2005;36:1056–1064. doi: 10.1016/j.bone.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Yamagiwa H, Hayami T. Human PTH (1–34) induces longitudinal bone growth in rats. J Bone Miner Metab. 2002;20:83–90. doi: 10.1007/s007740200011. [DOI] [PubMed] [Google Scholar]
- Ueda K, Yamanaka Y, Harada D, Yamagami E, Tanaka H, Seino Y. PTH has the potential to rescue disturbed bone growth in achondroplasia. Bone. 2007;41:13–18. doi: 10.1016/j.bone.2007.02.028. [DOI] [PubMed] [Google Scholar]
- Xie Y, Su N, Jin M. Intermittent PTH (1–34) injection rescues the retarded skeletal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Hum Mol Genet. 2012;21:3941–3955. doi: 10.1093/hmg/dds181. [DOI] [PubMed] [Google Scholar]
- Jin M, Yu Y, Qi H. A novel FGFR3-binding peptide inhibits FGFR3 signaling and reverses the lethal phenotype of mice mimicking human thanatophoric dysplasia. Hum Mol Genet. 2012;21:5443–5455. doi: 10.1093/hmg/dds390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool S, Jackson R, Pincus P, Dickinson I, Nurcombe V. Fibroblast growth factor receptor 4 (FGFR4) expression in newborn murine calvaria and primary osteoblast cultures. Int J Dev Biol. 2002;46:519–523. [PubMed] [Google Scholar]
- Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- Fei Y, Hurley MM. Role of fibroblast growth factor 2 and Wnt signaling in anabolic effects of parathyroid hormone on bone formation. J Cell Physiol. 2012;227:3539–3545. doi: 10.1002/jcp.24075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero A, Okada Y, Tomita M. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093. doi: 10.1172/JCI8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JF, Lopez A, Ros MA, Savage MP, Olwin BB, Simandl BK. FGF-2: apical ectodermal ridge growth signal for chick limb development. Science. 1994;264:104–107. doi: 10.1126/science.7908145. [DOI] [PubMed] [Google Scholar]
- Coffin JD, Florkiewicz RZ, Neumann J. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol Biol Cell. 1995;6:1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni M, Raz R, Coffin JD, Levy D, Basilico C. STAT1 mediates the increased apoptosis and reduced chondrocyte proliferation in mice overexpressing FGF2. Development. 2001;128:2119–2129. doi: 10.1242/dev.128.11.2119. [DOI] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ. Fibroblast growth factor 2 control of vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Sobue T, Esliger A. Disruption of the Fgf2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone. 2010;47:360–370. doi: 10.1016/j.bone.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue T, Naganawa T, Xiao L. Over-expression of fibroblast growth factor-2 causes defective bone mineralization and osteopenia in transgenic mice. J Cell Biochem. 2005;95:83–94. doi: 10.1002/jcb.20389. [DOI] [PubMed] [Google Scholar]
- Sabbieti MG, Agas D, Xiao L. Endogenous FGF-2 is critically important in PTH anabolic effects on bone. J Cell Physiol. 2009;219:143–151. doi: 10.1002/jcp.21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa T, Xiao L, Abogunde E. In vivo and in vitro comparison of the effects of FGF-2 null and haplo-insufficiency on bone formation in mice. Biochem Biophys Res Commun. 2006;339:490–498. doi: 10.1016/j.bbrc.2005.10.215. [DOI] [PubMed] [Google Scholar]
- Okada Y, Montero A, Zhang X. Impaired osteoclast formation in bone marrow cultures of Fgf2 null mice in response to parathyroid hormone. J Biol Chem. 2003;278:21258–21266. doi: 10.1074/jbc.M302113200. [DOI] [PubMed] [Google Scholar]
- Fei Y, Xiao L, Hurley MM. The impaired bone anabolic effect of PTH in the absence of endogenous FGF2 is partially due to reduced ATF4 expression. Biochem Biophys Res Commun. 2011;412:160–164. doi: 10.1016/j.bbrc.2011.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbieti MG, Agas D, Marchetti L. Signaling pathways implicated in PGF2alpha effects on Fgf2+/+ and Fgf2−/− osteoblasts. J Cell Physiol. 2010;224:465–474. doi: 10.1002/jcp.22143. [DOI] [PubMed] [Google Scholar]
- Xiao L, Liu P, Li X. Exported 18-kDa isoform of fibroblast growth factor-2 is a critical determinant of bone mass in mice. J Biol Chem. 2009;284:3170–3182. doi: 10.1074/jbc.M804900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Naganawa T, Lorenzo J, Carpenter TO, Coffin JD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and KLOTHO. J Biol Chem. 2010;285:2834–2846. doi: 10.1074/jbc.M109.030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Esliger A, Hurley MM. Nuclear fibroblast growth factor 2 (FGF2) isoforms inhibit bone marrow stromal cell mineralization through FGF23/FGFR/MAPK in vitro. J Bone Miner Res. 2013;28:35–45. doi: 10.1002/jbmr.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Boulet AM, Capecchi MR. Normal limb development in conditional mutants of Fgf4. Development. 2000;127:989–996. doi: 10.1242/dev.127.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M, Sun X, Martin GR. Fgf8 signalling from the AER is essential for normal limb development. Nat Genet. 2000;26:460–463. doi: 10.1038/82609. [DOI] [PubMed] [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267:246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- Mathijssen IM, van Leeuwen H, Vermeij-Keers C, Vaandrager JM. FGF-4 or FGF-2 administration induces apoptosis, collagen type I expression, and mineralization in the developing coronal suture. J Craniofac Surg. 2001;12:399–400. doi: 10.1097/00001665-200107000-00019. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rice DP, Kettunen PJ, Thesleff I. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development. 1998;125:1241–1251. doi: 10.1242/dev.125.7.1241. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Kasugai S, Oida S, Iimura T, Ohya K, Ohyama T. Anabolic effect of aminoterminally truncated fibroblast growth factor 4 (FGF4) on bone. Bone. 1999;25:431–437. doi: 10.1016/s8756-3282(99)00193-3. [DOI] [PubMed] [Google Scholar]
- Choi SC, Kim SJ, Choi JH, Park CY, Shim WJ, Lim DS. Fibroblast growth factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K–Akt and ERK1/2 signaling pathways. Stem Cells Dev. 2008;17:725–736. doi: 10.1089/scd.2007.0230. [DOI] [PubMed] [Google Scholar]
- Farre J, Roura S, Prat-Vidal C. FGF-4 increases in vitro expansion rate of human adult bone marrow-derived mesenchymal stem cells. Growth Factors. 2007;25:71–76. doi: 10.1080/08977190701345200. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim JH, Bae SC, Choi JY, Ryoo HM. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J Biol Chem. 2003;278:319–326. doi: 10.1074/jbc.M203750200. [DOI] [PubMed] [Google Scholar]
- Heikinheimo M, Lawshe A, Shackleford GM, Wilson DB, MacArthur CA. Fgf-8 expression in the post-gastrulation mouse suggests roles in the development of the face, limbs and central nervous system. Mech Dev. 1994;48:129–138. doi: 10.1016/0925-4773(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Mahmood R, Bresnick J, Hornbruch A. A role for FGF-8 in the initiation and maintenance of vertebrate limb bud outgrowth. Curr Biol. 1995;5:797–806. doi: 10.1016/s0960-9822(95)00157-6. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Minowada G, MacArthur CA, Martin GR. Roles for FGF8 in the induction, initiation, and maintenance of chick limb development. Cell. 1996;84:127–136. doi: 10.1016/s0092-8674(00)80999-x. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Moon AM, Capecchi MR. Fgf8 is required for outgrowth and patterning of the limbs. Nat Genet. 2000;26:455–459. doi: 10.1038/82601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulet AM, Moon AM, Arenkiel BR, Capecchi MR. The roles of Fgf4 and Fgf8 in limb bud initiation and outgrowth. Dev Biol. 2004;273:361–372. doi: 10.1016/j.ydbio.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Xu J, Lawshe A, MacArthur CA, Ornitz DM. Genomic structure, mapping, activity and expression of fibroblast growth factor 17. Mech Dev. 1999;83:165–178. doi: 10.1016/s0925-4773(99)00034-9. [DOI] [PubMed] [Google Scholar]
- Valta MP, Hentunen T, Qu Q. Regulation of osteoblast differentiation: a novel function for fibroblast growth factor 8. Endocrinology. 2006;147:2171–2182. doi: 10.1210/en.2005-1502. [DOI] [PubMed] [Google Scholar]
- Omoteyama K, Takagi M. FGF8 regulates myogenesis and induces Runx2 expression and osteoblast differentiation in cultured cells. J Cell Biochem. 2009;106:546–552. doi: 10.1002/jcb.22012. [DOI] [PubMed] [Google Scholar]
- Lin JM, Callon KE, Lin JS. Actions of fibroblast growth factor-8 in bone cells in vitro. Am J Physiol Endocrinol Metab. 2009;297:E142–E150. doi: 10.1152/ajpendo.90743.2008. [DOI] [PubMed] [Google Scholar]
- Uchii M, Tamura T, Suda T, Kakuni M, Tanaka A, Miki I. Role of fibroblast growth factor 8 (FGF8) in animal models of osteoarthritis. Arthritis Res Ther. 2008;10:R90. doi: 10.1186/ar2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht D, Zimmerman N, Bedford M, Avivi A, Yayon A. Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors. 1995;12:223–233. doi: 10.3109/08977199509036882. [DOI] [PubMed] [Google Scholar]
- Garofalo S, Kliger-Spatz M, Cooke JL. Skeletal dysplasia and defective chondrocyte differentiation by targeted overexpression of fibroblast growth factor 9 in transgenic mice. J Bone Miner Res. 1999;14:1909–1915. doi: 10.1359/jbmr.1999.14.11.1909. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Feldman B, Nadeau JH, Goldfarb M, Ornitz DM. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev Dyn. 1999;216:72–88. doi: 10.1002/(SICI)1097-0177(199909)216:1<72::AID-DVDY9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Hung IH, Yu K, Lavine KJ, Ornitz DM. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol. 2007;307:300–313. doi: 10.1016/j.ydbio.2007.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128:2095–2106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 2001;104:875–889. doi: 10.1016/s0092-8674(01)00284-7. [DOI] [PubMed] [Google Scholar]
- Fakhry A, Ratisoontorn C, Vedhachalam C. Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone. 2005;36:254–266. doi: 10.1016/j.bone.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Govindarajan V, Overbeek PA. FGF9 can induce endochondral ossification in cranial mesenchyme. BMC Dev Biol. 2006;6:7. doi: 10.1186/1471-213X-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M, Murakami H, Okawa A. FGF9 monomer-dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet. 2009;41:289–298. doi: 10.1038/ng.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XL, Gu MM, Huang L. Multiple synostoses syndrome is due to a missense mutation in exon 2 of FGF9 gene. Am J Hum Genet. 2009;85:53–63. doi: 10.1016/j.ajhg.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Liu G, Wang F. Generation of an Fgf9 conditional null allele. Genesis. 2006;44:150–154. doi: 10.1002/gene.20194. [DOI] [PubMed] [Google Scholar]
- Martin GR. The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 1998;12:1571–1586. doi: 10.1101/gad.12.11.1571. [DOI] [PubMed] [Google Scholar]
- Ohuchi H, Nakagawa T, Yamamoto A. The mesenchymal factor, FGF10, initiates and maintains the outgrowth of the chick limb bud through interaction with FGF8, an apical ectodermal factor. Development. 1997;124:2235–2244. doi: 10.1242/dev.124.11.2235. [DOI] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12:3156–3161. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879. doi: 10.1101/gad.965702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Dong SS, Clemens T, Alvarez J, Serra R. Co-ordination of TGF-beta and FGF signaling pathways in bone organ cultures. Mech Dev. 2005;122:557–571. doi: 10.1016/j.mod.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Shimoaka T, Ogasawara T, Yonamine A. Regulation of osteoblast, chondrocyte, and osteoclast functions by fibroblast growth factor (FGF)-18 in comparison with FGF-2 and FGF-10. J Biol Chem. 2002;277:7493–7500. doi: 10.1074/jbc.M108653200. [DOI] [PubMed] [Google Scholar]
- Reinhold MI, Abe M, Kapadia RM, Liao Z, Naski MC. FGF18 represses noggin expression and is induced by calcineurin. J Biol Chem. 2004;279:38209–38219. doi: 10.1074/jbc.M404855200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Lavine KJ, Hung IH, Ornitz DM. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol. 2007;302:80–91. doi: 10.1016/j.ydbio.2006.08.071. [DOI] [PubMed] [Google Scholar]
- Goetz R, Beenken A, Ibrahimi OA. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. 2008;237:18–27. doi: 10.1002/dvdy.21388. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta Y, Nakamura H, Konishi M. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150:4625–4633. doi: 10.1210/en.2009-0119. [DOI] [PubMed] [Google Scholar]
- Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–4940. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Wei W, Dutchak PA, Wang X. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proc Natl Acad Sci USA. 2012;109:3143–3148. doi: 10.1073/pnas.1200797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Levenson A, Kharitonenkov A, de Luca F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J Biol Chem. 2012;287:26060–26067. doi: 10.1074/jbc.M112.343707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicky RA, Wu S, Kharitonenkov A, de Luca F. Role of fibroblast growth factor 21 (FGF21) in undernutrition-related attenuation of growth in mice. Endocrinology. 2012;153:2287–2295. doi: 10.1210/en.2011-1909. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Mangelsdorf DJ. Fibroblast growth factor 21: from pharmacology to physiology. Am J Clin Nutr. 2010;91:254S–257S. doi: 10.3945/ajcn.2009.28449B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Bookout AL, Ding X. FGF21 contributes to neuroendocrine control of female reproduction. Nat Med. 2013;19:1153–1156. doi: 10.1038/nm.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–E49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- Yoshiko Y, Wang H, Minamizaki T. Mineralized tissue cells are a principal source of FGF23. Bone. 2007;40:1565–1573. doi: 10.1016/j.bone.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82:674–681. doi: 10.1210/jcem.82.2.3765. [DOI] [PubMed] [Google Scholar]
- Bianchine JW, Stambler AA, Harrison HE. Familial hypophosphatemic rickets showing autosomal dominant inheritance. Birth Defects Orig Artic Ser. 1971;7:287–295. [PubMed] [Google Scholar]
- Consortium A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- Shimada T, Muto T, Urakawa I. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–3182. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- Shimada T, Mizutani S, Muto T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001;98:6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riminucci M, Collins MT, Fedarko NS. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles KW, Halsey DL, Friedman NE, Lobaugh B. Correlations of serum concentrations of 1,25-dihydroxyvitamin D, phosphorus, and parathyroid hormone in tumoral calcinosis. J Clin Endocrinol Metab. 1988;67:88–92. doi: 10.1210/jcem-67-1-88. [DOI] [PubMed] [Google Scholar]
- Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- Araya K, Fukumoto S, Backenroth R. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:5523–5527. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- Larsson T, Yu X, Davis SI. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:2424–2427. doi: 10.1210/jc.2004-2238. [DOI] [PubMed] [Google Scholar]
- Shimada T, Urakawa I, Yamazaki Y. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004;314:409–414. doi: 10.1016/j.bbrc.2003.12.102. [DOI] [PubMed] [Google Scholar]
- Larsson T, Marsell R, Schipani E. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145:5269–5279. doi: 10.1210/en.2004-0233. [DOI] [PubMed] [Google Scholar]
- Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci USA. 1998;95:5372–5377. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Hasegawa H, Yamazaki Y. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Inves. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305. doi: 10.1016/j.molmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011;96:3541–3549. doi: 10.1210/jc.2011-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham BH, Joseph F, Bailey LM, Fraser WD. The association of circulating ferritin with serum concentrations of fibroblast growth factor-23 measured by three commercial assays. Ann Clin Biochem. 2007;44:463–466. doi: 10.1258/000456307781646102. [DOI] [PubMed] [Google Scholar]
- Clinkenbeard EL, Farrow EG, Summers LJ. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res. 2014;29:361–369. doi: 10.1002/jbmr.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow EG, Yu X, Summers LJ. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci USA. 2011;108:E1146–E1155. doi: 10.1073/pnas.1110905108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Okazaki R, Shibata M. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Yamashita T. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;349:505–506; author reply 505–506. [PubMed] [Google Scholar]
- Strom TM, Francis F, Lorenz B. Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum Mol Genet. 1997;6:165–171. doi: 10.1093/hmg/6.2.165. [DOI] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, Hesse M. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen C, Chen F, Flenniken AM. A novel Phex mutation in a new mouse model of hypophosphatemic rickets. J Cell Biochem. 2012;113:2432–2441. doi: 10.1002/jcb.24115. [DOI] [PubMed] [Google Scholar]
- Martin A, Liu S, David V. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. 2011;25:2551–2562. doi: 10.1096/fj.10-177816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JQ, Ward LM, Liu S. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz-Depiereux B, Bastepe M, Benet-Pages A. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–1250. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- Stubbs JR, He N, Idiculla A. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J Bone Miner Res. 2012;27:38–46. doi: 10.1002/jbmr.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles LD. The bone and beyond: ‘Dem bones’ are made for more than walking. Nat Med. 2011;17:428–430. doi: 10.1038/nm0411-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis. 2009;207:546–551. doi: 10.1016/j.atherosclerosis.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Gutierrez OM, Januzzi JL, Isakova T. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119:2545–2552. doi: 10.1161/CIRCULATIONAHA.108.844506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul C, Amaral AP, Oskouei B. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya N, Chong WH, Gafni RI, Collins MT. Fibroblast growth factor 23: state of the field and future directions. Trends Endocrinol Metab. 2012;23:610–618. doi: 10.1016/j.tem.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SL, Goddard JM, Capecchi MR. Mice homozygous for a targeted disruption of the proto-oncogene int-2 have developmental defects in the tail and inner ear. Development. 1993;117:13–28. doi: 10.1242/dev.117.1.13. [DOI] [PubMed] [Google Scholar]
- Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260–2268. doi: 10.1128/mcb.20.6.2260-2268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert JM, Rosenquist T, Gotz J, Martin GR. FGF5 as a regulator of the hair growth cycle: evidence from targeted and spontaneous mutations. Cell. 1994;78:1017–1025. doi: 10.1016/0092-8674(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Fiore F, Planche J, Gibier P, Sebille A, deLapeyriere O, Birnbaum D. Apparent normal phenotype of Fgf6−/− mice. Int J Dev Biol. 1997;41:639–642. [PubMed] [Google Scholar]
- Guo L, Degenstein L, Fuchs E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev. 1996;10:165–175. doi: 10.1101/gad.10.2.165. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]
- Scearce-Levie K, Roberson ED, Gerstein H. Abnormal social behaviors in mice lacking Fgf17. Genes Brain Behav. 2008;7:344–354. doi: 10.1111/j.1601-183X.2007.00357.x. [DOI] [PubMed] [Google Scholar]
- Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H. Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature. 2010;465:783–787. doi: 10.1038/nature09041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M, Schoorlemmer J, Williams A. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 2007;55:449–463. doi: 10.1016/j.neuron.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu QF, Yang L, Li S. Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell. 2012;149:1549–1564. doi: 10.1016/j.cell.2012.04.046. [DOI] [PubMed] [Google Scholar]
- Wang Q, Bardgett ME, Wong M. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35:25–38. doi: 10.1016/s0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Sasaki S, Konishi M. Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev Dyn. 2008;237:2947–2954. doi: 10.1002/dvdy.21726. [DOI] [PubMed] [Google Scholar]
- Barak H, Huh SH, Chen S. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev Cell. 2012;22:1191–1207. doi: 10.1016/j.devcel.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velocigene Alleles produced for the KOMP project by Velocigene (Regeneron Pharmaceuticals) MGI Direct Data Submission. 2008.
- Neve A, Corrado A, Cantatore FP. Osteocytes: central conductors of bone biology in normal and pathological conditions. Acta Physiol. 2012;204:317–330. doi: 10.1111/j.1748-1716.2011.02385.x. [DOI] [PubMed] [Google Scholar]
- Galli C, Passeri G, Macaluso GM. Osteocytes and WNT: the mechanical control of bone formation. J Dent Res. 2010;89:331–343. doi: 10.1177/0022034510363963. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Ferron M. The contribution of bone to whole-organism physiology. Nature. 2012;481:314–320. doi: 10.1038/nature10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G. Bone endocrine regulation of energy metabolism and male reproduction. C R Biol. 2011;334:720–724. doi: 10.1016/j.crvi.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- Sardiwal S, Magnusson P, Goldsmith DJ, Lamb EJ. Bone alkaline phosphatase in CKD-mineral bone disorder. Am J Kidney Dis. 2013;62:810–822. doi: 10.1053/j.ajkd.2013.02.366. [DOI] [PubMed] [Google Scholar]
- Clowes JA, Riggs BL, Khosla S. The role of the immune system in the pathophysiology of osteoporosis. Immunol Rev. 2005;208:207–227. doi: 10.1111/j.0105-2896.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- Mundy GR. Osteoporosis and inflammation. Nutr Rev. 2007;65:S147–S151. doi: 10.1111/j.1753-4887.2007.tb00353.x. [DOI] [PubMed] [Google Scholar]
- Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP, Rossant J. Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenetic movement through the primitive streak. Development. 1997;124:2829–2841. doi: 10.1242/dev.124.14.2829. [DOI] [PubMed] [Google Scholar]
- Xu X, Qiao W, Li C, Deng CX. Generation of Fgfr1 conditional knockout mice. Genesis. 2002;32:85–86. doi: 10.1002/gene.10028.abs. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Rousseau B, Dubayle D, Sennlaub F. Neural and angiogenic defects in eyes of transgenic mice expressing a dominant-negative FGF receptor in the pigmented cells. Exp Eye Res. 2000;71:395–404. doi: 10.1006/exer.2000.0892. [DOI] [PubMed] [Google Scholar]
- Hajihosseini MK, Lalioti MD, Arthaud S. Skeletal development is regulated by fibroblast growth factor receptor 1 signalling dynamics. Development. 2004;131:325–335. doi: 10.1242/dev.00940. [DOI] [PubMed] [Google Scholar]
- Revest JM, Spencer-Dene B, Kerr K. Fibroblast growth factor receptor 2-IIIb acts upstream of Shh and Fgf4 and is required for limb bud maintenance but not for the induction of Fgf8, Fgf10, Msx1, or Bmp4. Dev Biol. 2001;231:47–62. doi: 10.1006/dbio.2000.0144. [DOI] [PubMed] [Google Scholar]
- de Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal–epithelial signalling during mouse organogenesis. Development. 2000;127:483–492. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- Hajihosseini MK, Wilson S, de Moerlooze L, Dickson C. A splicing switch and gain-of-function mutation in FgfR2-IIIc hemizygotes causes Apert/Pfeiffer-syndrome-like phenotypes. Proc Natl Acad Sci USA. 2001;98:3855–3860. doi: 10.1073/pnas.071586898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S, Wei K, Flenniken A. The missense mutation W290R in Fgfr2 causes developmental defects from aberrant IIIb and IIIc signaling. Dev Dyn. 2010;239:1888–1900. doi: 10.1002/dvdy.22314. [DOI] [PubMed] [Google Scholar]
- Valverde-Franco G, Binette JS, Li W. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Hum Mol Genet. 2006;15:1783–1792. doi: 10.1093/hmg/ddl100. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Schlessinger J. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc Natl Acad Sci USA. 2007;104:3937–3942. doi: 10.1073/pnas.0700012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Xu X, Li C. Generation of Fgfr3 conditional knockout mice. Int J Biol. 2010;6:327–332. doi: 10.7150/ijbs.6.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Healy C, Babbs C. Skeletal analysis of the Fgfr3(P244R) mouse, a genetic model for the Muenke craniosynostosis syndrome. Dev Dyn. 2009;238:331–342. doi: 10.1002/dvdy.21790. [DOI] [PubMed] [Google Scholar]
- Pannier S, Couloigner V, Messaddeq N. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim Biophys Acta. 2009;1792:140–147. doi: 10.1016/j.bbadis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Iwata T, Li CL, Deng CX, Francomano CA. Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum Mol Genet. 2001;10:1255–1264. doi: 10.1093/hmg/10.12.1255. [DOI] [PubMed] [Google Scholar]
- Segev O, Chumakov I, Nevo Z. Restrained chondrocyte proliferation and maturation with abnormal growth plate vascularization and ossification in human FGFR-3(G380R) transgenic mice. Hum Mol Genet. 2000;9:249–258. doi: 10.1093/hmg/9.2.249. [DOI] [PubMed] [Google Scholar]
- Seitzer N, Mayr T, Streit S, Ullrich A. A single nucleotide change in the mouse genome accelerates breast cancer progression. Cancer Res. 2010;70:802–812. doi: 10.1158/0008-5472.CAN-09-3239. [DOI] [PubMed] [Google Scholar]
- Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmy-Susini B, Delmas E, Gourdy P. Role of fibroblast growth factor-2 isoforms in the effect of estradiol on endothelial cell migration and proliferation. Circ Res. 2004;94:1301–1309. doi: 10.1161/01.RES.0000127719.13255.81. [DOI] [PubMed] [Google Scholar]
- Azhar M, Yin M, Zhou M. Gene targeted ablation of high molecular weight fibroblast growth factor-2. Dev Dyn. 2009;238:351–357. doi: 10.1002/dvdy.21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Y, Alonso MT, Vendrell V. Requirements for FGF3 and FGF10 during inner ear formation. Development. 2003;130:6329–6338. doi: 10.1242/dev.00881. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Lenzner C, Leneuve P. Cre-mediated germline mosaicism: a method allowing rapid generation of several alleles of a target gene. Nucleic Acids Res. 2000;28:E92. doi: 10.1093/nar/28.21.e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EP, Noyes CA, Wang X, Wright TJ, Mansour SL. Fgf3 is required for dorsal patterning and morphogenesis of the inner ear epithelium. Development. 2007;134:3615–3625. doi: 10.1242/dev.006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness LD, Paxton CN, Wang X, Schoenwolf GC, Mansour SL. FGF signaling regulates otic placode induction and refinement by controlling both ectodermal target genes and hindbrain Wnt8a. Dev Biol. 2010;340:595–604. doi: 10.1016/j.ydbio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton MB, Colledge WH, Evans MJ. Crouzon-like craniofacial dysmorphology in the mouse is caused by an insertional mutation at the Fgf3/Fgf4 locus. Dev Dyn. 1998;212:242–249. doi: 10.1002/(SICI)1097-0177(199806)212:2<242::AID-AJA8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Zhang X, Mantela J, Ornitz DM, Ylikoski J. Fgf9 signaling regulates inner ear morphogenesis through epithelial-mesenchymal interactions. Dev Biol. 2004;273:350–360. doi: 10.1016/j.ydbio.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Behr B, Leucht P, Longaker MT, Quarto N. Fgf-9 is required for angiogenesis and osteogenesis in long bone repair. Proc Natl Acad Sci USA. 2010;107:11853–11858. doi: 10.1073/pnas.1003317107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Okawa A, Yoshida H, Nishikawa S, Moriya H, Koseki H. Elbow knee synostosis (Eks): a new mutation on mouse Chromosome 14. Mamm Genome. 2002;13:341–344. doi: 10.1007/s00335-001-2143-6. [DOI] [PubMed] [Google Scholar]
- Puk O, Moller G, Geerlof A. The pathologic effect of a novel neomorphic Fgf9(Y162C) allele is restricted to decreased vision and retarded lens growth. PLoS ONE. 2011;6:e23678. doi: 10.1371/journal.pone.0023678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui H, Shibayama M, Ohbayashi N, Konishi M, Takada S, Itoh N. Fgf18 is required for embryonic lung alveolar development. Biochem Biophys Res Commun. 2004;322:887–892. doi: 10.1016/j.bbrc.2004.07.198. [DOI] [PubMed] [Google Scholar]
- Longaker MT, Behr B, Sorkin M, Manu A, Lehnhardt M, Quarto N. Fgf-18 is required for osteogenesis but not angiogenesis during long bone repair. Tissue Eng Part A. 2011;17:2061–2069. doi: 10.1089/ten.tea.2010.0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura-Ueki M, Oda Y, Oki J. Hair cycle resting phase is regulated by cyclic epithelial FGF18 signaling. J Invest Dermatol. 2012;132:1338–1345. doi: 10.1038/jid.2011.490. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz M, Robbez-Masson L, Chioni AM, Cross B, Rosewell I, Grose R. Fibroblast growth factor 22 is not essential for skin development and repair but plays a role in tumorigenesis. PLoS ONE. 2012;7:e39436. doi: 10.1371/journal.pone.0039436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Raimann A, Ertl DA, Helmreich M, Sagmeister S, Egerbacher M, Haeusler G. Fibroblast growth factor 23 and Klotho are present in the growth plate. Connect Tissue Res. 2013;54:108–117. doi: 10.3109/03008207.2012.753879. [DOI] [PubMed] [Google Scholar]