

Abstract
Objective
Accumulation of myofibroblasts in fibrotic skin is a hallmark of systemic sclerosis (SSc; scleroderma), but the origins of these cells remain unknown. Because loss of intradermal adipose tissue is a consistent feature of cutaneous fibrosis, we sought to examine the hypothesis that myofibroblasts populating fibrotic dermis derive from adipocytic progenitors.
Methods
We performed genetic fate mapping studies to investigate the loss of intradermal adipose tissue and its potential role in fibrosis in mice with bleomycin-induced scleroderma. Modulation of adipocytic phenotypes ex vivo was investigated in adipose tissue–derived cells in culture.
Results
A striking loss of intradermal adipose tissue and its replacement with fibrous tissue were consistently observed in mice with bleomycin-induced fibrosis. Loss of adipose tissue and a decline in the expression of canonical adipogenic markers in lesional skin preceded the onset of dermal fibrosis and expression of fibrogenic markers. Ex vivo, subcutaneous adipocytes were driven by transforming growth factor β to preferentially undergo fibrogenic differentiation. Cell fate mapping studies in mice with the adiponectin promoter–driven Cre recombinase transgenic construct indicated that adiponectin-positive progenitors that are normally confined to the intradermal adipose tissue compartment were distributed throughout the lesional dermis over time, lost their adipocytic markers, and expressed myofibroblast markers in bleomycin-treated mice.
Conclusion
These observations establish a novel link between intradermal adipose tissue loss and dermal fibrosis and demonstrate that adiponectin-positive intradermal progenitors give rise to dermal myofibroblasts. Adipose tissue loss and adipocyte–myofibroblast transition might be primary events in the pathogenesis of cutaneous fibrosis that represent novel potential targets for therapeutic intervention.
Fibrosis of the skin and internal organs is the distinguishing pathologic hallmark of systemic sclerosis (SSc; scleroderma) and is responsible for its major complications (1,2). Skin fibrosis is caused by the presence of myofibroblasts, which are regarded as the primary fibrogenic effector cells in SSc driving excessive accumulation of fibrillar collagens and other extracellular matrix proteins in the dermis (3). Although multiple sources of myofibroblast precursors have been proposed, the origin of myofibroblasts within fibrotic lesions remains uncertain (4). In response to transforming growth factor β (TGFβ), Wnt ligands, biomechanical signals, and other profibrotic cues, fibroblasts can acquire a myofibroblast phenotype with prominent expression of α-smooth muscle actin (α-SMA) stress fibers (3). Myofibroblasts might also originate from bone marrow–derived progenitor cells or may arise via epithelial–mesenchymal transition (EMT) and endothelial–mesenchymal transition (endoMT) (3,5). The significance of these cell fate decisions, and their contribution to fibrosis, remain the subject of considerable debate.
A less well-appreciated pathologic hallmark of SSc is loss of intradermal adipose tissue (6). The lesional skin of patients with SSc and animals with experimentally induced fibrosis shows attenuation of intradermal adipose tissue subjacent to reticular dermis, with shrunken and misshapen adipocytes of variable diameter surrounded by fibrillar collagen (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract). The loss of intradermal adipose tissue accounts for the characteristic skin tethering in SSc. A similar phenomenon is noted in animal models of cutaneous fibrosis induced by bleomycin (7), HOCl (8), or angiotensin II (9), overexpression of constitutively active TGFβ receptor type I (TGFβRI) (10), or in sclerodermatous graft-versus-host disease (11). Mice with heritable cutaneous fibrosis, including Fra-2–transgenic (12), fibrillin-1–mutant (13), tsk1 (14), and tsk2 mice (15), display comparable atrophy of intradermal white adipose tissue. Moreover, we have shown that dermal fibrosis in transgenic mice with adipocyte-specific expression of Wnt-10b is accompanied by complete disappearance of intradermal adipose tissue (16).
Adipocytes exert a regulatory influence on connective tissue metabolism through both cell-autonomous and paracrine mechanisms (17). However, despite the prominence of intradermal adipose tissue loss in patients with SSc and in experimental murine models, the characteristics and functional sequelae of this phenomenon are unknown. In the current study, we sought to investigate the process of intradermal adipose loss and to determine the origin of dermal myofibroblasts in cutaneous fibrosis. Our results show that intradermal adipose tissue loss preceded the onset of dermal fibrosis and was accompanied by a decline in the expression of peroxisome proliferator–activated receptor γ (PPARγ), the adipogenic master regulator, followed by the expression of myofibroblast-associated genes.
Cell fate mapping studies in mice revealed that dermal fibrosis was associated with the recruitment of adipocyte-derived mesenchymal cells into lesional dermis. These cells accounted for a great majority of the accumulating myofibroblasts. Ex vivo treatment of subcutaneous adipocytes with TGFβ resulted in loss of morphologic and biochemical adipocytic characteristics and acquisition of myofibroblast features in the absence of significant proliferation. This process was accompanied by large-scale transcriptional reprograming, with decreasing expression of many adipogenic genes and a reciprocal increase in the expression of pivotal fibrogenic genes. Taken together, these observations indicate that the majority of myofibroblasts populating fibrotic skin originate from intradermal adipocytic progenitors through a process termed adipocyte–myofibroblast transition (AMT). Intradermal adipose tissue loss might therefore represent a primary event in the pathogenesis of cutaneous fibrosis.
MATERIALS AND METHODS
Animals
Previously described adiponectin-Cre (AdipoP-Cre)–transgenic mice were maintained on a C57BL/6J genetic background (18,19). C57BL/6J mice and Ai14 reporter mice, which harbor the tandem dimer tdTomato fluorophore inserted into the Gt(ROSA)26Sor locus, were obtained from The Jackson Laboratory (stock nos. 000664 and 007914, respectively). All experimental procedures complied with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the protocols were approved by the Northwestern University Institutional Animal Care and Use Committee.
Adipocyte lineage tracing
To trace the fate of intradermal adipocytes, AdipoP-Cre–transgenic mice were mated with Ai14 reporter (tdTomato) mice. The experiments focused on mice with the genotype AdipoP-Cre+;tdTomato+/f. Mice were genotyped by polymerase chain reaction (PCR) using genomic DNA isolated from tail biopsy specimens. All genotypes were reconfirmed when the experimental procedures were completed.
Bleomycin-induced cutaneous fibrosis
Young (8–15 weeks old) and old (20–30 weeks old) male or female AdipoP-Cre+;tdTomato+/f mice were administered 100 mM phosphate buffered saline (PBS; 100 μl) or bleomycin (1 mg/ml in PBS; 10 mg/kg/day) by daily subcutaneous injections for up to 14 days and killed 5, 8, 14, or 21 days later (7). In other experiments, 8–12-week-old female C57BL/6J mice received daily injections of PBS or bleomycin and were killed at the indicated time points. Serum, skin, and visceral fat specimens were harvested and processed for analysis (7). Each experimental group consisted of 3–5 mice, and the experiments were repeated 2–3 times with consistent results.
Determination of circulating adiponectin
Levels of total adiponectin in serum were determined by enzyme-linked immunosorbent assay according to the manufacturer’s instructions (catalog no. EZMADP-60K; Millipore).
Histopathologic analysis
Samples of skin were fixed in 4% phosphate buffered paraformaldehyde (PFA; pH 7.4), embedded in paraffin, and 4-μm–thick sections were stained with hematoxylin and eosin (H&E). Dermal thickness (distance from the epidermal–dermal junction to the dermal–fat junction) and intradermal adipose tissue layer thickness (distance from the dermal–fat junction to the fat–muscle junction) were determined in 5 randomly selected regions per high-power field (hpf) using ImageJ software (http://rsbweb.nih.gov/ij/download.html) (7). To evaluate collagen deposition and organization in the dermis, sections were stained with Masson’s trichrome. In some experiments, freshly harvested tissue was cryoprotected overnight at 4°C in graded (15–30%) sucrose, embedded in OCT, and sectioned at 12-μm thickness.
Cell culture and ex vivo differentiation assays
Human subcutaneous tissue stem cells (adipose-derived stem cells [ADSC; Lonza]) from 2 healthy adult donors were maintained in ADSC growth medium (ADSC-GM; Lonza). At low passage (passages 2–4), cells were seeded in 12-well plates (1 × 105 cells/well) and incubated in growth medium (preadipocyte basal medium-2 [PBM-2]; Lonza), followed by adipocyte differentiation medium (PBM-2/PDM; Lonza), for 10 days. Cells displaying characteristic adipocyte morphology by phase-contrast microscopy were incubated in media with 5 ng/ml of human recombinant TGFβ2 (PeproTech) for up to 72 hours. Experiments were performed in triplicate and repeated at least 3 times with consistent results. In other experiments, fibroblasts were explanted from the interscapular skin of AdipoP-Cre+;tdTomato+/f–transgenic mice, and low-passage confluent cultures were incubated with TGFβ2 (5 ng/ml), rosiglitazone (10 μM; Cayman Chemical), or bleomycin (1 μM; APP Pharmaceuticals) for 7 days.
Immunocytochemical analysis
At the end of each experiment, cells were fixed in 4% PFA and incubated with goat anti–type I collagen (1:200; SouthernBiotech), rabbit antiadiponectin (1:250; Abcam), rabbit anti–α-SMA (1:1,000; Abcam), or goat anti–perilipin A (1:50; Abcam) primary antibodies for 60 minutes. Species-appropriate secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (all from Invitrogen) were used to localize primary antibody binding. Negative controls were stained without primary antibody. Nuclei were detected using DAPI. Cells were visualized using a Nikon A1 confocal laser scanning microscope.
Immunohistochemical analysis
Frozen or paraffin-embedded sections of lesional skin were incubated with goat anti–perilipin A (1:250; Abcam), rabbit anti–α-SMA (1:200; Abcam), rabbit anti-S100A4 (1:1,000; Abcam), rat anti-F4/80 (1:1,000; eBioscience), goat anti-CD31 (1:500; Santa Cruz Biotechnology), rat anti-CD45 (1:250; PharMingen), or rabbit anti–cleaved caspase 3 (1:50; Cell Signaling Technology) primary antibodies. Species-appropriate secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (Invitrogen) were used to localize primary antibody binding. Nuclei were detected using Hoechst 33342. Negative controls stained without primary antibody were used to confirm specificity.
For immunofluorescence analysis, tissue specimens were evaluated under a Nikon A1 confocal laser scanning microscope. The proportion of immunopositive cells was determined in a blinded manner by 2 independent observers (RGM and BK) by scoring cells in 5 randomly selected hpf areas throughout the dermis in 3–4 mice (n = 2 independent experiments). When quantifying myofibroblasts, cells with distinct capillary morphology were excluded from analysis. For quantification of F4/80 immunostaining, slides were first scanned using TissueFAX software (TissueGnostics), and both the intensity and percentage of immunopositive cells were determined in 10 randomly chosen regions within the intradermal adipose tissue layer, using HistoQuest software (TissueG-nostics). To quantify apoptosis, the number of cells positive for both cleaved caspase 3 and perilipin A were determined.
Adipocyte size and number
The number and size of intradermal adipocytes were evaluated in H&E-stained sections at 100× magnification. Adipocyte diameter (n = 100) was determined in 3 noncontiguous hpf areas per mouse (n = 3–4 mice per group) using ImageJ software, and the data were used to generate diameter–frequency histograms (20).
RNA isolation and real-time quantitative PCR (qPCR)
Skin samples were immersed in RNAlater (Ambion) and stored at −80°C. The samples were homogenized, and RNA was isolated using TRIzol reagent (Invitrogen) (7). In cell culture experiments, RNA was isolated using a Quick-RNA MiniPrep Kit (Zymo Research) and processed for microarray analysis. Real-time qPCR was performed using SYBR Green Master Mix (Applied Biosystems) in an ABI Prism 7900 sequence detection system (Applied Biosystems). The oligonucleotide primer sequences used for gene expression analysis are available upon request. All samples were normalized to GAPDH gene expression, and results are expressed as the fold change of Ct values (mean of 3 replicates) relative to controls, using the 2−ΔΔCt formula.
Western blot analysis
Whole cell lysates were isolated and subjected to electrophoresis in 4–15% Tris–glycine gradient gels. After protein transfer, membranes were treated with the blocking buffer followed by incubation with primary antibodies to type I collagen (1:400 dilution; Southern Biotech), α-SMA (1:5,000; Sigma), fibronectin (1:500; Calbiochem), fatty acid binding protein 4, perilipin (1:1,500; Abcam), PPARγ (1:200; Santa Cruz Biotechnology), or GAPDH (1:5,000; Invitrogen). Electrophoretic bands were visualized using enhanced chemiluminescence (Pierce Biotechnology) on a LAS-3000 Luminescent Image Analyzer (Fujifilm Medical Systems).
DNA microarray hybridization, data processing, and differential expression analysis
RNA was isolated at various time points and processed for hybridization using an Agilent SurePrint G3 Human Gene Expression 8x60K Microarray kit (product no. G4851A) (21). Quality control was performed using an Agilent 2100 Bioanalyzer; all samples had RNA integrity values of >9. Agilent Feature Extraction image analysis software version 10.7.3 was used to extract data from raw microarray image files. Microarray data were log2 lowess–normalized and filtered for probes with intensity of ≥1.5-fold over local background in the Cy3 or Cy5 channel. Expression values were multiplied by −1 to convert to log2 (Cy3:Cy5) ratios. After probes with >20% missing data were excluded, 41,589 probes passed the filtering criteria. The probes were median-centered across all arrays. Missing values were imputed based on the k nearest neighbor algorithm (k = 10) using Euclidean distance as the distance metric. Differentially expressed genes were selected by Significance Analysis of Microarrays analysis using a 2-class unpaired t-test. Expression data for 1,143 probes differentially expressed between untreated and TGFβ-treated samples were selected at a false discovery rate of 1.1%.
Statistical analysis
Results are presented as the mean ± SEM. For in vivo studies, Wilcoxon–Mann-Whitney test or Student’s t-test was used for comparisons between 2 groups, and analysis of variance was used for comparisons between multiple groups. The analyses were performed using GraphPad Prism 6. P values less than 0.05 were considered significant.
RESULTS
Loss of intradermal fat precedes the onset of dermal fibrosis
Loss of intradermal adipose tissue is consistently observed in lesional skin biopsy specimens from patients with SSc (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract) and patients with other forms of dermal fibrosis, as well as from mice with experimentally induced fibrosis. To evaluate the process of adipose tissue loss in fibrogenesis, a well-characterized model of dermal fibrosis was investigated (7). C57BL/6J mice challenged with daily subcutaneous injections of bleomycin showed a time-dependent progressive increase in dermal thickness and collagen deposition (Figure 1A; see also Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract). Dermal fibrosis was associated with marked attenuation of the intradermal adipose tissue layer (Figure 1A). Significantly, time-course studies revealed that attenuation of intradermal adipose tissue was evident prior to the appearance of dermal fibrosis. For instance, by day 5, >50% of the adipose tissue layer was lost (P = 0.02), whereas significant dermal thickening was not detected until day 14 (P = 0.002) (Figure 1B). These results were consistent across multiple experiments (n = 3). In lesional skin, a relatively early decline in the expression of adipogenic genes preceded the increase in the expression of fibrogenic genes (Figure 2; see also Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract).
Figure 1.

Loss of intradermal fat precedes dermal fibrosis in bleomycin-treated mice. C57BL/6J mice were given daily subcutaneous injections of bleomycin (bleo) or phosphate buffered saline (control) for up to 14 days. Skin specimens were obtained at the indicated time points. A, Masson’s trichrome–stained sections showing decreased intradermal adipose tissue on day 5 and increased collagen deposition on day 21 in bleomycin-treated mice. Bars = 100 μm. B, Changes in thickness of the dermis and intradermal adipose tissue over time in bleomycin-treated mice, as determined in 5 high-power fields (hpf)/mouse, showing that decreased intradermal adipose tissue thickness occurs prior to dermal thickening. Values are the fold change relative to control mice (n = 3–4 mice). Symbols represent individual mice; bars show the mean ± SEM. * = P ≤ 0.001 versus day 0. C, Numbers of intradermal adipocytes in the skin of bleomycin-treated mice, as counted in 2–3 hpf per tissue section. Values are the mean ± SEM (n = 2–3 mice/group). * = P ≤ 0.05 versus day 0. a and b, Representative hematoxylin and eosin–stained intradermal adipose tissue sections from control and bleomycin-treated mice, respectively, on day 5. Original magnification × 63. D, Changes in the size of adipocytes over time in control and bleomycin-treated mice, as determined in >200 adipocytes from 3–4 mice in each group. Epi = epidermis.
Figure 2.
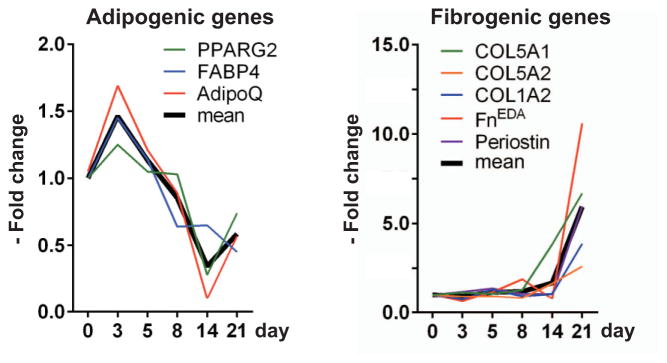
Gene expression changes during cutaneous fibrogenesis. C57BL/6J mice were given daily subcutaneous injections of bleomycin or phosphate buffered saline for up to 14 days. Skin tissue samples isolated from the areas of injection were harvested at the indicated time points, and mRNA levels were determined. Results were normalized to GAPDH and are representative of triplicate determinations (n = 3–4 mice/group). * = P ≤ 0.001 versus day 0. FnEDA = cellular fibronectin.
Changes in intradermal adipose tissue were examined by detailed morphometric analysis of the lesional skin. A significant decrease in both the number and size of intradermal adipocytes was apparent by day 5 (Figures 1C and D). These changes were progressive until day 14, after which partial regeneration was noted. Attenuation of intradermal adipose tissue in bleomycin-treated mice was not due to loss of body fat, because these changes were not observed in nonlesional skin (i.e., abdomen and thigh) (data not shown). Moreover, subcutaneous or perirenal fat depots showed no significant alterations. Despite the marked loss of intradermal fat in bleomycin-treated mice, levels of circulating adiponectin were comparable in bleomycin-treated mice and PBS-treated mice across all time points.
Bleomycin induced rapid accumulation of F4/80-positive macrophages in lesional skin, most prominently surrounding remnants of intradermal adipocytes, where they formed characteristic crown-like structures (results not shown). By day 3, a 4-fold increase in the number of macrophages (P = 0.003) was detected, peaking on day 8 and remaining significantly elevated for up to day 28 (data not shown). Double immunofluorescence staining with antibodies to cleaved caspase 3 and perilipin showed a significant increase in the number of apoptotic adipocytes within intradermal adipose tissue. Increased apoptosis was apparent by day 3 (24%), peaked on day 5 (37%), and then declined. These observations indicate that bleomycin induced a relatively early localized loss of intradermal adipose tissue that preceded the onset of dermal fibrosis and was accompanied by a transient increase in apoptosis and inflammatory cell infiltration.
Myofibroblasts populating the fibrotic dermis originate from adiponectin-positive intradermal progenitors
The extent of intradermal apoptosis observed in bleomycin-treated mice does not account for the progressive loss of intradermal adipose tissue. We therefore hypothesized that loss of intradermal adipose tissue might contribute to dermal fibrosis through an alternate process involving transdifferentiation. To examine whether intradermal adipocytic cells might give rise to myofibroblasts populating the fibrotic dermis, we generated adipocyte-specific lineage-tracing mice (Figure 3A). The 5.4-kb promoter fragment of the ap2 gene (aP2-Cre) has been thought to drive Cre recombinase expression specifically in adipocytes (18). However, aP2- Cre–transgenic mice showed prominent tdTomato reporter expression in multiple cell types throughout the dermis (see Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract) and therefore could not be used for adipocyte-specific lineage tracing. In contrast, AdipoP-Cre–transgenic mice showed robust tdTomato reporter expression that was restricted to the intradermal adipose tissue compartment (Figure 3B). Expression of tdTomato completely colocalized with the adipocyte marker perilipin, confirming the lineage specificity of the AdipoP promoter (19).
Figure 3.

Selective expression of adiponectin-Cre in adipocytes. A, Adiponectin-Cre–transgenic (AdipoP-Cre) mice were crossed with Ai14 (tandem dimer Tomato [tdTomato; tdT]) reporter mice to generate progeny expressing tdTomato fluorescent protein restricted to adipocytes. B, Skin harvested from the interscapular region of 6-week-old female AdipoP-Cre−;tdTomato+/f and AdipoP-Cre+;tdTomato+/f mice was immunostained with antibodies to perilipin (green). No tdTomato expression from the unrecombined Ai14 allele was observed in mice lacking Cre-recombinase (AdipoP-Cre−). In AdipoP-Cre+;tdTomato+/f mice, 100% colocalization of the tdTomato reporter with perilipin-labeled adipocytes was observed. Hoechst 33342 (blue) counterstained; bars = 50 μm. Dotted lines outline the dermis.
To trace the fate of intradermal adipocytes during cutaneous fibrogenesis, bleomycin was used to induce fibrosis in AdipoP-Cre–tdTomato–transgenic mice. By day 5 of bleomycin injections, substantial attenuation of the intradermal adipose tissue (mean ± SEM 34 ± 10%; P = 0.03) was evident. At this time point, the thickness of the dermis was unchanged (see Supplementary Figure 4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract), and no tdTomato-positive cells could be detected within the dermis (see Supplementary Figure 4). By day 8, multiple triple-positive (transitional) cells were observed within the intradermal adipose tissue that coexpressed the myofibroblast marker α-SMA and the adipocyte-specific marker perilipin, along with the lineage tracer tdTomato (Figure 4). At later time points (day 14), numerous cells double-positive for tdTomato and α-SMA but no longer expressing perilipin were observed in the lower dermis.
Figure 4.
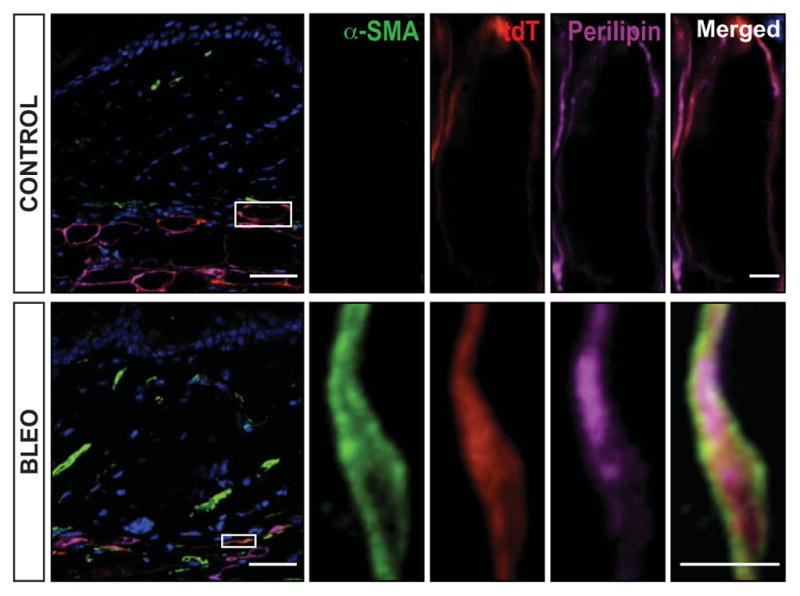
Triple-positive transitional cells are present in intradermal fat in early fibrogenesis. AdipoP-Cre+;tdTomato+/f mice were given daily subcutaneous injections of bleomycin (bleo) or phosphate buffered saline (control). Lesional skin harvested on day 8 was immunostained with antibodies to α-smooth muscle actin (α-SMA) (green) and perilipin (purple) and examined by confocal microscopy. Endogenous tdTomato label (red) was restricted to the intradermal fat layer. Numerous lineage-labeled adipocytes (red + purple) also expressed α-SMA. Hoechst 33342 (blue) counterstained; bars = 50 μm (inset = 5 μm). Boxed areas show higher-magnification views. Images are representative of 3 mice/group. See Figure 3 for other definitions.
On day 21, a significant increase in dermal thickness (P = 0.02) and an accumulation of densely packed collagen bundles were evident. Notably, a substantial proportion of spindle-shaped cells populating the fibrotic dermis were tdTomato positive (Figure 5A). These adipocyte-derived cells were localized primarily within the reticular dermis adjacent to the intradermal adipose tissue and failed to be recognized by antibodies to perilipin or markers for macrophages, lymphocytes, or endothelial cells (see Supplementary Figure 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract).
Figure 5.

Myofibroblasts populating the fibrotic dermis are derived from adipocytes. AdipoP-Cre+;tdTomato+/f mice were given daily subcutaneous injections of bleomycin (bleo) or phosphate buffered saline (control) for 14 days. Skin tissue isolated from the areas of injection was harvested on day 21 and immunostained with antibodies to α-smooth muscle actin (α-SMA). A, Representative split image photomicrographs showing immunolabeling for endogenous tdTomato (red) and α-SMA (green). Hoechst 33342 (blue) counterstained; bar = 20 μm. Dotted lines represent the border between the epidermis (epi) and dermis (bottom). B, Quantification of tdTomato-positive cells (top), α-SMA–positive cells (middle), and the production of double-positive cells in the dermis. Values are the mean ± SEM (n = 3–5 mice/treatment group) and are representative of 3 independent experiments. * = P < 0.01 versus control. hpf = high-power field (see Figure 3 for other definitions).
In striking contrast, virtually all tdTomato-positive cells within the dermis expressed α-SMA (Figure 5A) as well as the putative fibroblast marker S100A4 (see Supplementary Figure 6, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38990/abstract). These results indicate that myofibroblasts accumulating within the fibrotic dermis originate from adiponectin-positive intradermal progenitors. The results were consistent in multiple independent experiments with both female and male transgenic mice as well as young (mean ± SEM 10.9 ± 3.2 weeks) and old (22.6 ± 5.8 weeks) transgenic mice. The possibility that aberrant tdTomato reporter activation in fibroblasts might account for the presence of tdTomato-positive myofibroblasts in fibrotic dermis was examined using AdipoP-Cre–tdTomato–positive skin fibroblasts in culture. Incubation with bleomycin, TGFβ, or rosiglitazone for up to 7 days showed that although rosiglitazone induced robust induction of tdTomato expression, as expected, no tdTomato expression was detected in AdipoP-Cre–tdTomato–positive fibroblasts exposed to bleomycin or TGFβ (data not shown).
Transition of adipocytes into myofibroblasts ex vivo
To investigate how adipocyte phenotypes are modulated ex vivo, human adipose tissue–derived progenitor cells were used. Upon incubation in differentiation media for 10 days, these cells acquired characteristic adipocyte morphology, with a rounded shape and accumulation of prominent intracytoplasmic lipid droplets, and were immunopositive for both adiponectin and perilipin (Figure 6A). Incubation with TGFβ led to striking time-dependent changes in cell morphology (Figure 6A). After 24 hours, cells coexpressing both perilipin and α-SMA (transitional cells) could be observed. At later time points, progressive loss of adipocytic markers was accompanied by accumulation of intracellular type I collagen and expression of α-SMA–positive stress fibers characteristic of myofibroblasts (Figure 6A). Consistent results were obtained with cultures of adipocytes derived from 2 donors (Figure 6B).
Figure 6.

Transforming growth factor β (TGFβ) induces myofibroblast differentiation from adipocytes. Human subcutaneous adipose tissue–derived progenitor cells (ADSCs) were incubated in differentiation media for 10 days, followed by incubation with TGFβ (5 ng/ml) for up to 72 hours. A, Immunofluorescence analysis using antibodies to adiponectin (green) and type I collagen (Cng I; red) (top row) or perilipin (green) and α-smooth muscle actin (α-SMA) (red) (second row from top and bottom row). The 3 top rows show results at 72 hours, and the bottom row shows results at 0 and 24 hours. DAPI was used for nuclear staining (blue). Bars = 50 μm. B, Representative Western blots of whole cell lysates. ADSC 1 = donor 1; FnEDA = cellular fibronectin. C and D, Heatmaps showing the expression of adipogenic genes (C) and fibrogenic genes (D), organized by hierarchical clustering. Total RNA was hybridized to Agilent microarrays. Genes with expression levels above the mean are shown in red, and those with expression levels below the mean are shown in green. The first pair of columns (from left) show the mRNA profile of a sample and its technical replicate, and the second pair of columns show the profiles of a biologic replicate sample and its technical replicate.
Genome-wide expression profiling of these cells showed that TGFβ induced substantial reciprocal changes in the expression of fibrogenic (up-regulated) and adipogenic (down-regulated) genes (Figures 6C and D). Genes that were up-regulated in adipocytes in response to TGFβ included secreted Frizzled-related protein 1 (SFRP2), a Wnt family inhibitor; Wnt-5a (WNT5A), which is implicated in the regulation of fibroblast proliferation; microfibrillar-associated protein 4 (MFAP4); plasminogen activator inhibitor 1 (PAI1); Colα1(I) (COL1A1); Colα2(I) (COL1A2); Colα5(I) (COL5A1); tenascin C (TNC); and fibronectin 1 (FN1). Interestingly, among the most highly up-regulated genes were antiadipogenic transcription factors (nuclear receptor corepressor 2 [NCOR2] and Kruppel-like factor 7 [KLF7]). In contrast, expression of multiple adipogenic genes, including FABP5, LPIN1, EBF2, and AGT, was significantly down-regulated in TGFβ-treated cultures. These results underscore the phenotypic plasticity of fully differentiated human adipocytes that, in response to TGFβ, undergo morphologic and transcriptional reprogramming.
DISCUSSION
Myofibroblasts actively participate in physiologic and pathologic connective tissue remodeling (3). Persistent accumulation of α-SMA–positive myofibroblasts is prominent in SSc, but despite the pivotal role of myofibroblasts, their origin in fibrotic dermis is unknown (22). Cutaneous fibrosis in patients with SSc and in animal models of scleroderma is commonly accompanied by substantial loss of intradermal fat (6–15). A similar phenomenon is also observed in heritable and inducible forms of cutaneous fibrosis in mice. Nevertheless, despite its prominence in SSc and in experimentally induced fibrosis, the role of intradermal adipose tissue loss in dermal fibrosis remains unknown.
Adipocytes, which are distributed widely throughout the body, display remarkable functional pleiotropy and phenotypic plasticity (23). Intradermal adipose tissue represents a unique white adipose tissue depot that is distinct from inguinal, visceral, mesenteric, pericardial, and perirenal adipose tissue depots (24,25). Lying anatomically between the reticular dermis and skeletal muscle, the intradermal adipose tissue layer is composed of 6–8 parallel layers of relatively uniformly sized round adipocytes, with each containing a single large cytoplasmic lipid droplet. Although the origins, precise physiologic role, and secreted adipokine repertoire of intradermal adipose tissue remain poorly understood, there is emerging recognition of its importance in skin homeostasis and remodeling (26,27).
Using a combination of genetic and pharmacologic approaches to disrupt adipogenesis, Schmidt and Horsley recently demonstrated an essential functional role for intradermal adipocytes in wound healing (28). In the current study, we sought to characterize intradermal adipose tissue loss during cutaneous fibrogenesis and to investigate its sequelae in genetically engineered lineage-tracing mice. Using a TGFβ and Wnt/β-catenin–dependent model of cutaneous fibrosis (7), we observed that loss of intradermal adipose tissue preceded the onset of dermal fibrosis. Intradermal adipose tissue loss was accompanied by early down-regulation of PPARγ and other adipocyte-associated genes, followed by subsequent expression of myofibroblast-associated genes. Adipose tissue apoptosis was an early and transient event that did not fully account for progressive loss of intradermal adipose tissue. Cell fate mapping indicated that bleomycin caused recruitment of tdTomato-positive mesenchymal cells that accounted for a great majority of the myofibroblasts populating the fibrotic dermis. Complementary ex vivo studies with human subcutaneous adipocytes indicated that TGFβ induced the loss of adipocytic characteristics and reciprocal acquisition of myofibroblast features in these cells. In view of the parallels between the phenotypic cell fate transitions we describe and the much-studied EMT and endoMT, we refer to this process as AMT and propose that it might play an important role in cutaneous fibrogenesis.
Adipogenesis is a dynamic process, and its dysregulation is implicated in obesity, insulin resistance, and the metabolic syndrome (24). Adipocytes store lipids for energy and provide thermal and structural support. The complex roles of adipose tissue as a progenitor cell niche and endocrine organ with potent cell-autonomous and paracrine effects on cancer, inflammation, and tissue remodeling are increasingly being recognized (17,24). Loss of adipose tissue is linked with pathologic tissue fibrosis in a variety of human diseases and in experimental animal models. In addition to SSc, this phenomenon has been reported in LMNA-associated inherited laminopathies and other mutations that underlie generalized and familial partial lipodystrophies (29). Adipose tissue loss is also associated with fibrosis in acquired lipodystrophies secondary to panniculitis, autoimmune diseases, restrictive dermopathy, scarring alopecia, anorexia, and cancer cachexia, as well as antiviral therapy with protease inhibitors (27,30–32).
The link between adipose tissue loss and fibrosis is particularly striking in animal models. Examples include mice with adipocyte-targeted deletion of PPARγ resulting in lipodystrophy associated with dermal fibrosis (33) and mouse models of cancer cachexia characterized by fibrous replacement of adipose tissue (20,32). Transgenic mice with aP2-Cre–driven Wnt overexpression develop a scleroderma phenotype associated with progressive disappearance of intradermal adipocytes and their replacement by dense fibrosis (16). Similarly, ectopic expression of TGFβ using the PEPCK promoter Cre or of constitutively active β-catenin using either aP2-Cre or PPARγ-Cre resulted in replacement of white adipose tissue with fibrotic tissue (34,35). Corollary studies demonstrated that cutaneous fibrosis in mice is commonly accompanied by atrophy of the adjacent intradermal adipose tissue. For instance, dermal fibrosis in mice harboring mutant Fbn1, which causes stiff skin syndrome, is accompanied by marked loss of intradermal adipose tissue (13). Intradermal adipose tissue loss is also prominent in mice transgenic for Col1a2-TGFβRICA as well as in tsk2 mutant mice (10,15). Taken together with the findings of our current study, these results highlight a functional relationship linking intradermal adipose tissue with myofibroblast differentiation and cutaneous fibrosis. Potential mechanisms include depletion of adipose tissue–derived paracrine factors such as adiponectin and endotrophin that directly influence fibroblast proliferation, differentiation, and function, and phenotypic transformation of adipocytic cells into matrix-producing myofibroblasts.
To examine the possibility that intradermal adipocytic cells might serve as myofibroblast progenitors, we used transgenic lineage-tracing mice to track the fate of genetically labeled mature adipocytes. We first generated reporter mice by crossing aP2-Cre mice with tdTomato-transgenic mice (18). Unexpectedly, these mice showed widespread tdTomato expression in the skin that was not confined to intradermal adipose tissue. This lack of specificity of the putative adipocyte marker aP2 has impeded progress in understanding the regulation of adipose tissue development (36–38). In contrast, AdipoP-Cre–transgenic mice showed efficient and highly specific labeling of intradermal adipocytic cells (19,37).
In skin from untreated and PBS-treated AdipoP-Cre–transgenic mice, tdTomato-labeled cells were observed only within the intradermal adipose tissue compartment and showed complete colocalization with the adipocyte marker perilipin. In contrast, bleomycin treatment resulted in progressive accumulation of tdTomato-labeled perilipin-negative cells throughout the fibrotic dermis. Triple-positive “transitional cells” expressing tdTomato along with perilipin and α-SMA could be observed transiently within the intradermal adipose tissue and subsequently lost perilipin expression during accumulation within the dermis. Significantly, 80% of tdTomato-positive mesenchymal cells within the dermis of bleomycin-treated mice were positive for α-SMA, identifying them as myofibroblasts. In contrast, none of those cells expressed markers of macrophages, lymphocytes, or endothelial cells. Because adiponectin is a marker specific for mature adipocytes (19,36,37,39), our findings suggest that myofibroblasts within the fibrotic dermis were derived from intradermal adipocytic cells that had undergone phenotypic transformation. It is also plausible that adiponectin-positive progenitors differentiate into multipotent mesenchymal cells, which then redifferentiate into myofibroblasts.
Our findings are broadly consistent with other progenitor cell fate switches implicated in skin fibrogenesis, such as the emergence of myofibroblasts from SOX2-positive progenitors in bleomycin-treated mice (40) or ADAM-12–positive progenitors in a model of acute skin injury (41), and of activated fibroblasts originating from PPARγ-labeled adipocytic cells harboring constitutively active β-catenin in transgenic mice in which cutaneous fibrosis and lipoatrophy develop spontaneously (35). Our ex vivo studies confirm that adipocytic cells undergo reprogramming into activated myofibroblasts upon treatment with TGFβ. Such adipocyte plasticity also occurs during wound healing and within fibrous stroma in breast cancer (42,43). Moreover, phenotypic transformation of terminally differentiated cells via EMT and similar processes has been implicated in fibrosis affecting the lungs, liver, kidneys, bone marrow, and spinal cord (44–47). Although comparable cell fate switches in SSc have not been well studied to date, our results provide strong evidence for an adipocyte origin of activated myofibroblasts in fibrotic dermis.
Taken together, our observations suggest that the process of AMT, a phenotypic transition akin to EMT and endoMT, might play a previously unrecognized important role in cutaneous fibrosis. In view of the similarities in cutaneous pathology in mice with bleomycin-induced scleroderma and in patients with SSc, our findings may have significant implications for understanding the pathogenesis of and therapy for this still untreatable disease.
Supplementary Material
Acknowledgments
We thank the Northwestern University Cell Imaging Facility and Northwestern University Mouse Histology and Phenotyping Laboratory, which were supported in part by the NIH (National Cancer Institute grant P30-CA-060553).
Supported by the NIH (grants OD-010945 and CA-060553 to Dr. Tourtellotte and grant AR-42309 to Dr. Varga). Dr. Whitfield’s work was supported by the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases [NIAMS] grant P30-AR-061271). Dr. Graham’s work was supported in part by the NIH (NIAMS grant T32-AR-060710).
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Tourtellotte and Varga had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Marangoni, Korman, Wei, Whitfield, Scherer, Tourtellotte, Varga.
Acquisition of data. Marangoni, Korman, Wood, Graham, Whitfield, Tourtellotte, Varga.
Analysis and interpretation of data. Marangoni, Korman, Wei, Wood, Graham, Whitfield, Scherer, Tourtellotte, Varga.
Dr. Whitfield has received consulting fees, speaking fees, and/or honoraria from GlaxoSmithKline, EMD Serono, and Biogen Idec (less than $10,000 each).
References
- 1.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 2.Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–50. [PubMed] [Google Scholar]
- 3.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–55. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504:277–81. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–80. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleischmajer R, Damiano V, Nedwich A. Scleroderma and the subcutaneous tissue. Science. 1971;171:1019–21. doi: 10.1126/science.171.3975.1019. [DOI] [PubMed] [Google Scholar]
- 7.Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-γ. Am J Pathol. 2009;174:519–33. doi: 10.2353/ajpath.2009.080574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Servettaz A, Goulvestre C, Kavian N, Nicco C, Guilpain P, Chereau C, et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol. 2009;182:5855–64. doi: 10.4049/jimmunol.0803705. [DOI] [PubMed] [Google Scholar]
- 9.Stawski L, Han R, Bujor AM, Trojanowska M. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther. 2012;14:R194. doi: 10.1186/ar4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, et al. Postnatal induction of transforming growth factor β signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007;56:334–44. doi: 10.1002/art.22328. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, McCormick LL, Desai SR, Wu C, Gilliam AC. Murine sclerodermatous graft-versus-host disease, a model for human scleroderma: cutaneous cytokines, chemokines, and immune cell activation. J Immunol. 2002;168:3088–98. doi: 10.4049/jimmunol.168.6.3088. [DOI] [PubMed] [Google Scholar]
- 12.Maurer B, Distler JH, Distler O. The Fra-2 transgenic mouse model of systemic sclerosis. Vascul Pharmacol. 2013;58:194–201. doi: 10.1016/j.vph.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503:126–30. doi: 10.1038/nature12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manne J, Markova M, Siracusa LD, Jimenez SA. Collagen content in skin and internal organs of the tight skin mouse: an animal model of scleroderma. Biochem Res Int. 2013;2013:436053. doi: 10.1155/2013/436053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christner PJ, Peters J, Hawkins D, Siracusa LD, Jimenez SA. The tight skin 2 mouse: an animal model of scleroderma displaying cutaneous fibrosis and mononuclear cell infiltration. Arthritis Rheum. 1995;38:1791–8. doi: 10.1002/art.1780381212. [DOI] [PubMed] [Google Scholar]
- 16.Wei J, Melichian D, Komura K, Hinchcliff M, Lam AP, Lafyatis R, et al. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum. 2011;63:1707–17. doi: 10.1002/art.30312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18:470–7. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–7. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang ZV, Deng Y, Wang QA, Sun K, Scherer PE. Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology. 2010;151:2933–9. doi: 10.1210/en.2010-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bing C, Russell S, Becket E, Pope M, Tisdale MJ, Trayhurn P, et al. Adipose atrophy in cancer cachexia: morphologic and molecular analysis of adipose tissue in tumour-bearing mice. Br J Cancer. 2006;95:1028–37. doi: 10.1038/sj.bjc.6603360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PloS One. 2008;3:e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol. 2012;8:42–54. doi: 10.1038/nrrheum.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12:722–34. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Driskell RR, Jahoda CA, Chuong CM, Watt FM, Horsley V. Defining dermal adipose tissue. Exp Dermatol. 2014;23:629–31. doi: 10.1111/exd.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berry DC, Stenesen D, Zeve D, Graff JM. The developmental origins of adipose tissue. Development. 2013;140:3939–49. doi: 10.1242/dev.080549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rivera-Gonzalez G, Shook B, Horsley V. Adipocytes in skin health and disease. Cold Spring Harb Perspect Med. 2014;4:a015271. doi: 10.1101/cshperspect.a015271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt BA, Horsley V. Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Development. 2013;140:1517–27. doi: 10.1242/dev.087593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bereziat V, Cervera P, Le Dour C, Verpont MC, Dumont S, Vantyghem MC, et al. the Lipodystrophy Study Group. LMNA mutations induce a non-inflammatory fibrosis and a brown fat-like dystrophy of enlarged cervical adipose tissue. Am J Pathol. 2011;179:2443–53. doi: 10.1016/j.ajpath.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bing C, Trayhurn P. New insights into adipose tissue atrophy in cancer cachexia. Proc Nutr Soc. 2009;68:385–92. doi: 10.1017/S0029665109990267. [DOI] [PubMed] [Google Scholar]
- 31.Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–34. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 32.Karnik P, Tekeste Z, McCormick TS, Gilliam AC, Price VH, Cooper KD, et al. Hair follicle stem cell-specific PPARγ deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243–57. doi: 10.1038/jid.2008.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S, Huang LW, Snow KJ, Ablamunits V, Hasham MG, Young TH, et al. A mouse model of conditional lipodystrophy. Proc Natl Acad Sci U S A. 2007;104:16627–32. doi: 10.1073/pnas.0707797104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-β1 transgenic mice. J Clin Invest. 1997;100:2697–713. doi: 10.1172/JCI119815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeve D, Seo J, Suh JM, Stenesen D, Tang W, Berglund ED, et al. Wnt signaling activation in adipose progenitors promotes insulin-independent muscle glucose uptake. Cell Metab. 2012;15:492–504. doi: 10.1016/j.cmet.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang S, Kong X, Rosen ED. Adipocyte-specific transgenic and knockout models. Methods Enzymol. 2014;537:1–16. doi: 10.1016/B978-0-12-411619-1.00001-X. [DOI] [PubMed] [Google Scholar]
- 37.Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–74. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang QA, Scherer PE, Gupta RK. Improved methodologies for the study of adipose biology: insights gained and opportunities ahead. J Lipid Res. 2014;55:605–24. doi: 10.1194/jlr.R046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeffery E, Berry R, Church CD, Yu S, Shook BA, Horsley V, et al. Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte. 2014;3:206–11. doi: 10.4161/adip.29674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S, Herault Y, Pavlovic G, Leask A. Skin progenitor cells contribute to bleomycin-induced skin fibrosis. Arthritis Rheumatol. 2014;66:707–13. doi: 10.1002/art.38276. [DOI] [PubMed] [Google Scholar]
- 41.Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12+ perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18:1262–70. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- 42.Bochet L, Lehuede C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73:5657–68. doi: 10.1158/0008-5472.CAN-13-0530. [DOI] [PubMed] [Google Scholar]
- 43.Desai VD, Hsia HC, Schwarzbauer JE. Reversible modulation of myofibroblast differentiation in adipose-derived mesenchymal stem cells. PloS One. 2014;9:e86865. doi: 10.1371/journal.pone.0086865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Ann Rev Pathol. 2013;8:241–76. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–83. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–42. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 47.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–53. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.