

Abstract
Rosiglitazone is an insulin-sensitizing thiazolidinedione (TZD) that activates the transcription factor peroxisome proliferator-activated receptor gamma (PPARγ). Although rosiglitazone effectively treats type II diabetes mellitus (T2DM), it carries substantial complications, including increased fracture risk. This predisposition to fracture is consistent with the fact that PPARγ preferentially promotes formation of adipocytes at the cost of osteoblasts. Rosiglitazone-activated PPARγ, however, also stimulates osteoclast formation. A new TZD analog with low affinity for binding and activation of PPARγ but whose insulin-sensitizing properties mirror those of rosiglitazone has been recently developed. Because of its therapeutic implications, we investigated the effects of this new TZD analog (MSDC-0602) on skeletal homeostasis, in vitro and in vivo. Confirming it activates the nuclear receptor in osteoclasts, rosiglitazone enhances expression of the PPARγ target gene, CD36. MSDC-0602, in contrast, minimally activates PPARγ and does not alter CD36 expression in the bone-resorptive cells. Consistent with this finding, rosiglitazone increases receptor activator of NF-κB ligand (RANKL)-induced osteoclast differentiation and number, whereas MSDC-0602 fails to do so. To determine if this new TZD analog is bone sparing, in vivo, we fed adult male C57BL/6 mice MSDC-0602 or rosiglitazone. Six months of a rosiglitazone diet results in a 35% decrease in bone mass with increased number of osteoclasts, whereas that of MSDC-0602–fed mice is indistinguishable from control. Thus, PPARγ sparing eliminates the skeletal side effects of TZDs while maintaining their insulin-sensitizing properties.
Keywords: TYPE II DIABETES MELLITUS, THIAZOLIDINEDIONE, OSTEOCLASTS, BONE HISTOMORPHOMETRY, BONE μCT
Introduction
Type II diabetes mellitus (T2DM) is characterized by resistance to insulin and its reduced secretion. Thiazolidinediones (TZDs) are insulin sensitizers that preserve β-cell function and are thus effective in treating this disease.(1) These drugs are high-affinity ligands for peroxisome proliferator-activated receptor gamma (PPARγ), a member of the nuclear receptor superfamily of transcription factors.(2) PPARγ is highly expressed in adipose tissue and regulates transcriptional events mediating adipogenesis, lipid metabolism, inflammation, and metabolic homeostasis.(3,4) The antidiabetic actions of TZDs are believed mediated via PPARγ,(5) but TZD activation of this transcription factor is associated with substantial side effects, including weight gain, fluid retention, and predisposition to fracture.
Bone is constantly remodeled by tethering of the activities of osteoclasts and osteoblasts. Osteoclasts arise from hematopoietic progenitors of monocyte/macrophage lineage, which acquire the bone-resorptive phenotype under the influence of receptor activator of NF-κB ligand (RANKL).(6) Osteoblasts, on the other hand, are derived from mesenchymal progenitors.(7) Bone mass is stable in physiologic conditions because the activities of the two cells are balanced, whereas in osteoporosis bone resorption outpaces formation.(8)
CD36 is a membrane glycoprotein present on many cells including macrophages. It functions as a scavenger receptor by recognizing specific lipids,(9,10) thus regulating their accumulation in phagocytic cells.(11) Recognition of endogenous lipids by CD36 also plays a role in IL-4-induced fusion of macrophages but not in osteoclast formation.(12) Importantly in the context of the present exercise, CD36 is a PPARγ target gene because ligand activation of the nuclear receptor induces CD36 expression in myeloid cells.(11,13,14) Osteoblasts and adipocytes are derived from a common mesenchymal precursor whose commitment is dictated by PPARγ. Specifically, the transcription factor promotes adipogenesis at the cost of osteogenesis.(15–17) PPARγ is also expressed in osteoclast precursors, and its activation has a stimulatory effect on formation of the bone-resorptive cell, an event mediated by the AP-1 transcription factor, c-fos.(18) Thus, TZDs may negatively impact bone mass by both reducing formation and enhancing resorption. Although the contribution of each component is unclear, these observations are in keeping with the experimental bone loss induced by the drugs and, most importantly, the enhanced fracture risk experienced by TZD-treated patients.(18–24)
The concept that the beneficial effects of TZDs on the metabolic syndrome are mediated by PPARγ activation has recently been challenged.(25) This hypothesis prompted development of a novel TZD analog (MSDC-0602) with low affinity for the transcription factor. Despite failure to meaningfully activate PPARγ, MSDC-0602 improves multi-organ insulin sensitivity, adipose tissue inflammation, hepatic lipogenesis, and gluconeogenesis in obese mice as effectively as pioglitazone and rosiglitazone.(25) Given the negative effects of PPARγ activation on bone mass, we hypothesized that MSDC-0602 will also reduce the osteopenic effects of TZDs. In fact, we find that MSDC-0602 does not activate PPARγ in osteoclasts and unlike rosiglitazone does not accelerate osteoclast differentiation or enhance their number in vitro. More importantly, MSDC-0602 does not promote bone loss, whereas rosiglitazone significantly diminishes skeletal mass by inducing osteoclastogenesis while not effecting osteoblast function in vivo. Thus, the skeletal complications of TZDs that are mediated by enhanced resorption may be avoided by insulin-sensitizing, PPARγ-independent drugs.
Materials and Methods
Materials
MSDC-0602 was provided by Metabolic Solutions Development Company (Kalamazoo, MI, USA). Rosiglitazone was obtained from Cayman Chemical (Ann Arbor, MI, USA). Calcein and alizalin 3-methyl iminodiatic acid were purchased from Sigma (St. Louis, MO, USA).
Macrophage isolation and osteoclast culture
Isolated bone marrow macrophages (BMMs) were differentiated into mature, multinucleated osteoclasts as described.(26) Briefly, marrow was extracted from femurs and tibias of 6- to 8-week-old mice with α-MEM and cultured in α-MEM containing 10% fetal bovine serum (FBS), 100 IU/mL penicillin and streptomycin with 1:10 CMG (conditioned medium supernatant containing recombinant macrophage colony-stimulating factor [M-CSF]) in bacterial dishes.(21) For osteoclast generation, 1.5 × 104 cells were cultured in 500 μL α-MEM containing 10% FBS and various amounts of RANKL, 1:50 CMG and vehicle (DMSO), rosiglitazone (1 μM), MSDC-0602 (1 μM), or MSDC-1473 (1 μM) in 48-well tissue culture plates for 5 days. Cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) activity (kit 387-A; Sigma).
Animals and treatment
Male C57BL/6 mice were obtained from the Jackson Laboratory. All mice were housed in the animal care unit of the Washington University School of Medicine and maintained according to the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care. All animal experimentation was approved by the Animal Studies Committee of Washington University School of Medicine.
For in vivo drug treatment, 6-month-old mice, average 31 g body weight, were divided into 3 groups of 10 each and respectively fed repelleted Purina rodent chow (C13513; Research Diets Inc., New Brunswick, NJ, USA) alone or containing 200 ppm rosiglitazone (C13844; Research Diets), or 331 ppm MSDC-0602 (C13845; Research Diets) for 6 months. The administered amount of MDSC-O602 provides ~30 mg/kg/d of the TZD, sufficient to normalize insulin sensitivity in obese mice.(25) Food intake and body weights of individual animals were monitored.
Serum biomarkers
At death, serum insulin was determined using the Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem Inc., Downers Grove, IL, USA). Serum osteocalcin was determined with the Mouse Osteocalcin EIA kit (Biomedical Technologies Inc., Stoughton, MA, USA).
Bone densitometry
Whole-body bone mineral density (BMD) and % fat of anesthetized, prostrate mice were assessed using dual-energy X-ray absorptiometry (DXA, PIXImus Lunar-GE, Madison, WI, USA). The DXA machine was calibrated daily. One investigator performed all scans.(27)
Micro–computed tomography (μCT) analysis
The trabecular volume in the distal femoral metaphysis was measured using a Scanco μCT40 scanner (Scanco Medical AG, Basserdorf, Switzerland). A threshold of 211 was used for evaluation of all scans, and 100 slices were analyzed, beginning with an image in which condyles and primary spongiosa were no longer visible.
Histology and histomorphometry
Mice were injected with calcein (20 mg/kg body weight). Fourteen days later, they were injected with alizalin 3-methyl iminodiatic acid (30 mg/kg body weight). Femurs and tibias were harvested after 2 days, fixed in 70% ethanol, and embedded in methylmethacrylate for sectioning. All histomorphometric parameters were assessed using BioQuant OsteoII (BioQuant Image Analysis Corporation, Nashville, TN, USA). Dynamic assessment of trabecular bone formation was determined based on dual calcein-alizarin labeling. Adjacent sections were stained for TRAP activity. Adipocyte number in bone marrow of tibia was measured on five different microscopic fields at 200× magnification.(20)
RNA isolation and quantitative real-time RT-PCR analysis
Immediately after euthanization, epididymal white adipose tissue (WAT) was dissected and tibias were cleaned of all remaining soft tissue. The tissue was snap-frozen in liquid nitrogen for storage at −80°C. WAT and bones were homogenized using a Bullet Blender (Next Advance Inc., Averill Park, NY, USA), and total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and purified using RNeasy kits (Qiagen, Valencia, CA, USA) with DNase I treatment, according to the manufacturer’s directions. An amount of 1 μg total RNA was reverse transcribed using an iScript cDNA Synthesis kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Real-time PCR was performed using the SYBR Green Master Mix kit and gene-specific primers. Quantitative PCR reaction was performed on ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA). All reactions were performed in triplicate, and relative mRNA levels were calculated by the comparative threshold cycle method using cyclophilin as an internal control. The primer sequences used in quantitative real-time PCR analyses are provided in Table 1.
Table 1.
Primer Sequences for Quantitative Real-Time RT-PCR
| Gene name | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| CD36 | GCTGTGTTTGGAGGCATTCT | CCTTGATTTTGCTGCTGTTC |
| PGC-1β | CTCCAGGCAGGTTCAACCC | GGGCCAGAAGTTCCCTTAGG |
| β3-integrin | TTCGACTACGGCCAGATGATT | GGAGAAAGACAGGTCCATCAAGT |
| Cathepsin K | AGGCAGCTAAATGCAGAGGGTACA | AGCTTGCATCGATGGACACAGAGA |
| TRAP | CAGCTCCCTAGAAGATGGATTCAT | GTCAGGAGTGGGAGCCATATG |
| Calcitonin receptor | CAAGAACCTTAGCTGCCAGAG | CAAGCACGCGGACAATGTTG |
| Adiponectin | GTCAGTGGATCTGACGACACCAA | ATGCCTGCCATCCAACCTG |
| Cyclophilin | AGCATACAGGTCCTGGCATC | TTCACCTTCCCAAAGACCAC |
Statistical analysis
Statistical significance was determined by one-way ANOVA followed by post hoc analysis by Tukey-Kramer. Data are represented as mean ± SD.
Results
MSDC-0602 does not activate PPARγ in osteoclasts
MSDC-0602 has minimal stimulatory effect on PPARγ in hepatocytes.(11) To determine if the same occurs in osteoclasts, we exposed precursor cells, in the form of bone marrow macrophages (BMMs), to low-dose (25 ng/mL) RANKL and M-CSF in the presence of rosiglitazone, MSDC-0602, or carrier (DMSO) for 5 days, after which CD36 mRNA was measured by qPCR. As shown in Fig. 1A, rosiglitazone but not MSDC-0602 increases expression of the PPARγ target gene. Similarly, rosiglitazone stimulates expression of peroxisome proliferator-activated receptor-γ coactivator 1β (PGC-1β), a PPARγ-induced co-activator, whereas MSDC-0602 has no such effect(28,29) (Fig. 1B). Thus, despite its ability to normalize glucose homeostasis as effectively as rosiglitazone,(25) MSDC-0602 does not activate PPARγ in osteoclasts.
Fig. 1.
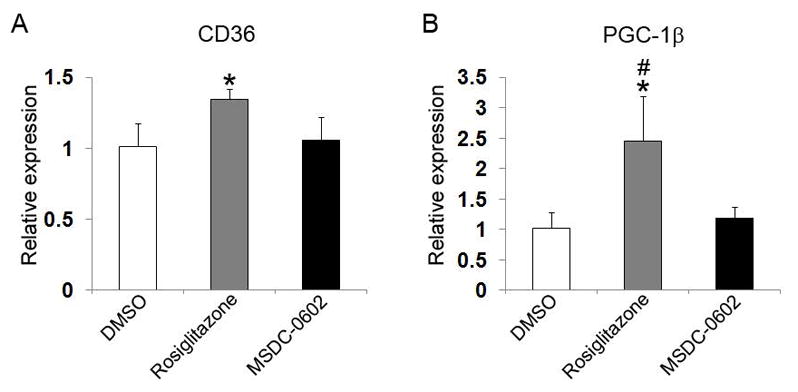
MSDC-0602 does not activate PPARγ. (A) BMMs were cultured in M-CSF and low-dose RANKL and treated with vehicle (DMSO) or TZDs for 5 days. CD36 was determined by qPCR. (B) BMMs were cultured in M-CSF and low-dose RANKL and treated with vehicle (DMSO) or TZDs for 5 days. The PPARγ co-activator, PGC-1β, was determined by qPCR. *p < 0.05 versus DMSO; #p < 0.05 versus MSDC-0602.
MSDC-0602 does not influence osteoclast differentiation in vitro
Rosiglitazone stimulates osteoclast differentiation in a PPARγ-dependent manner.(18) To assess the osteoclastogenic properties of MSDC-0602, we again cultured BMMs with M-CSF and low-dose RANKL in the presence or absence of rosiglitazone or MSDC-0602. Five days later, the cells were stained for TRAP activity and osteoclastogenic marker mRNA measured. Rosiglitazone doubles osteoclast number and increases expression of β3 integrin, cathepsin K, TRAP, and calcitonin receptor fourfold (Fig. 2). MSDC-0602, on the other hand, has no impact on osteoclastogenesis.
Fig. 2.

MSDC-0602 does not affect osteoclast differentiation. (A) BMMs were cultured in M-CSF and RANKL and treated with vehicle (DMSO) or TZDs for 5 days. The cells were stained for TRAP activity. Scale bars = 200 μm. (B) Quantitation of TRAP-expressing multinucleated cells (MNCs) (more than three nuclei per cell) generated from BMMs treated with vehicle or TZDs for 5 days. (C) BMMs were cultured in M-CSF and RANKL and treated with vehicle or TZDs for 5 days. Osteoclast differentiation markers β3 integrin subunit, cathepsin K, TRAP, and calcitonin receptor were determined by qPCR. **p < 0.01 versus DMSO; ##p < 0.01 versus MSDC-0602.
MSDC-0602 does not promote bone loss
To determine the effects of MSDC-0602 in vivo, adult male C57BL/6 mice were fed a rodent chow diet, alone or supplemented with MSDC-0602 or rosiglitazone. Six months of feeding yielded no differences in average daily consumption of chow (Fig. 3A). Body weight increased approximately 11% in all groups (Fig. 3B, C). TZDs increase insulin sensitivity by stimulating expression of beneficial adipokines, and as previously noted(25) both rosiglitazone and MSDC-0602 enhanced adiponectin concentration in white adipose tissue (Fig. 3D). Although circulating insulin tended to diminish in both TZD-treated groups, the change was not significant relative to control (Fig. 3E).
Fig. 3.

Effects of TZDs on food intake, body weight, insulin levels, and adiponectin expression. (A) Average food intake per mouse per day during experiment. (B) Average body weight measured at euthanization. (C) Average change of body weight. (D) Adiponectin mRNA in epididymal WAT measured at euthanization. (E) Serum insulin measured at euthanization. White bars = control; gray bars = rosiglitazone; black bars = MSDC-0602. **p < 0.01 versus control.
Despite similar metabolic properties, rosiglitazone and MSDC-0602 have distinct skeletal effects. In keeping with its fracture-promoting properties, DXA analysis of whole-body bone mineral density (BMD) revealed that after 6 months of oral intake, rosiglitazone but not MSDC-0602 diminished total body BMD (Fig. 4A). Similarly, μCT analysis established that only rosiglitazone-fed mice exhibited a significant decline in trabecular bone volume and consistent changes in trabecular number, thickness, and separation (Fig. 4B, C). Similar changes were determined by histomorphometry (Fig. 4D, E). In keeping with the osteoporotic effects of rosiglitazone reflecting accelerated resorption, osteoclast number per mm bone surface and the percentage of bone surface covered by resorptive cells increased in rosiglitazone-treated mice. In contrast, MSDC-0602 produced no evidence of enhanced osteoclastogenesis. Although MSDC-0602 also increased marrow adipocytes approximately threefold, their number was considerably lower than that of rosiglitazone-treated mice. In contrast to the stimulatory impact of rosiglitazone on bone resorption, neither it nor MSDC-0602 effected bone formation as determined by histomorphometry and circulating levels of osteocalcin (Fig. 4F–I, Supplemental Fig. S1). Thus, rosiglitazone promotes skeletal loss via accelerated bone resorption, an event spared by a TZD, which fails to significantly activate PPARγ.
Fig. 4.

MSDC-0602 does not affect bone. Six-month-old mice were fed native chow or that supplemented with rosiglitazone or MSDC-0602. All determinations were made after 6 months. (A) Whole-body BMD. (B) μCT image of distal femur. (C) μCT determination of BV/TV, trabecular number, trabecular thickness, and trabecular separation of distal femoral. (D) TRAP-stained histological sections of proximal tibia of control, rosiglitazone-treated, and MSDC-0602-treated mice. (E) Histomorphometrically determined BV/TV, total number of osteoclasts normalized to bone surface (Oc.N/BS), the percentage of bone surface to which osteoclasts are juxtaposed (Oc.S/BS), and adipocyte number/field. (F–H) Mice were administered time-spaced courses of calcein and alizarin. Bone formation rate (BFR) and mineral apposition rate (MAR) were histomorphometrically quantitated. (I) Serum osteocalcin determination. White bars = control; gray bars = rosiglitazone; black bars = MSDC-0602. **p < 0.01 versus control; *p < 0.05 versus control; ##p < 0.01 versus rosiglitazone; #p < 0.05 versus <zaq;3>. Scale bars = 1 mm (B); 500 μm (D, top panels); 50 μm (D, bottom panels, and F).
Discussion
PPARγ is a nuclear receptor that exerts its transcriptional effects by complexing RXRα on specific gene promoters. It primarily targets adipocytes, accelerating their differentiation and inclusion of free fatty acids and triglycerides.(30) In T2DM, fat fails to store these lipogenic molecules, which incorporate into liver and muscle, thereby prompting insulin resistance. This model is buttressed by the appearance of lipodystrophy and impaired glucose homeostasis in states of PPARγ dysfunction.(31)
Decades ago, TZDs were found to promote insulin sensitivity by enigmatic mechanisms.(2,32,33) Subsequent discoveries established that TZDs, such as rosiglitazone, normalize glucose homeostasis by accelerating adipocyte differentiation and stimulating synthesis and release of insulin-sensitizing adipokines such as adiponectin.(30) Because TZDs bind and transcriptionally activate PPARγ, it has been assumed that the insulin-sensitizing effects of the drugs are mediated via the nuclear receptor.(34,35) Recent evidence indicates, however, that the antidiabetic effects of PPARγ-liganding drugs may not reflect their agonistic effects on the nuclear receptor. For example, PPARγ-binding compounds exist that fail to induce its transcriptional activity but retain antidiabetic actions, likely by blocking Cdk5-mediated phosphorylation of serine 273.(36,37) The insulin-sensitizing effects of these agents are unassociated with the adipogenic properties characteristic of TZDs. Furthermore, TZDs suppress inflammation and, importantly, normalize hyperglycemia and insulin sensitivity, independent of PPARγ recognition.(38)
TZDs bind to mitochondrial membranes, and the drugs’ PPARγ-independent properties are likely mediated by altered mitochondrial function via pyruvate transport.(39,40) Thus, there is uncertainty regarding the means by which PPARγ and TZDs exert their insulin-sensitizing effects and if, in fact, they are mechanistically related. Furthermore, these observations challenge the concept that the antidiabetic properties of TZDs reflect their activation of PPARγ.
Recognition of the likelihood that many of the advantageous effects of TZDs are likely independent of PPARγ and that, to a significant degree, their side effects reflect PPARγ activation prompted development of TZDs, which recognize the nuclear receptor with low affinity and thus fail to activate it while retaining beneficial metabolic properties. Such an agent, MSDC-0602, improves multi-organ insulin sensitivity in obese mice, including suppression of hepatolipogenesis and gluconeogenesis.(25) Because of evidence suggesting the negative effects of TZDs on the skeleton are also mediated by PPARγ, we hypothesized that MSDC-0602 would obviate this complication.
To address this issue, we fed mice an unaltered chow diet or that supplemented with rosiglitazone or MSDC-0602. We chose C56/BL6 mice because they exhibit strain-specific sensitivity to rosiglitazone-induced bone loss.(41) We found a 6-month diet of rosiglitazone causes a 35% loss of trabecular bone, which is greater than that noted in other studies, a difference we attribute to the substantially longer duration of our study.(13,14,19,21,41) In contrast, quantities of MSDC-0602 that promote insulin sensitivity have no impact on DXA- or μCT-determined skeletal mass or parameters of trabecular bone architecture. Our animals exhibit no change in body weight regardless of diet. In keeping with its promotion of insulin sensitivity, MSDC-0602 is as effective as rosiglitazone in inducing adiponectin expression in these non-obese mice.(42,43)
Bone mass reflects the relative activities of osteoclasts and osteoblasts, both of which are influenced by PPARγ. In fact, conditional deletion of PPARγ in osteoclast lineage cell arrests bone resorption, resulting in osteopetrosis.(18) Although rosiglitazone stimulates PPARγ in BMMs, as evidenced by expression of CD36 and the co-activating protein PGC1β, MSDC-0602 has no such effect on these cells, suggesting the PPARγ-sparing TZD may not induce osteoclastogenesis. In contrast to the osteoclast-differentiating effects of rosiglitazone, such proved to be the case, in vitro and in vivo.
PPARγ promotes osteoclast formation in a cell-autonomous manner, but there is controversial evidence that its activation by TZDs does so, indirectly, by stimulating osteoblast lineage cells to express RANKL.(20) More compelling, however, are experiments indicating PPARγ governs bone formation by regulating differentiation of mesenchymal precursors common to osteoblasts and adipocytes.(15) Thus, the nuclear receptor dictates differentiation of these cells into fat at the cost of osteoblastogenesis. It is, therefore, postulated that the predisposition to fracture induced by TZDs reflects, at least in part, suppression of bone formation. In fact, PPARγ overexpression, specifically in osteoblasts, lowers bone mass and enhances estrogen-deficient osteoporosis. Given the evidence that PPARγ suppresses osteoblastogenesis, we were surprised that rosiglitazone-induced bone loss, in our experiments, appears exclusively the result of accelerated resorption with no evidence of inhibited formation. This apparent discrepancy may reflect the controversy surrounding the mechanisms of TZD-suppressed bone formation. Although some find direct inhibition of osteoblasts, others maintain the drugs actually stimulate differentiation of the cell and exert their anti-osteogenic effects by promoting apoptosis of osteocytes and osteoblasts.(19,22,44) Moreover, the state of differentiation of the targeted cell and the age of treated mice appear central to the inhibitory effects of TZDs. Specifically, partially differentiated osteoblast precursors and older mice are resistant to the anti-osteogenic effects of TZDs.(19,20) Because our mice were 1 year old at the time of euthanization, it is possible that suppressed formation contributed to the osteoporotic phenotype induced by rosiglitazone earlier in the course of therapy but not at termination of the experiment. This posture is in keeping with the enhanced marrow adipogenesis in rosiglitazone-treated mice, whereas the increase in those receiving MSDC-0602 is much less substantial. In any event, our findings are consistent with the hypothesis that TZD-induced bone loss in older individuals is predominantly an osteoclast-mediated event.
Our data support the concept that TZD-induced bone loss and enhanced insulin resistance are mechanistically distinct. The fact that MSDC-0602, which fails to substantially activate PPARγ yet normalizes the metabolic defects of obese mice, does not carry the osteoporotic complications of rosiglitazone indicates that the beneficial effects of TZDs occur by a PPARγ-independent mechanism such as mitochondrial pyruvate transport. Alternatively, rosiglitazone-induced bone loss is mediated via activation of the nuclear receptor. Thus, the availability of TZDs, which minimally activate PPARγ, hold promise that diabetic patients may be treated with this family of drugs without the fracture-promoting complications of the parent compounds.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 AR032788, R01 AR057037, R01 AR046523 and P30 AR057235.
Footnotes
Disclosures
JRC is the president and chief scientific officer of Metabolic Solutions Development Company. All other authors state that they have no conflicts of interest.
Additional Supporting Information may be found in the online version of this article.
References
- 1.DeFronzo RA, Abdul-Ghani MA. Preservation of beta-cell function: the key to diabetes prevention. J Clin Endocrinol Metab. 2011;96(8):2354–66. doi: 10.1210/jc.2011-0246. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270(22):12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 3.Ahmadian M, Suh JM, Hah N, et al. PPAR gamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–66. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355–61. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 5.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARgamma agonists: time for a reassessment. Trends Endocrinol Metab. 2012;23(5):205–15. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170(2):427–35. doi: 10.2353/ajpath.2007.060834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 8.Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457–84. doi: 10.1146/annurev.pathmechdis.3.121806.151431. [DOI] [PubMed] [Google Scholar]
- 9.Collot-Teixeira S, Martin J, McDermott-Roe C, Poston R, McGregor JL. CD36 and macrophages in atherosclerosis. Cardiovasc Res. 2007;75(3):468–77. doi: 10.1016/j.cardiores.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 10.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2(72):re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7(1):48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 12.Helming L, Winter J, Gordon S. The scavenger receptor CD36 plays a role in cytokine-induced macrophage fusion. J Cell Sci. 2009;122(Pt 4):453–9. doi: 10.1242/jcs.037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–52. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 14.Ballesteros I, Cuartero MI, Pradillo JM, et al. Rosiglitazone-induced CD36 up-regulation resolves inflammation by PPARgamma and 5-LO-dependent pathways. J Leukoc Biol. 2014;95(4):587–98. doi: 10.1189/jlb.0613326. [DOI] [PubMed] [Google Scholar]
- 15.Akune T, Ohba S, Kamekura S, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113(6):846–55. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barak Y, Nelson MC, Ong ES, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4(4):585–95. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 17.Wan Y. PPARgamma in bone homeostasis. Trends Endocrinol Metab. 2010;21(12):722–8. doi: 10.1016/j.tem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13(12):1496–503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- 19.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146(3):1226–35. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 20.Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148(6):2669–80. doi: 10.1210/en.2006-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Pan LC, Simmons HA, et al. Surface-specific effects of a PPARgamma agonist, darglitazone, on bone in mice. Bone. 2006;39(4):796–806. doi: 10.1016/j.bone.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Soroceanu MA, Miao D, Bai XY, Su H, Goltzman D, Karaplis AC. Rosiglitazone impacts negatively on bone by promoting osteoblast/osteocyte apoptosis. J Endocrinol. 2004;183(1):203–16. doi: 10.1677/joe.1.05723. [DOI] [PubMed] [Google Scholar]
- 23.Sottile V, Seuwen K, Kneissel M. Enhanced marrow adipogenesis and bone resorption in estrogen-deprived rats treated with the PPARgamma agonist BRL49653 (rosiglitazone) Calcif Tissue Int. 2004;75(4):329–37. doi: 10.1007/s00223-004-0224-8. [DOI] [PubMed] [Google Scholar]
- 24.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145(1):401–6. doi: 10.1210/en.2003-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Vigueira PA, Chambers KT, et al. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor gamma-sparing thiazolidinedione. J Biol Chem. 2012;287(28):23537–48. doi: 10.1074/jbc.M112.363960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP. High dose M-CSF partially rescues the Dap12−/− osteoclast phenotype. J Cell Biochem. 2003;90(5):871–83. doi: 10.1002/jcb.10694. [DOI] [PubMed] [Google Scholar]
- 27.Craft CS, Zou W, Watkins M, et al. Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J Biol Chem. 2010;285(31):23858–67. doi: 10.1074/jbc.M110.113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii KA, Fumoto T, Iwai K, et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15(3):259–66. doi: 10.1038/nm.1910. [DOI] [PubMed] [Google Scholar]
- 29.Wei W, Wang X, Yang M, et al. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010;11(6):503–16. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 31.Duan SZ, Ivashchenko CY, Whitesall SE, et al. Hypotension, lipodystrophy, and insulin resistance in generalized PPARgamma-deficient mice rescued from embryonic lethality. J Clin Invest. 2007;117(3):812–22. doi: 10.1172/JCI28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kletzien RF, Foellmi LA, Harris PK, Wyse BM, Clarke SD. Adipocyte fatty acid-binding protein: regulation of gene expression in vivo and in vitro by an insulin-sensitizing agent. Mol Pharmacol. 1992;42(4):558–62. [PubMed] [Google Scholar]
- 33.Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med. 1994;331(18):1188–93. doi: 10.1056/NEJM199411033311803. [DOI] [PubMed] [Google Scholar]
- 34.Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet Med. 1999;16(3):179–92. doi: 10.1046/j.1464-5491.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- 35.Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev. 2002;18(Suppl 2):S10–5. doi: 10.1002/dmrr.249. [DOI] [PubMed] [Google Scholar]
- 36.Choi JH, Banks AS, Kamenecka TM, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477(7365):477–81. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi JH, Banks AS, Estall JL, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466(7305):451–6. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinstein DL, Spagnolo A, Akar C, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70(2):177–88. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 39.Bricker DK, Taylor EB, Schell JC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337(6090):96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Divakaruni AS, Wiley SE, Rogers GW, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci USA. 2013;110(14):5422–7. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ackert-Bicknell CL, Shockley KR, Horton LG, Lecka-Czernik B, Churchill GA, Rosen CJ. Strain-specific effects of rosiglitazone on bone mass, body composition, and serum insulin-like growth factor-I. Endocrinology. 2009;150(3):1330–40. doi: 10.1210/en.2008-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee B, Shao J. Adiponectin and energy homeostasis. Rev Endocr Metab Disord. 2014;15(2):149–56. doi: 10.1007/s11154-013-9283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saltiel AR. You are what you secrete. Nat Med. 2001;7(8):887–8. doi: 10.1038/90911. [DOI] [PubMed] [Google Scholar]
- 44.Bruedigam C, Eijken M, Koedam M, et al. A new concept underlying stem cell lineage skewing that explains the detrimental effects of thiazolidinediones on bone. Stem Cells. 2010;28(5):916–27. doi: 10.1002/stem.405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.