

Abstract
Osteoarthritis (OA) is a degenerative joint disease and a major cause of pain and disability in older adults. We have previously identified epidermal growth factor receptor (EGFR) signaling as an important regulator of cartilage matrix degradation during epiphyseal cartilage development. To study its function in OA progression, we performed surgical destabilization of the medial meniscus (DMM) to induce OA in two mouse models with reduced EGFR activity, one with genetic modification (EgfrWa5/+ mice) and the other one with pharmacological inhibition (gefitinib treatment). Histological analyses and scoring at 3 months post-surgery revealed increased cartilage destruction and accelerated OA progression in both mouse models. TUNEL staining demonstrated that EGFR signaling protects chondrocytes from OA-induced apoptosis, which was further confirmed in primary chondrocyte culture. Immunohistochemistry showed increased aggrecan degradation in these mouse models, which coincides with elevated amounts of ADAMTS5 and matrix metalloproteinase 13 (MMP13), the principle proteinases responsible for aggrecan degradation, in the articular cartilage after DMM surgery. Furthermore, hypoxia-inducible factor 2α (HIF2α), a critical catabolic transcription factor stimulating MMP13 expression during OA, was also upregulated in mice with reduced EGFR signaling. Taken together, our findings demonstrate a primarily protective role of EGFR during OA progression by regulating chondrocyte survival and cartilage degradation.
Introduction
Osteoarthritis (OA) is a progressive and degenerative disorder of the joint primarily characterized by the destruction of articular cartilage. Articular cartilage is predominantly composed of collagen, proteoglycans and other matrix proteins. Loss of large proteoglycan, aggrecan, decreases cartilage compressive stiffness and precedes the damage to the collagen fibrillar network, which is responsible for tensile properties of the tissue. The structural and functional integrity of articular cartilage relies on the balance between anabolic and catabolic activities of chondrocytes. The pathology of OA in cartilage involves the production of pro-inflammatory cytokines, inflammation, degradation of extracellular matrix (ECM) by proteases, chondrocyte hypertrophy and apoptosis.1 Moreover, the subchondral bone is inevitably altered with cortical plate thickening, enhanced bone remodeling and osteophyte formation.2 The past decade has witnessed significant advances in deciphering the basic mechanism by which OA develops using genetically modified and surgical animal models, but there are as yet no disease modifying treatments available. It remains imperative to understand what drives cartilage degeneration during OA.
Growth factors, such as transforming growth factor beta (TGF-β), insulin-like growth factors, bone morphogenic proteins and fibroblast growth factors, regulate synthesis and maintenance of cartilage ECM and therefore, play important roles in cartilage homeostasis and OA development. We recently demonstrated that epidermal growth factor receptor (EGFR), a tyrosine kinase receptor, and one of its cognate ligands, TGF-α, are important for cartilage matrix degradation during endochondral ossification.3,4 Deficiency of EGFR activity either globally or specifically in chondrocytes causes expansion of the hypertrophic zone in the growth plate3,5 and delayed formation of secondary ossification center in long bones at early postnatal stage.6 Consistently, TGF-α null mice exhibited similar developmental phenotypes.4 Mechanistic studies revealed that activation of EGFR signaling stimulates the expression of matrix metalloproteinases (MMPs), including MMP9, 13 and 14, and receptor activator of nuclear factor-κB ligand (RANKL) in hypertrophic chondrocytes via β-catenin-dependent and -independent pathways, resulting in increased cartilage matrix degradation by chondrocytes and osteoclasts at the chondro-osseous junctions.6 EGFR ligands are membrane-bound proteins requiring ectodomain shedding to release the functional peptides. The major sheddase for TGF-α is TNF-α-converting enzyme, also known as a disintegrin and metalloprotease 17 (ADAM17).7 Studies of cartilage-specific TNF-α-converting enzyme knockout mice revealed a similar growth plate phenotype, providing further evidence that EGFR signaling stimulates the degradation of cartilage matrix.8,9
Since cartilage ECM degradation is a critical step toward OA pathogenesis, we hypothesized that EGFR and its ligands constitute another growth factor signaling pathway regulating OA development. Indeed, previous reports implied that this pathway might play a catabolic role in OA pathology. It was reported that the amount of TGF-α is elevated by articular chondrocytes in experimentally induced OA and human OA.10,11 Disruption of mitogen-inducible gene 6 (mig-6), a negative feedback inhibitor of EGFR activity, resulted in a degenerative joint OA-like disease in mice.12–14 Moreover, mice deficient in MMP13 activity either globally15 or conditionally in chondrocytes16 were protected from OA development induced by surgery. In the present study, in order to directly investigate the role of EGFR in OA development, we performed destabilization of the medial meniscus (DMM) to induce OA in mouse models with reduced EGFR activity. This OA surgery is well characterized and it induces progressive cartilage degeneration with little synovial inflammation.17 Surprisingly, we found that these mice show more severe cartilage destruction compared to control mice. Furthermore, histological analyses revealed that loss of EGFR activity accelerates chondrocyte apoptosis and increases cartilage matrix degradation, which coincide with the upregulation of proteinases (ADAMTS5 and MMP13), and a catabolic transcription factor, HIF2α. These results uncover an important function of EGFR in protecting articular cartilage during OA progress and suggest this protein as a potential target for OA treatment.
Materials and methods
Surgical induction of OA by DMM in mice with deficient EGFR activity
Two mouse models with deficient EGFR activity were used in our study. In the first model, mice were heterozygous for a dominant negative allele of EGFR, Wa5.18 These mice (EgfrWa5/+) on a 129S1/SvImJ background were generated by breeding EgfrWa5/+ and wild-type (WT, Egfr+/+) mice, and were identified by their wavy hair appearance. Both EgfrWa5/+ (n=9) and their WT siblings (n=10) at 3 months of age were subjected to DMM surgery as detailed below. In the second model, WT 129S2 mice were purchased from Charles River (Malvern, PA, USA) at the age of 2 months. A month later, they received DMM surgery followed by either vehicle (0.5% methyl cellulose, n=8) or gefitinib (100 mg⋅kg−1, n=9, an EGFR inhibitor; LC Laboratories, Woburn, MA, USA) treatment via oral gavage immediately after surgery, once every other day, for 12 weeks. In both models, there was no significant difference in body weight between control and EGFR deficient group at the beginning of surgery and at 3 months after surgery when the knee joints were harvested.
Experimental OA was induced by DMM surgery and performed by a single surgeon (XZ) in these two mouse models as described previously.17 Only male mice were used in our study because male 129 mice develop more severe OA than females.19 Briefly, under general anesthesia, the right knee joint capsule was exposed and the medial meniscotibial ligament was transected under microscope to give destabilization of the medial meniscus. A sham operation was performed on the left knee joint in which the ligament was visualized but not transected. Mice were maintained in their preoperative groups, allowed unrestricted cage exercise, and were weighed weekly until they were euthanized 12 weeks after surgery. All animal works performed in this study were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Histological analysis of OA cartilage
After euthanasia, bilateral knee joints were harvested free of soft tissues, fixed in 4% paraformaldehyde overnight, decalcified in 0.5 mol⋅L−1 ethylene diamine tetraacetic acid (EDTA) (pH 7.4) for 4 weeks at 4 °C and embedded in paraffin. Serial 5-μm-thick sagittal sections were cut across the medial compartment of the joint. Two sections within every consecutive six sections were stained with Safranin O/Fast green and hematoxylin and the one with better morphology was used for quantification. Each knee yielded about 15 sections for scoring by two blinded observers (XZ and ME-I) using modified Mankin’s methods.20 In brief, each section was assigned a score, which is the sum of cartilage structure (0–5), chondrocytes (0–3), Safranin-O staining (0–5) and tidemark (0–1). Each knee received a score from the section with the maximal score. The results of Mankin score represent the mean of maximal scores in each group. To characterize the loss of articular cartilage, cartilage area (total) and Safranin O-stained area (uncalcified) were outlined and quantified on projected images of about 15 sections per bone selected as above followed by averaging. Thickness of each area was determined by averaging five thicknesses evenly distributed across the entire cartilage. To quantify changes in hypertrophic chondrocytes in the sham-operated knees, the number of hypertrophic chondrocytes in tibial articular cartilage was counted based on their morphology (enlarged chondrocyte lacunae with a lack of Safranin-O stain around a collapsed cells) and divided by articular cartilage area.
Immunohistochemistry and TUNEL staining
Serial sections adjacent to those with representative Safranin O-stained images were dewaxed in xylene and rehydrated through a graded series of alcohols for immunohistochemical evaluation. Briefly, after antigen retrieval with boiling in sodium citrate buffer, endogenous peroxidase activity was quenched in 3% H2O2 for 15 min and washed in phosphate buffered saline (PBS). Sections were then blocked in serum for 30 min followed by incubation with the primary antibody in humidified chamber at 4 °C overnight. Biotinylated secondary antibody was added for 30 min on the second day, followed by an avidin-biotinylated horseradish peroxidase complex according to the manufacturer’s directions (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA, USA). Finally, peroxidase activity was revealed by immersion in DAB substrate (Dako, Glostrup, Denmark). The following primary antibodies were used: rabbit anti-aggrecanase generated-aggrecan neoepitope NITEGE, rabbit anti-MMP-generated aggrecan-neoepitope DIPEN (gifts from Dr John S Mort), rabbit anti-MMP13 (ab75606; Abcam, Cambridge, MA, USA), rabbit anti-ADAMTS5 (ab41037; Abcam), rabbit anti-HIF2α (NB100–122; Novus Biologicals, Littleton, CO, USA). TUNEL staining was carried out on paraffin sections using ApopTag Plus Peroxidase in Situ Apoptosis Detection Kit (S7101; Millipore, Billerica, MA, USA) according to the manufacturer’s instructions.
Primary chondrocytes, qRT-PCR and apoptosis assay
Primary epiphyseal chondrocytes were obtained from rat newborn pups as described previously.3 Cells were plated at a density of 4×104 per cm2 in 12-well plates in chondrogenic medium (DMEM/F12 medium with 5% fetal bovine serum, 50 µg⋅mL−1 of L-ascorbic acid, 1% glutamine, 100 µg⋅mL−1 of streptomycin and 100 U⋅mL−1 of penicillin). Four days later, when cells reached 90% confluence, cultures were given fresh medium overnight before adding 50 ng⋅mL−1 of recombinant human TGF-α (Pepro-Tech, Rocky Hill, NJ, USA). RNA was harvested after 48 h for qRT-PCR. The primer sequences are: adamts5 (forward 5′-CGTTCCTGCAGTGTCATACCCT-3′, reverse 5′-TTTGACTCCTTTTGCATCGGAC-3′) and hif2α (forward 5′-GTGGTCTGTGGGCAATCAGAGCG-3′, reverse 5′-GGAGACATGAGGCGGGGTGC-3′).
To study the survival effect of TGF-α, primary rat chondrocytes were seeded in 12-well plates at 4×104 cells per cm2 and serum-starved overnight. The next day, cells were pretreated with either vehicle or 25 ng⋅mL−1 tumor necrosis factor α (TNF-α) (Pepro-Tech) before the addition of 50 ng⋅mL−1 TGF-α. Two days later, cells were washed with PBS and stained with 5 μg⋅mL−1 ethidium bromide and 5 μg⋅mL−1 acridine orange in 1×PBS as described previously.21 Apoptotic cells were identified as cells having condensed chromatin (green at early apoptotic stage and red at late apoptotic stage). Living cells had a characteristic green chromatin staining with a normal morphology of the nucleus. The number of apoptotic and total cells was counted in three fields per well under a fluorescence microscope.
Statistic analysis
All statistical analysis was performed by independent Student’s t-test assuming equal variances in each group or two-way analysis of variance with a Bonferroni’s post hoc test. For cell culture experiments, all results are derived from experiments being repeated independently at least three times. A value of P<0.05 was considered significant. All data are expressed as mean±s.e.m.
Results
Mice with reduced EGFR activity exhibit accelerated OA development
Two mouse models with reduced EGFR activity were studied. In the first model, we compared OA development between Egfrwa5/+ and their WT siblings after DMM surgery. Egfrwa5 codes for a kinase-dead, dominant negative receptor. Mice homozygous for Wa5 are embryonic lethal, but the heterozygotes are viable and show no major pathological changes,18 allowing for long-term studies. We recently reported that primary chondrocytes derived from Egfrwa5/+ mice shows a slightly decrease in extracellular signal-regulated kinases (ERK) phosphorylation after being treated with epidermal growth factor (EGF) compared to WT cells, suggesting that the EGFR activity in chondrocytes from Egfrwa5/+ mice is modestly suppressed.6 In the second model, we compared OA development in WT mice treated with either vehicle or gefitinib, an EGFR kinase inhibitor.22 Previously, we have demonstrated that similar inhibitor treatment results significant bone loss at both trabecular and cortical sites in young mice.23
DMM surgery was performed in the right knees of 3-month-old skeletally mature mice in both models to induce OA. The left knees received sham operation. Three months after surgery, knee joints were harvested for histological analyses. The sham-operated knees appeared normal in morphology and their Mankin scores representing the severity of OA were negligible (less than 1) in all groups (data not shown). In DMM-operated knees, while WT mice exhibited typical OA features, EgfrWa5/+ mice had marked increases in cartilage surface fibrillation, clefting and erosion down to the tidemark (Figure 1a). Moreover, total cartilage area and thickness, particularly the uncalcified area and thickness, at both medial femoral condyle and medial tibial plateau were reduced in EgfrWa5/+ mice in comparison with WT mice (Figure 1b). Further scoring by Mankin’s method revealed significant 19% and 36% increases of OA scores at medial femoral condyle and medial tibial plateau, respectively, in EgfrWa5/+ mice compared to those in WT mice (Figure 1c).
Figure 1.

EgfrWa5/+ mice exhibit accelerated osteoarthritis progression after DMM surgery. (a) Representative Safranin O/Fast Green staining images of mouse knee joints show increased articular cartilage degradation in EgfrWa5/+ mice 3 months after DMM surgery. Bottom panels are magnified images of top panels. (b) Total articular cartilage, uncalcified articular cartilage, and their respective thickness were quantified at both femoral condyle and tibial plateau regions. (c) Mankin score graded by blinded observers confirmed more articular cartilage destruction in EgfrWa5/+ mice (n=9) compared to WT (n=10) mice after DMM surgery. * P<0.05; ** P<0.01; *** P<0.001.
In gefitinib-treated mice, OA damages on the articular cartilage was relatively milder than those in EgfrWa5/+ mice, but still more severe than their vehicle-treated controls with increased fibrillation, clefting and erosion at the surface of articular cartilage (Figure 2a). While we did not observe overall loss of total articular cartilage in gefitinib-treated mice, the area and thickness of uncalcified cartilage were remarkably decreased in those mice compared to their vehicle controls (Figure 2b). Mankin scores revealed a significant 28% increase at medial femoral condyle and a trend of 22% increase at medial tibial plateau in these mice compared to controls (Figure 2c). Taken together, these results demonstrate that mice with reduced EGFR activity exhibit more cartilage destruction and accelerated OA progression.
Figure 2.

Mice treated with gefitinib exhibit accelerated osteoarthritis progression after DMM surgery. (a) Representative Safranin O/Fast Green staining images of mouse knee joints show increased articular cartilage degradation in gefitinib-treated mice 3 months after DMM surgery. Bottom panels are magnified images of top panels. (b) Total articular cartilage, uncalcified articular cartilage, and their respective thickness were quantified at both femoral condyle and tibial plateau regions. (c) Mankin score graded by blinded observers confirmed more articular cartilage destruction in gefitinib-treated mice (n=9) compared to vehicle-treated (n=8) mice after DMM surgery. * P<0.05.
Chondrocyte hypertrophy is routinely seen in the development of osteoarthritis. Since our previous work demonstrated that EGFR deficiency induces expansion of the hypertrophic zone of the growth plate in young rats and neonatal mice,3 we quantified the number of hypertrophic chondrocytes in the articular cartilage and normalized it against the total cartilage area. In the sham-operated contralateral joints, we observed a significant 111% increase of hypertrophic chondrocytes in EgfrWa5/+ mice (WT: 0.12±0.03 cells per mm2; EgfrWa5/+: 0.25±0.02 cells per mm2, n=5 in each group, P=0.01, Figure 3a) and a similar trend of increase in gefitinib-treated mice (vehicle-treated: 0.19±0.02 cells per mm2; gefitinib-treated: 0.24±0.03 cells per mm2, n=5 in each group, P=0.17, Figure 3b) compared to their respective controls. We found that it was difficult to perform similar measurement at the DMM knees because of severe erosion and/or loss of articular cartilage in the EGFR deficient group. These findings indicate that EGFR prevents chondrocyte hypertrophy and may explain why mice with reduced EGFR signaling are predisposed to the development of osteoarthritis.
Figure 3.
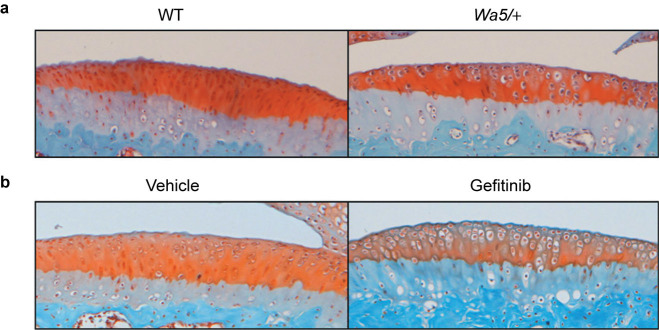
Mice with reduced EGFR activity have more hypertrophic chondrocytes in the articular cartilage. Representative Safranin O/Fast Green staining images of sham-operated tibial plateau from EgfrWa5/+ mice (a) and gefitinib-treated mice (b) and their respective controls.
Reduced EGFR activity promotes apoptosis in cartilage chondrocytes after DMM surgery
Hypertrophic chondrocytes undergo cell death and are replaced by bone in the process of endochondral ossification. However, this process at the articular surface could lead to osteoarthritis. Chondrocyte apoptosis has been observed during OA progression and the apoptotic rate is positively correlated with the severity of cartilage damage.24,25 Indeed, the contribution of apoptosis to OA was further demonstrated by using caspase inhibitors, which block cell death and decrease severity of cartilage lesions in posttraumatic OA.26 EGFR has multifaceted actions on cells from a variety of tissues and is capable of regulating their proliferation, differentiation, survival, migration, and adhesion. To investigate the role of EGFR in chondrocyte apoptosis during OA development, TUNEL staining was performed on knee joints at 3 months post-surgery in mouse models with reduced EGFR activity. In DMM-operated knees from control mice, only 8%–12% of chondrocytes in articular cartilage underwent apoptosis and most of these apoptotic chondrocytes were located in the calcified zone (Figure 4a and 4b). In contrast, 30% and 24% of chondrocytes in DMM-operated knees from Egfrwa5/+ mice and gefitinib-treated mice, respectively, were TUNEL-positive and many of them extended into the middle and deep zones (Figure 4a and 4b), suggesting that EGFR protects chondrocytes from OA-induced cell death.
Figure 4.

EGFR signaling protects articular chondrocytes from OA-induced apoptosis. (a) TUNEL staining showed increased number of apoptotic chondrocytes in knee joints from EgfrWa5/+ mice at 3 months post-surgery compared to that from WT mice. The percentage of TUNEL-positive chondrocytes was quantified (n=4 in each group, right panel). ⋆⋆⋆ P<0.001. (b) The same TUNEL staining was performed with gefitinib- and vehicle-treated mice and quantification showed similar results (n=4 in each group, right panel). ** P<0.01. (c) TGF-α, an EGFR ligand, suppresses chondrocyte apoptosis induced by serum depletion and TNF-α. Primary chondrocytes were serum starved for 1 day followed by addition of 10% serum (FBS), vehicle (con), 50 ng⋅mL−1 TGF-α, 25 ng⋅mL−1 TNF-α and TNF-α plus TGF-α. Two days later, ethidium bromide/acridine orange staining was performed to quantify the percentage of apoptotic cells (bottom panel). Arrow points to a late apoptotic cell. ss, serum starvation. ** P<0.01; *** P<0.001.
To confirm this finding in vitro, we isolated epiphyseal chondrocytes from neonatal pups and used serum starvation together with TNF-α, a pro-inflammatory cytokine and a well-known inducer of chondrocyte apoptosis in OA,27 to induce cell death in these cells. Ethidium bromide/acridine orange staining showed that removal of serum alone increased the percentage of apoptotic cells from 2% to 14% and that addition of TNF-α further elevated it to 21% (Figure 4c). However, cotreatment with TGF-α completely abrogated the apoptotic signals of serum depletion and TNF-α, reducing the percentage of apoptotic cells to the level similar to those grown in normal serum-containing medium (Figure 4c). Taken together, these in vivo and in vitro results clearly indicate that EGFR transduces survival signals to chondrocytes during OA progression.
Aggrecan degradation is accelerated in OA knee joints from mice with reduced EGFR activity
Proteolytic cleavage of aggrecan weakens the cartilage matrix and is a key event in OA pathogenesis.28,29 Next, we examined aggrecan degradation products in articular cartilage after DMM surgery by immunohistochemistry. Staining of aggrecan-neoepitopes NITEGE (aggrecanase-generated aggrecan cleavage fragment) and DIPEN (MMP-generated aggrecan cleavage fragment) showed that aggrecan degradation products generated by either aggrecanases or MMPs were significantly enhanced in both femoral and tibial articular cartilage areas in EgfrWa5/+ mice and gefitinib-treated mice compared to their respective control mice (Figure 5). These results are in line with much more severe loss of Safranin O staining observed in knee joints of mice with reduced EGFR activity (Figure 1a and 2a), indicating that activation of EGFR signaling pathway suppresses cartilage matrix degradation during OA development.
Figure 5.
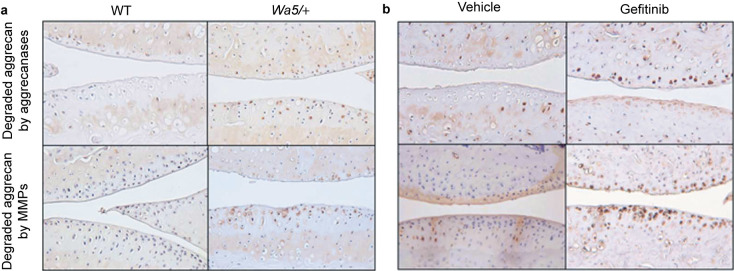
Aggrecan degradation is elevated in osteoarthritic cartilage from mice with reduced EGFR activity. Immunohistological assay revealed increased amounts of degraded aggrecan products generated either by aggrecanases (top panels) or by MMPs (bottom panels) in knee joints from WT and EgfrWa5/+ mice (a) and vehicle- and gefitinib-treated mice (b) after DMM surgery.
ADAMTS5 is the major aggrecanase in mouse cartilage and deletion of this proteinase blocks cartilage degradation and prevents OA progression.30,31 Immunostaining revealed that ADAMTS5 amounts were markedly increased in articular cartilage after DMM surgery in both mouse models with reduced EGFR activity, compared to those in their respective control mice (Figure 6a and 6b, right panels). Similar observations were also obtained in sham-operated knees (Figure 6a and 6b, left panels), implying that EGFR signaling regulates adamts5 expression in chondrocytes. Indeed, qRT-PCR experiment with primary chondrocytes further demonstrated that activation of EGFR in chondrocytes inhibits the mRNA expression of adamts5 (Figure 6c).
Figure 6.

The protein amounts of ADAMTS5 and MMP13 are increased in osteoarthritic cartilage from mice with reduced EGFR activity. (a, b) Immunostaining of ADAMTS5 and MMP13 in sham- (left panels) and DMM-operated (right panels) knee joints from WT and EgfrWa5/+ mice (a) and vehicle- and gefitinib-treated mice (b). For ADAMTS5, we observed increased staining in both sham and DMM knees from mice with reduced EGFR activity. However, for MMP13, we observed decreased staining in sham knees but increased staining in DMM knees from mice with reduced EGFR activity. The percentages of ADAMTS5- and MMP13-positive chondrocytes were quantified. * P<0.05; *** P<0.001. (c) qRT-PCR shows that EGFR signaling inhibits chondrogenic adamts5 expression. Primary chondrocytes were serum starved overnight, treated with vehicle (control) or TGF-α (50 ng⋅mL−1) for 1 day, and harvested for mRNA quantification. ** P<0.01.
MMP13 is another critical proteinase responsible for cartilage degradation during OA development.15,16,32 Our previous work found that activation of EGFR signaling induces Mmp13 expression in chondrocytes, while inhibition of EGFR signal by gefitinib attenuates its expression in the growth plate.3 Immunostaining of MMP13 was weak and mostly located below the tidemark in sham-operated knees. In line with the above growth plate results, we found that MMP13 amount was further reduced in articular cartilage from EgfrWa5/+ mice and gefitinib-treated mice (Figure 6a and 6b, left panels). In contrast, MMP13 immunostaining occurred in the uncalcified cartilage above the tide marker in DMM knee joints of EgfrWa5/+ mice and gefitinib-treated mice and its amounts in those mice were significantly elevated compared to their respective control mice (Figure 6a and 6b, right panels). Taken together, our results indicate that inhibition of EGFR signaling promotes aggrecan degradation during OA development by increasing the amounts of important catabolic enzymes, such as ADAMTS5 and MMP13.
To understand the mechanism for the opposite regulation of Mmp13 expression by EGFR in sham- and DMM-operated knee joints, we next analyzed the expression of hif2a, a transcription factor highly expressed in OA development and essential for Mmp13 expression in chondrocytes,33–35 in the articular cartilage of those mouse models after DMM surgery. Immunostaining revealed a considerably increased number of HIF2α-positive cells in articular cartilage from EgfrWa5/+ and gefitinib-treated mice at 12 weeks after surgery, compared to their respective control mice (Figure 7a and 7b). To confirm these in vivo results, we evaluated the regulation of HIF2α expression by EGFR signaling in primary epiphyseal chondrocytes culture. qRT-PCR demonstrated that TGF-α significantly decreased hif2α expression in primary chondrocytes (Figure 7c). Hence, our results suggest that one mechanism for EGFR to suppress Mmp13 expression is likely through inhibiting hif2α expression during OA progression. In normal knees, since the basal level of HIF2α is very low in the cartilage,34 EGFR signaling must use alternative transcription factor(s) to activate the expression of Mmp13.
Figure 7.

Deficiency of EGFR signaling increases HIF2α amount in osteoarthritic cartilage. (a, b) Immunostaining of HIF2α in DMM-operated knee joints from WT and EgfrWa5/+ mice (a) and vehicle- and gefitinib-treated mice (b). The percentage of HIF2α-positive chondrocytes was quantified (n=4–6 in each group). * P<0.05; *** P<0.001. (c) qRT-PCR shows that EGFR signaling inhibits chondrogenic hif2α expression. Primary chondrocytes were serum starved overnight, treated with vehicle (control) or TGF-α (50 ng⋅mL−1) for 1 day, and harvested for mRNA quantification. * P<0.05.
Discussion
The present study identifies a novel role of EGFR signaling in articular cartilage homeostasis and OA progression. We demonstrated that a genetically modified mouse model (EgfrWa5/+ ) and a pharmacological mouse model (gefitinib treatment), both of which have reduced EGFR activity, exhibit accelerated articular cartilage destruction in knee joints after surgical induction of OA. This observation is correlated with increased chondrocyte apoptosis and aggrecan degradation. Further mechanistic studies suggest that this is likely due to augmented expression of catabolic proteinases (ADAMTS5 and MMP13) and a transcription factor (HIF2α) targeting cartilage matrix degradation after suppressing EGFR activity.
Our findings initially appeared surprising to us because they are not consistent with our previous conclusion that chondrogenic EGFR signaling stimulates cartilage ECM degradation by upregulating MMPs and stimulating osteoclastogenesis at the chondro-osseous junction at postnatal developmental stage.3 However, different from metabolically active epiphyseal chondrocytes at developmental stage, chondrocytes in adult cartilage are normally quiescent with very low activities of synthesizing and degrading cartilage matrix. In response to mechanical stress and pro-inflammatory cytokines, cartilage homeostasis is disrupted with increased ECM remodeling, hypertrophy-like maturation, cartilage calcification and chondrocyte apoptosis. Therefore, both anabolic and catabolic pathways are elevated during OA progression and imbalance between these two pathways toward more catabolic activity leads to this debilitating joint disease. Indeed, we and others have found that activating EGFR signaling executes both anabolic and catabolic actions on chondrocytes. It has been recognized for a long time that EGF is a potent mitogen for cultured chondrocytes.36,37 Interestingly, recent studies with either limb or cartilage targeted deletion of the intracellular EGFR inhibitor mig-6 revealed remarkably thickened articular cartilage in those mice at early adult stage due to enhanced chondrocyte proliferation.12,13 Although we did not notice a reduction in cartilage thickness in the sham-operated knees in mice with reduced EGFR activity, a close examination did reveal an increase in the number of hypertrophic chondrocytes, suggesting that EGFR prevents chondrocyte hypertrophy. Together with our data that EGFR signaling prevents chondrocyte apoptosis both in vitro and in vivo, these findings demonstrate a strong anabolic role of EGFR signaling in articular cartilage by increasing the number of articular chondrocytes, particularly proliferative and undifferentiated chondrocytes, which eventually leads to more cartilage matrix synthesis. Meanwhile, activating EGFR signaling transduces strong catabolic signals to chondrocytes, such as enhancing the expression of MMPs and RANKL3,4,9 and stimulating cartilage ECM degradation.3,4 Hence, we propose that the final outcomes in terms of OA progression in mouse with altered EGFR activity is combined effects of both anabolic and catabolic actions of EGFR pathway determined by the level of EGFR activity. According to this hypothesis, the severe OA phenotypes observed in mig-6 knockout mice is likely due to highly elevated EGFR activity. Note that our EgfrWa5/+ mouse model has modest decrease in chondrogenic EGFR activity compared to WT mice while a chondrogenic EGFR knockout mouse model (col2-cre EgfrWa5/flox) we established previously has more drastic decrease in EGFR activity.6 We are currently studying the OA progression in this mouse model.
Excessive proteolysis has long been recognized as a major contributory factor to cartilage matrix degradation in OA. A variety of proteases have been implicated, ranging from lysosomal cysteine proteases to MMP family members. Among them, ADAMTS5 and MMP13 are the most critical ones in mediating cartilage ECM degradation because their expression is greatly enhanced in osteoarthritic knees38–41 and because mice deficient in either ADAMTS5 or MMP13 have diminished OA progression after DMM surgery.15,16,30,42 Interestingly, while ADAMTS5 amounts were similarly increased in both sham and DMM knee joints from mice with reduced EGFR activity, MMP13 amounts were decreased in sham knees, which is consistent with our previous report that EGFR signaling stimulates MMP13 expression in chondrocytes, but were increased in DMM knee joints from mice with reduced EGFR activity compared to their controls. The apparent contradiction could be explained by enhanced hif2α expression in DMM knee joints after suppression of EGFR activity. Previous reports33–35 have shown that HIF2α is a major regulator for Mmp13 but not adamts5 genes in chondrocytes. Elevated HIF2α amount thus surpasses the other pathway(s) which mediates the stimulatory effect of EGFR signaling on Mmp13 expression.
In conclusion, in the present study, we provide the first direct evidence for the involvement of EGFR signaling in the OA development. Modestly decreased EGFR activity in our two mouse models accelerates articular cartilage destruction, which is associated with increased chondrocyte apoptosis and cartilage matrix degradation by ADAMTS5 and MMP13. This study lays the groundwork for understanding the role of EGFR and its cognate ligands in the maintenance of articular cartilage and in the pathogenesis of OA. Further studies to maximize its anabolic actions while minimizing its catabolic actions in cartilage will shed light on targeting this novel pathway in OA treatment.
Acknowledgments
The authors would like to thank John S. Mort at Shriners Hospital for Children (Montreal, Canada) for kindly providing antibodies for anti-aggrecanase generated and anti-MMP generated aggrecan cleavage fragments. This study was supported by ASBMR Research Career Enhancement Award (to LQ), NIH grants AR060991 (to LQ) and AR062908 (to ME-I).
The authors declare no conflict of interest.
References
- Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann NY Acad Sci. 2010;1192:230–237. doi: 10.1111/j.1749-6632.2009.05240.x. [DOI] [PubMed] [Google Scholar]
- Sharma AR, Jagga S, Lee SS, Nam JS. Interplay between cartilage and subchondral bone contributing to pathogenesis of osteoarthritis. Int J Mol Sci. 2013;14:19805–19830. doi: 10.3390/ijms141019805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Siclari VA, Lan S. The critical role of the epidermal growth factor receptor in endochondral ossification. J Bone Miner Res. 2011;26:2622–2633. doi: 10.1002/jbmr.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmani SE, Pest MA, Kim G, Ohora SN, Qin L, Beier F. Transforming growth factor alpha controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone. 2012;51:131–141. doi: 10.1016/j.bone.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Wang K, Yamamoto H, Chin JR, Werb Z, Vu TH. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J Biol Chem. 2004;279:53848–53856. doi: 10.1074/jbc.M403114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhu J, Li Y. Epidermal growth factor receptor (EGFR) signaling regulates epiphyseal cartilage development through beta-catenin-dependent and -independent pathways. J Biol Chem. 2013;288:32229–32240. doi: 10.1074/jbc.M113.463554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KC, Hill D, Otero M. ADAM17 controls endochondral ossification by regulating terminal differentiation of chondrocytes. Mol Cell Biol. 2013;33:3077–3090. doi: 10.1128/MCB.00291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Horiuchi K, Kimura T. Conditional inactivation of TNFalpha-converting enzyme in chondrocytes results in an elongated growth plate and shorter long bones. PLoS One. 2013;8:e54853. doi: 10.1371/journal.pone.0054853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 2007;56:1854–1868. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- Appleton CT, Usmani SE, Bernier SM, Aigner T, Beier F. Transforming growth factor alpha suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis Rheum. 2007;56:3693–3705. doi: 10.1002/art.22968. [DOI] [PubMed] [Google Scholar]
- Shepard JB, Jeong JW, Maihle NJ, O’Brien S, Dealy CN. Transient anabolic effects accompany epidermal growth factor receptor signal activation in articular cartilage in vivo. Arthritis Res Ther. 2013;15:R60. doi: 10.1186/ar4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal B, Williams BO, Beier F, Vande Woude GF, Zhang YW. Cartilage-specific deletion of Mig-6 results in osteoarthritis-like disorder with excessive articular chondrocyte proliferation. Proc Natl Acad Sci USA. 2014;111:2590–2595. doi: 10.1073/pnas.1400744111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Su Y, Lanning N. Targeted disruption of Mig-6 in the mouse genome leads to early onset degenerative joint disease. Proc Natl Acad Sci USA. 2005;102:11740–11745. doi: 10.1073/pnas.0505171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little CB, Barai A, Burkhardt D. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Sampson ER, Jin H. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15:R5. doi: 10.1186/ar4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061–1069. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Lee D, Cross SH, Strunk KE. Wa5 is a novel ENU-induced antimorphic allele of the epidermal growth factor receptor. Mamm Genome. 2004;15:525–536. doi: 10.1007/s00335-004-2384-2. [DOI] [PubMed] [Google Scholar]
- Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. 2007;15:695–700. doi: 10.1016/j.joca.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Aigner T, Cook JL, Gerwin N. Histopathology atlas of animal model systems—overview of guiding principles. Osteoarthritis Cartilage. 2010;18 (Suppl 3):S2–S6. doi: 10.1016/j.joca.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Chandra A, Lan S, Zhu J, Siclari V, Qin L. Epidermal Growth Factor Receptor (EGFR) signaling promotes proliferation and survival in osteoprogenitors by increasing Early Growth Response Protein (Egr2) expression. J Biol Chem. 2013;288:20488–20498. doi: 10.1074/jbc.M112.447250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari PM, Allen GW, Bonner JA. Biology of interactions: antiepidermal growth factor receptor agents. J Clin Oncol. 2007;25:4057–4065. doi: 10.1200/JCO.2007.11.8984. [DOI] [PubMed] [Google Scholar]
- Zhang X, Tamasi J, Lu X. Epidermal growth factor receptor plays an anabolic role in bone metabolism in vivo. J Bone Miner Res. 2011;26:1022–1034. doi: 10.1002/jbmr.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif M, Whitehouse A, Sharman P, Perry M, Adams M. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 2004;50:507–515. doi: 10.1002/art.20020. [DOI] [PubMed] [Google Scholar]
- Thomas CM, Fuller CJ, Whittles CE, Sharif M. Chondrocyte death by apoptosis is associated with the initiation and severity of articular cartilage degradation. Int J Rheum Dis. 2011;14:191–198. doi: 10.1111/j.1756-185X.2010.01578.x. [DOI] [PubMed] [Google Scholar]
- D’Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54:1814–1821. doi: 10.1002/art.21874. [DOI] [PubMed] [Google Scholar]
- Malemud CJ, Islam N, Haqqi TM. Pathophysiological mechanisms in osteoarthritis lead to novel therapeutic strategies. Cells Tissues Organs. 2003;174:34–48. doi: 10.1159/000070573. [DOI] [PubMed] [Google Scholar]
- Heinegard D, Saxne T. Macromolecular markers in joint disease. J Rheumatol Suppl. 1991;27:27–29. [PubMed] [Google Scholar]
- Lohmander LS, Neame PJ, Sandy JD. The structure of aggrecan fragments in human synovial fluid. Evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum. 1993;36:1214–1222. doi: 10.1002/art.1780360906. [DOI] [PubMed] [Google Scholar]
- Glasson SS, Askew R, Sheppard B. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- Stanton H, Rogerson FM, East CJ. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- Neuhold LA, Killar L, Zhao W. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata M, Kugimiya F, Fukai A. C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum Mol Genet. 2012;21:1111–1123. doi: 10.1093/hmg/ddr540. [DOI] [PubMed] [Google Scholar]
- Saito T, Fukai A, Mabuchi A. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med. 2010;16:678–686. doi: 10.1038/nm.2146. [DOI] [PubMed] [Google Scholar]
- Tamiya H, Ikeda T, Jeong JH. Analysis of the Runx2 promoter in osseous and non-osseous cells and identification of HIF2A as a potent transcription activator. Gene. 2008;416:53–60. doi: 10.1016/j.gene.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Gospodarowicz D, Mescher AL. A comparison of the responses of cultured myoblasts and chondrocytes to fibroblast and epidermal growth factors. J Cell Physiol. 1977;93:117–127. doi: 10.1002/jcp.1040930115. [DOI] [PubMed] [Google Scholar]
- Kato Y, Gospodarowicz D. Growth requirements of low-density rabbit costal chondrocyte cultures maintained in serum-free medium. J Cell Physiol. 1984;120:354–363. doi: 10.1002/jcp.1041200314. [DOI] [PubMed] [Google Scholar]
- Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002;46:2648–2657. doi: 10.1002/art.10531. [DOI] [PubMed] [Google Scholar]
- Flannelly J, Chambers MG, Dudhia J. Metalloproteinase and tissue inhibitor of metalloproteinase expression in the murine STR/ort model of osteoarthritis. Osteoarthritis Cartilage. 2002;10:722–733. doi: 10.1053/joca.2002.0818. [DOI] [PubMed] [Google Scholar]
- Kevorkian L, Young DA, Darrah C. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004;50:131–141. doi: 10.1002/art.11433. [DOI] [PubMed] [Google Scholar]
- Yang S, Kim J, Ryu JH. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16:687–693. doi: 10.1038/nm.2153. [DOI] [PubMed] [Google Scholar]
- Majumdar MK, Askew R, Schelling S. Double-knockout of ADAMTS-4 and ADAMTS-5 in mice results in physiologically normal animals and prevents the progression of osteoarthritis. Arthritis Rheum. 2007;56:3670–3674. doi: 10.1002/art.23027. [DOI] [PubMed] [Google Scholar]