

Abstract
Drugs with prolonged, on-target residence time often show superior efficacy, yet general strategies for optimizing drug-target residence time are lacking. Here, we demonstrate progress toward this elusive goal by targeting a noncatalytic cysteine in Bruton's tyrosine kinase (BTK) with reversible covalent inhibitors. Utilizing an inverted orientation of the cysteine-reactive cyanoacrylamide electrophile, we identified potent and selective BTK inhibitors that demonstrate biochemical residence times spanning from minutes to 7 days. An inverted cyanoacrylamide with prolonged residence time in vivo remained bound to BTK more than 18 hours after clearance from the circulation. The inverted cyanoacrylamide strategy was further utilized to discover fibroblast growth factor receptor (FGFR) kinase inhibitors with residence times of several days, demonstrating generalizability of the approach. Targeting noncatalytic cysteines with inverted cyanoacrylamides may serve as a broadly applicable platform that facilitates “residence time by design”, the ability to modulate and improve the duration of target engagement in vivo.
Drug discovery programs historically have sought to optimize molecules by maximizing the thermodynamic affinity (i.e., Kd or IC50) of the drug-target interaction. Less attention has been given to drug residence time (τ)1, which is defined as the inverse of the off-rate (koff) such that τ = 1/koff. Recently, it has become clear that drug-target residence time, rather than affinity, often drives pharmacodynamic activity and disease efficacy in vivo2-4. Hence, residence time should be a key focus during the drug discovery process5-7. A current lack of systematic approaches to design molecules with slow off-rates has hindered efforts to discover drugs with prolonged residence time.
Inhibitors that form a reversible covalent bond with a noncatalytic cysteine of their target may provide a widely applicable strategy for obtaining prolonged residence time. An approach to discover such inhibitors was recently reported8,9. In this work, a cyanoacrylamide electrophile attached to a kinase-recognition scaffold was designed to form a covalent but fully reversible bond with Cys436 in the C-terminal kinase domain of RSK2. These cyanoacrylamide inhibitors provided sustained engagement of RSK2, with biochemical residence times up to 6 hours9. Despite these promising results, it remained unclear how residence time could be systematically modulated. Moreover, characterization of cyanoacrylamide-based kinase inhibitors has to date focused on biochemical and cellular systems, and whether such inhibitors demonstrate prolonged residence time in vivo after oral administration has not been established.
Reversible covalent drugs have at least two theoretical advantages over their irreversible counterparts. First, relative to reversible covalent drugs, drugs that rely on intrinsically irreversible chemistry (e.g., acrylamides) are more likely to form permanent covalent adducts10-12 with off-target proteins, including closely related targets (e.g., off-target kinases with a homologous cysteine) as well as unrelated targets with hyper-reactive cysteines13. Second, and equally important, reversible cysteine engagement may enable fine tuning of inhibitor residence time, a feature that would facilitate utilization not only in therapeutic applications requiring sustained target engagement, but also in applications where more rapid target disengagement is preferred2. Since many drug targets, including kinases14-16, have an accessible cysteine in their binding site, there is broad opportunity to discover reversible covalent inhibitors for use across many therapeutic areas.
Bruton's tyrosine kinase (BTK) contains a noncatalytic cysteine (Cys481) that has been targeted by several inhibitors17-23. Among these compounds, ibrutinib17 is particularly noteworthy and was recently approved for treatment of the B cell malignancies chronic lymphocytic leukemia24 and mantle cell lymphoma25. Selective BTK inhibitors have also shown activity in collagen-induced arthritis and other rodent models of inflammation17,19,26,27, providing a rationale for BTK inhibition in autoimmune diseases. Given that irreversible acrylamide-based kinase inhibitors, including ibrutinib, have been shown to form permanent covalent adducts with kinase and non-kinase off-target proteins28, it is not clear whether such compounds have the requisite selectivity profile for use in autoimmune diseases, which typically require chronic dosing and a high safety margin.
Here, we explored the possibility of engaging BTK Cys481 with reversible covalent inhibitors. Inverting the orientation of the cyanoacrylamide relative to the kinase active site and perturbing the steric and electronic environment of the electrophilic carbon led to the discovery of inhibitors with remarkably slow off-rates. These inhibitors exhibit biochemical residence times up to 7 days and illustrate the feasibility of fine-tuning inhibitor-BTK residence times across a wide dynamic range. An inhibitor that was orally bioavailable also demonstrated sustained BTK occupancy in vivo. The prolonged residence time and excellent kinase selectivity profile suggest potential applications of these inhibitors in chronic inflammatory diseases. The inverted cyanoacrylamide approach was further applied to an entirely different kinase and cysteine position (FGFR1 Cys486) and also resulted in inhibitors with prolonged and tunable residence time, illustrating broad applicability of the strategy to numerous drug targets.
Results
Design of reversible covalent BTK inhibitors
Structure-based design was used to identify lead molecules starting with scaffolds for which BTK crystallographic binding modes had been reported (Supplementary Results, Supplementary Fig. 1)29,30. A key challenge was identifying an accessible vector to Cys481 that could accommodate a reversible cyanoacrylamide-based electrophile. A previous design strategy8,9 – linking the electrophilic β-carbon directly to a kinase-recognition scaffold – appeared suboptimal, as Cys481 is located outside the ATP binding site. To solve this problem, the cyanoacrylamide was inverted relative to the scaffold, orienting the electrophilic β-carbon toward the protein surface, distal to the active site. This novel orientation provided unexpected opportunities for modulating residence time. First, it allowed exploration of multiple vectors to a common pyrazolopyrimidine scaffold by coupling geometrically diverse, amine-containing linkers to the cyanoacrylamide carboxyl group. Second, it opened a previously unavailable path for manipulating the steric and electronic environment of the electrophilic β-carbon through the use of branched-alkyl capping groups (Fig. 1a).
Figure 1. Reversible covalent BTK inhibitors based on inverted cyanoacrylamides.
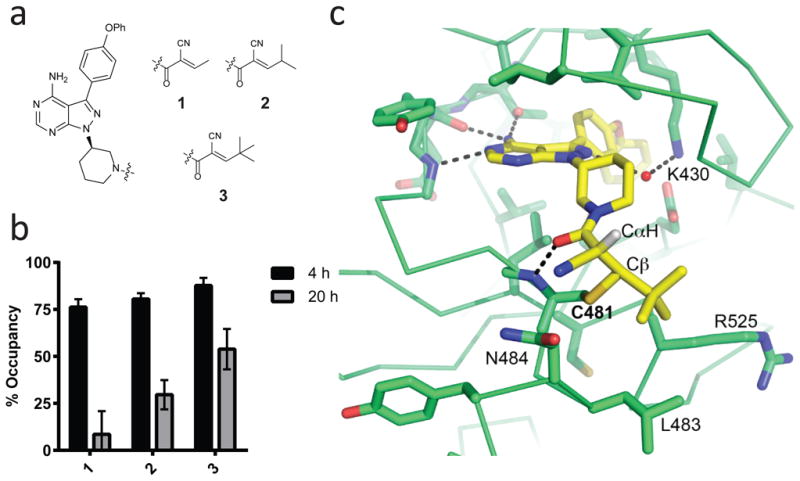
(a) Cyanoacrylamides, attached via a piperidine linker to a pyrazolopyrimidine scaffold, are capped with alkyl groups of increasing steric demand (1-3). (b) Inhibitors 1-3 have distinct levels of BTK durability in cells following inhibitor washout. Ramos B cells were treated with DMSO or 1-3 (1 μM) for 1 hr, washed 3 times with PBS, and incubated in compound-free media at 37 °C. After 4 or 20 hrs, cells were treated with 1 μM PP-BODIPY (the irreversible probe that labels BTK Cys481) for 1 hr and then lysed and analyzed by in-gel fluorescence. Occupancy was calculated from the normalized in-gel fluorescence intensity divided by the DMSO control value and subtracted from 100% (mean ± SD, n = 3). (c) Co-crystal structure of 3 bound to BTK at 2.2 Å resolution. The covalent bond between Cys481 and Cβ, newly formed CαH, and select hydrogen bonds are indicated. Two of the capping group methyls form hydrophobic contacts with Leu483 and Arg525, while the third is solvent exposed.
To test whether alkyl capping groups with increased branching and steric size confer prolonged residence time, cyanoacrylamides 1-3 bearing a piperidine linker and a pyrazolopyrimidine scaffold were prepared (Fig. 1a). Cyanoacrylamides with branched-alkyl capping groups are unique since the vast majority of cyanoacrylamides contain aromatic or heteroaromatic β-substituents (including previously reported reversible covalent kinase inhibitors8,9). Despite having similar potency in a cellular BTK occupancy assay under continuous exposure (Supplementary Fig. 2), compounds 1-3 exhibited distinct BTK residence times. This was assessed after extensively washing compound-treated Ramos B cells prior to labeling with an irreversible, pyrazolopyrimidine fluorescent probe (referred to here as PP-BODIPY) previously shown to penetrate cells and covalently bind BTK with high selectivity17. The tert-butyl-capped variant 3 had the longest residence time, showing 55% BTK occupancy 20 hrs post-washout (Fig. 1b). The other inhibitors showed significantly reduced (2) or negligible (1) BTK occupancy after 20 hrs, demonstrating durability can be modulated by alterations to the capping group.
To provide molecular insight into the prolonged occupancy of 3, its co-crystal structure bound to the BTK kinase domain was solved at 2.2 Å resolution. The electron density enabled unambiguous ligand placement (Supplementary Fig. 3), revealing strong density for the covalent bond and formation of two sp3-hybridized carbons resulting from Cys481 addition to Cβ and protonation of Cα (Fig. 1c). Examination of the torsion angles and interactions surrounding this region revealed structural features that likely contribute to the prolonged residence time. To minimize 1,3-torsional strain and steric clashes with the protein, both the tert-butyl capping group and the piperidine amide are oriented in a manner that shields the critical proton attached to Cα, which must be removed prior to (or during) elimination of the Cys481 thiol (Fig. 1c). Moreover, this conformation prevents overlap between the Cα-H bond and the carbonyl π-system and therefore should reduce its kinetic and thermodynamic acidity31 and hence the rate of Cys481 elimination. Interactions between the tert-butyl capping group and a hydrophobic patch near Cys481, as well as a hydrogen bond between the amide carbonyl of 3 and the backbone NH of Cys481, may further stabilize the covalent complex. Consistent with the concept that these and other noncovalent interactions with the folded kinase domain play an essential role in kinetically trapping the complex, guanidine-mediated unfolding resulted in rapid regeneration of 3 in 88 ± 10% yield (Supplementary Fig. 4). Finally, the structure revealed that one of the tert-butyl methyl groups is solvent exposed, providing a site for attaching polar substituents that could potentially improve aqueous solubility.
BTK residence time modulated over a wide dynamic range
Although 3 exhibits sustained BTK occupancy in both cellular and biochemical assays (Supplementary Fig. 5a) and is highly selective over other kinases with a structurally homologous cysteine (>1,000-fold selectivity over EGFR and JAK3; 60-fold over ITK, Supplementary Fig. 5b), its low solubility and poor oral bioavailability prompted exploration of structural modifications throughout the molecule, with a primary focus on the linker and capping group. These efforts, which included substitutions on the scaffold (e.g., mono-fluorination) and installation of a more flexible linker, led to the identification of 4 as a starting point for modulating BTK residence time (Fig. 2a). To bracket both extremes of the residence time spectrum, an acrylamide variant of 4 expected to bind irreversibly (5, referred to hereafter as PP-ir) and a non-electrophilic variant expected to have a rapid off-rate (6, referred to hereafter as PP-ne) were also synthesized (Fig. 2a).
Figure 2. Prolonged and tunable residence time of reversible covalent BTK inhibitors.
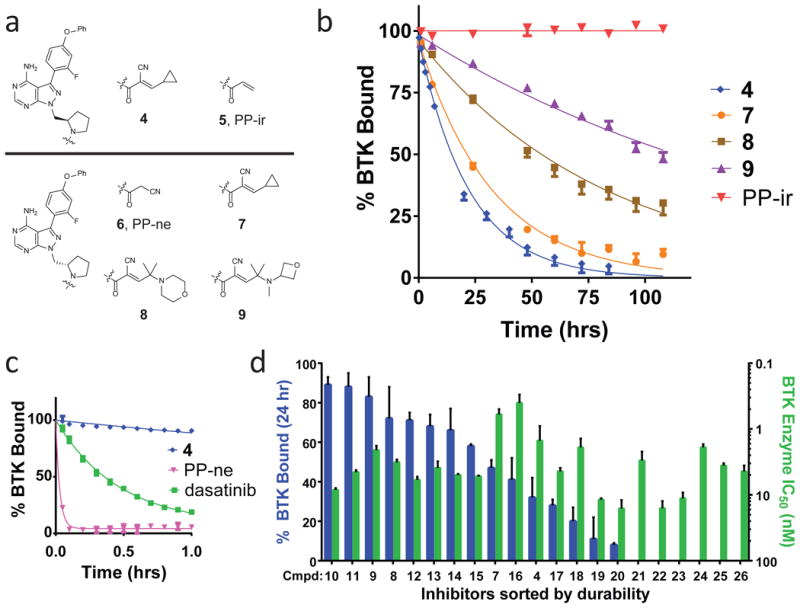
(a) BTK inhibitors with methylpyrrolidine linkers characterized during residence time-based optimization. Compounds contain R (4-5, top) and S (6-9, bottom) linker configurations. Inhibitors 4, 7, 8, 9 contain a cyanoacrylamide electrophile, PP-ir contains an acrylamide electrophile, and PP-ne has no electrophile. (b) Dissociation curves of BTK inhibitors. Shown is the percent BTK with inhibitor bound (% BTK Bound) versus time (hrs) for a 108-hour dissociation experiment. Residence times, τ, were determined as follows: 4: 22 ± 3 hrs; 7: 34 ± 5 hrs; 8: 83 ± 14 hrs; 9: 167 ± 21 hrs; PP-ir: >200 hrs. Data from representative experiments are shown (mean ± SD, n = 2). (c) A 1-hour inhibitor dissociation study. Residence times, τ, were determined as follows: PP-ne: <0.1 hrs; dasatinib: 0.46 ± 0.6 hrs. Data from representative experiments are shown (mean ± SD, n = 2). (d) Prolonged residence time is tunable and not predicted by enzyme IC50 potency. A series of 21 cyanoacrylamide-containing pyrazolopyrimidines with pyrrolidine linkers were characterized for both occupancy after 24 hrs in the biochemical off-rate assay (blue) and for potency in a BTK enzymatic activity assay (green). The occupancy at 24 hrs in the off-rate assay is plotted side-by-side with the IC50 for the same compound (mean ± SD, n ≥ 2). Compounds were sorted left-to-right in descending order of durability, also see Supplementary Table 1.
To facilitate detection of subtle differences in inhibitor residence times, a kinetic competition assay was developed using recombinant, full-length BTK (Supplementary Fig. 6). The assay measures the time an inhibitor prevents binding of a high-affinity fluorescent tracer added in large excess32-34. Inhibitor potency was also determined using a conventional enzymatic activity assay. Inhibitor 4 exhibited high potency (IC50 = 1.4 ± 0.2 nM, Supplementary Fig. 7) and gradually dissociated from BTK over many hours at room temperature (τ = 22 ± 3 hrs) (Fig. 2b), suggesting that it forms a covalent but reversible bond with Cys481. In contrast, the acrylamide-containing inhibitor PP-ir showed no evident dissociation, suggesting irreversible cysteine engagement. The compound lacking an electrophile, PP-ne, was less potent (IC50 = 170 ± 50 nM) and dissociated within 5 minutes (Fig. 2c). Dasatinib, a potent noncovalent BTK inhibitor35 (IC50 = 0.5 ± 0.1 nM), had a residence time of only 0.46 ± 0.06 hrs, indicating that high potency does not necessarily predict slow dissociation.
A comparison of both enantiomers of the methylpyrrolidine linker (4 vs. 7) revealed that the S-configuration provided greater durability (Fig. 2b). Maintaining the S-methylpyrrolidine linker, the capping group was revisited. This led to the discovery that inhibitors containing polar, branched-alkyl capping substituents, exemplified by morpholine 8 and oxetane 9 (Fig. 2a), exhibited further increases in residence time. With a residence time of 167 ± 21 hours (one week) (Fig. 2b), 9 is to our knowledge among the most durable reversible inhibitors yet identified2,3. Trypsinization of the complex formed between BTK and 9 led to quantitative recovery of unaltered 9, confirming complete reversibility (Supplementary Fig. 8).
Examination of 21 cyanoacrylamide-containing pyrazolopyrimidines encompassing 4 and 7-26 (Supplementary Table 1) revealed a graded continuum of BTK occupancy 24 hrs after adding the competitive tracer (Fig. 2d); discrete differences in BTK occupancy were also evident at earlier time points (Supplementary Table 1). Thus, residence time was tuned across a wide dynamic range via subtle structural perturbations to the linker and branched-alkyl capping group. Little correlation was observed between durability and IC50 values in the BTK enzyme assay, consistent with the general inability of IC50 measurements to capture the true binding affinity of slow off-rate compounds (Fig. 2c). It is unlikely that inhibitors like 9 would have been discovered using a strategy focused solely on optimizing IC50 values, rather than residence time.
Several approaches were employed to corroborate the prolonged residence time observed with 9. First, the rate at which BTK enzymatic activity recovered after incubation with compound followed by dialysis was measured. Compound 9 demonstrated durable BTK inhibition, showing only 25% recovery of activity after 3 days of dialysis at room temperature (Supplementary Fig. 9). Compound 4 was also durable but to a lesser extent, reflecting the same rank order as the off-rate assay. In separate experiments, 9 demonstrated time-dependent inhibition of BTK enzymatic activity (Supplementary Figs. 10-11), as expected for an inhibitor with an extremely slow off-rate. kinact/Ki values were determined for compounds 4-9, and similar to the IC50 values, kinact/Ki did not correlate with residence time (Supplementary Fig. 11).
Durability of reversible covalent BTK inhibitors in cells
Cellular potency and durability were assessed using the ability of inhibitors to block binding of probe PP-BODIPY to BTK. Pre-incubation of Ramos B cells with 4, 7, 9, and PP-ir blocked BTK labeling in a dose-dependent manner (Fig. 3a; Supplementary Figs. 12-13). To test for durable BTK occupancy, cells were incubated with 9, extensively washed to remove unbound inhibitor, and subsequently incubated with PP-BODIPY 4 hrs after removal of 9. Inhibition of PP-BODIPY labeling by 9 was equally effective with and without washout (Fig. 3a), indicating durable interaction. The ability of 4-9 to block PP-BODIPY in Ramos cells was next compared 4 and 18 hrs post-washout, and a correlation was observed between biochemical residence time and durable occupancy in cells (Fig. 3b; Supplementary Fig. 14). A higher-throughput method for determining cellular BTK occupancy was developed based on an irreversible, biotin-labeled probe (27, referred to as PP-biotin, see Supplementary Fig. 15a) and AlphaScreen technology36 (Supplementary Fig. 15b). Inhibitors 4, 7, 8, 9, and PP-ir blocked PP-biotin binding to BTK with IC50 values from 5 ± 1 nM to 27 ± 11 nM (Supplementary Fig. 15c). Similar to biochemical residence time measurements, a washout experiment in Ramos cells using Alphascreen detection indicated that reversible covalent BTK inhibitors exhibit varying degrees of durability, with 9 being the most durable (59% ± 17% occupancy 18 hrs post-washout, Fig. 3c). Co-treatment with the protein synthesis inhibitor cycloheximide indicated that much of the apparent recovery of unoccupied BTK during the 18 hr washout period is due to BTK turnover and re-synthesis in Ramos B cells rather than compound dissociation (Supplementary Fig. 16).
Figure 3. Long-term cellular durability of reversible covalent BTK inhibitors.
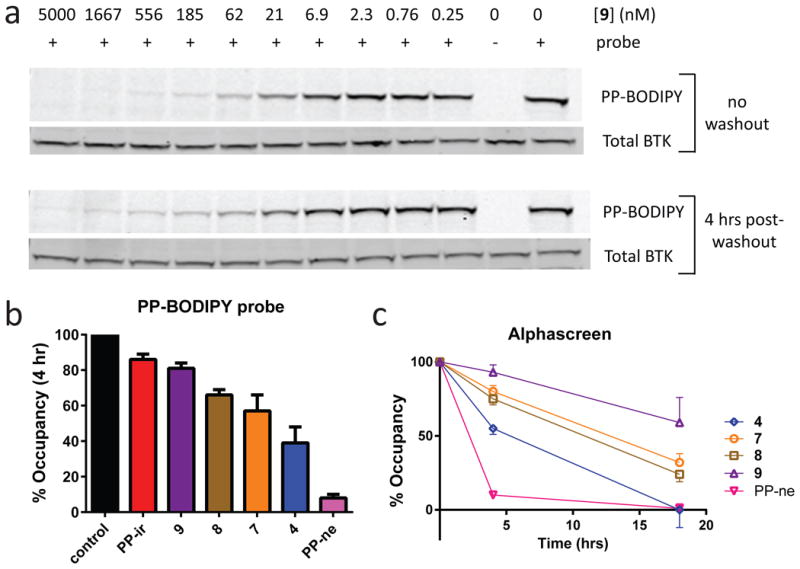
(a) Sustained BTK occupancy of compound 9 in cells 4 hrs following inhibitor washout. Ramos B cells were incubated with various concentrations of 9 and either treated with 1 μM PP-BODIPY directly (top) or washed 3 times and returned to culture for 4 hrs prior to adding 1 μM PP-BODIPY (bottom). Lysates were evaluated both for binding of the PP-BODIPY fluorescent probe to BTK (PP-BODIPY) and total BTK by Western blot (Total BTK) (n = 2). See Supplementary Fig. 25 for full gel images. (b) Reversible covalent BTK inhibitors exhibit distinct levels of durability in cells. Ramos B cells were incubated with 1 μM inhibitor, washed 3 times, and returned to culture for 4 hrs prior to addition of 1 μM PP-BODIPY. Lysates were evaluated both for binding of the PP-BODIPY probe and total BTK to calculate % Occupancy (mean ± SD, n ≥ 2) of the inhibitor (See Supplementary Fig. 14). (c) BTK occupancy of reversible covalent inhibitors 4 hrs and 18 hrs following cell washout (mean ± SD, n ≥ 2) evaluated using Alphascreen technology (Supplementary Fig. 15).
BTK occupancy led to diminished signaling downstream of the B cell receptor, as revealed by a flow cytometry-based CD69 expression assay. In human whole blood, BTK inhibitors including 9 blocked CD69 expression following stimulation of the B cell receptor with anti-IgM (Supplementary Figs. 17-18). Potent BTK inhibition in cells also required Cys481 as a C481S BTK mutant transfected into HEK293 cells was >50-fold less sensitive to 9 than wild-type BTK (Supplementary Fig. 19).
BTK inhibitor selectivity and in vivo residence time
To determine kinase selectivity, 9 was evaluated at concentrations of 1 μM and 0.1 μM in a 254-kinase panel with subsequent enzyme IC50 determinations (Fig. 4; Supplementary Fig. 20). Because BTK Cys481 is conserved in only 10 other kinases (BLK, BMX, EGFR, ERB-B2, ERB-B4, ITK, MKK7, JAK3, TEC, and TXK), we hypothesized that 9 would inhibit a subset of these kinases without affecting kinases that lack this cysteine. Indeed, only 6 of 254 kinases were inhibited by >90% at 1 μM of 9 (Fig. 4a; Supplementary Table 2); the sensitive kinases (BTK, BLK, BMX, ERB-B4, TEC, TXK) share the conserved cysteine as well as a threonine in the gatekeeper position. Furthermore, only BTK and BMX were inhibited by >90% at 0.1 μM of 9 (Fig. 4a; Supplementary Table 3). Importantly, other kinases that have this cysteine and are known to play critical tissue-specific homeostatic roles in vivo (EGFR, ERB-B2, ITK, JAK3) were unaffected by 9 (IC50 >3 μM, Fig. 4b). Compound 9 failed to inhibit signaling downstream of EGFR and the T cell receptor (Fig. 4c), indicating selectivity in cells for the B cell receptor pathway.
Figure 4. Kinase selectivity of inhibitor 9.
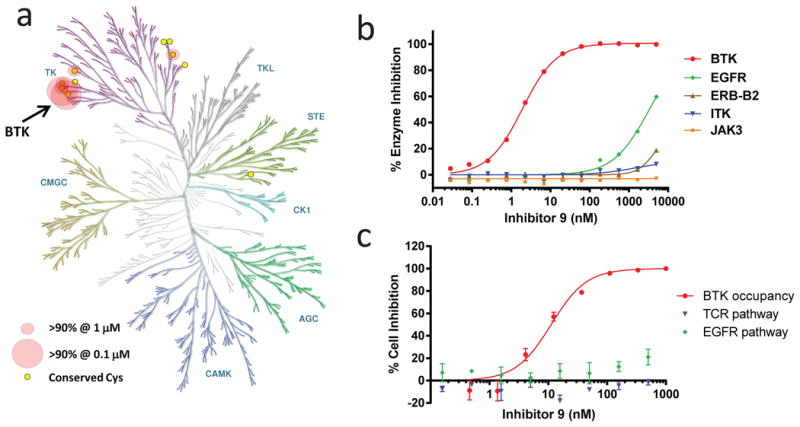
(a) Kinase selectivity of 9 evaluated at 1 μM and 0.1 μM in the Nanosyn 254 kinase panel. Circles indicate the 6 kinases that showed >90% inhibition at 1 μM. Large circles indicate the 2 kinases (BTK and BMX) that also demonstrated >90% inhibition at 0.1 μM. The figure was reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com). (b) Enzymatic IC50 curves determined using inhibitor 9 toward BTK and other kinases with a homologous cysteine with known tissue-specific homeostatic roles in vivo (EGFR, ERB-B2, ITK, JAK3) (mean ± SD, n ≥ 2). IC50 values and curves toward the other related cysteine-containing kinases are shown in Supplementary Fig. 20. (c) Cellular selectivity of inhibitor 9. The activity of 9 in Ramos B cells using a BTK occupancy assay (see Supplementary Fig. 15) was compared with the activity toward ITK and EGFR in T cell receptor (TCR) and EGFR cellular reporter assays, respectively (mean ± SD, n ≥ 2). In the TCR assay, 9 was studied in a Jurkat T cell line stimulated with anti-CD3 plus anti-CD28 (to activate the T cell receptor pathway) and NFAT reporter expression was measured. In the EGFR assay, 9 was studied in ME-180 cervical carcinoma cells stimulated with EGF (to activate the EGFR receptor pathway) and AP-1 reporter expression was measured.
We asked whether the sustained residence time of 9 in biochemical and cellular experiments translated into a prolonged pharmacodynamic effect in rodents. PP-BODIPY was used to determine the level of BTK target engagement in rat peripheral blood mononuclear cells (PBMC) at multiple times following an oral 40 mg/kg dose of 9. Levels of 9 in plasma were simultaneously determined to assess whether BTK occupancy was driven by prolonged residence time or high levels of 9 in circulation. The plasma level of 9 peaked at 104 ± 66 ng/ml 1 hr after dosing and fell to 24 ± 17 ng/ml at 6 hrs and 3 ± 3 ng/ml at 14 hrs (Fig. 5). Since the IC50 of 9 in human whole blood is 146 ± 10 ng/ml (corresponding to 240 ± 20 nM, Supplementary Fig. 18), it seemed unlikely that the low plasma levels observed after 6 hrs could drive BTK target engagement. Assessment of BTK occupancy revealed that 74 ± 9%, 82 ± 5%, 57 ± 9%, and 41 ± 15% of BODIPY probe labeling was blocked at 1 hr, 6 hrs, 14 hrs, and 24 hrs after dosing, respectively (Fig. 5). Hence, despite being cleared from the circulation, 9 exhibits significant target engagement 24 hrs after oral dosing, reflecting its slow dissociation from BTK in vivo.
Figure 5. Extended pharmacodynamic effect of an orally bioavailable BTK inhibitor.
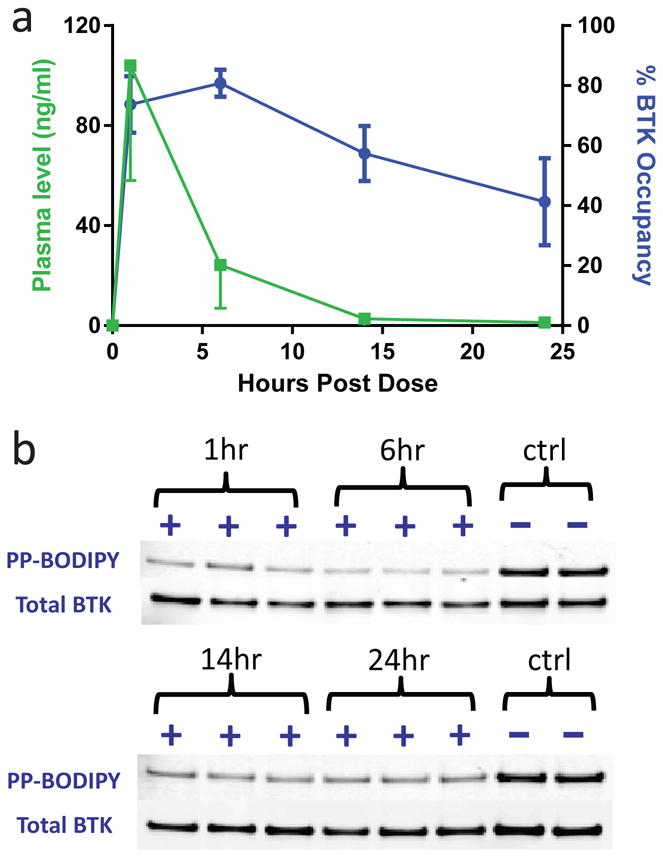
(a) PK/PD relationship for 9 in rats dosed orally at 40 mg/kg. The percent BTK occupancy (in blue) and the concentration of 9 in plasma (in green) at each time point following dosing is shown (mean ± SD, n = 3). The potency of 9 in the human whole blood CD69 expression assay was 146 ± 10 ng/ml. (b) The gel-based data used to calculate the BTK occupancy of 9. Occupancy was determined with in-gel fluorescence using rat PBMC collected from 3 separate animals at each time point. The % BTK occupancy was determined from the percentage block of BODIPY probe compared to vehicle treated animals (ctrl) with all samples normalized for the total amount of BTK as evaluated by Western blotting (n = 2). See Supplementary Fig. 25 for full gel images.
Prolonged, tunable residence time of FGFR inhibitors
We sought to show that the strategy of tuning inhibitor residence time by varying cyanoacrylamide capping groups is generalizable beyond BTK. Cys486 of FGFR1, located in the kinase P loop, has previously been targeted with acrylamide-based irreversible inhibitors utilizing a pyrimidopyridine scaffold37. This cysteine is found in an active site region distinct from both Cys481 of BTK and Cys436 of RSK2 (Fig. 6a), providing an opportunity to demonstrate cyanoacrylamide-based cysteine targeting in a third distinct protein environment. A series of FGFR inhibitors were synthesized that all contain the same pyridopyrimidinone core and phenyl linker but a variety of electrophile capping groups. Inhibitors were all potent toward FGFR1 in an enzyme activity assay (IC50 ≤ 6 nM; Supplementary Table 4) but varied dramatically in their residence times, as exemplified by 28, 29, and 30 (Fig. 6b and 6c). Compound 28, a highly durable inhibitor containing a spirocyclic capping group, maintained FGFR1 occupancy throughout the 24 hour assay period (τ > 150 hrs) whereas the equally potent but noncovalent FGFR inhibitor BGJ39838 dissociated much more rapidly (τ = 1.9 ± 0.4 hrs; IC50 = 0.9 ± 0.1 nM) (Fig. 6c). Likewise, although both 28 and BGJ398 potently blocked FGF-stimulated ERK phosphorylation in human umbilical vein endothelial cells, only 28 maintained inhibition following extensive cell washout, indicating prolonged residence time in cells (Supplementary Fig. 21). The FGFR inhibitor series, encompassing compounds 28 through 38 (Supplementary Table 4), demonstrated a wide continuum of FGFR occupancy levels at 24 hours in the kinetic competition assay (Fig. 6d; Supplementary Table 4), indicating that simple modifications in the capping group and linker discretely modulated FGFR1 residence time over several orders of magnitude, analogous to the cyanoacrylamide pyrazolopyrimidine series toward BTK (Fig. 2c). Excellent kinase selectivity for the FGFR family was also obtained (Supplementary Fig. 22; Supplementary Tables 5 and 6). Many of the capping groups that provided prolonged residence time toward FGFR were distinct from those that provided prolonged residence time toward BTK, indicating that the unique protein environment of each cysteine residue determines which capping groups provide the greatest durability (Supplementary Figs. 23 and 24).
Figure 6. Inverted cyanoacrylamide FGFR inhibitors with prolonged, tunable residence time.
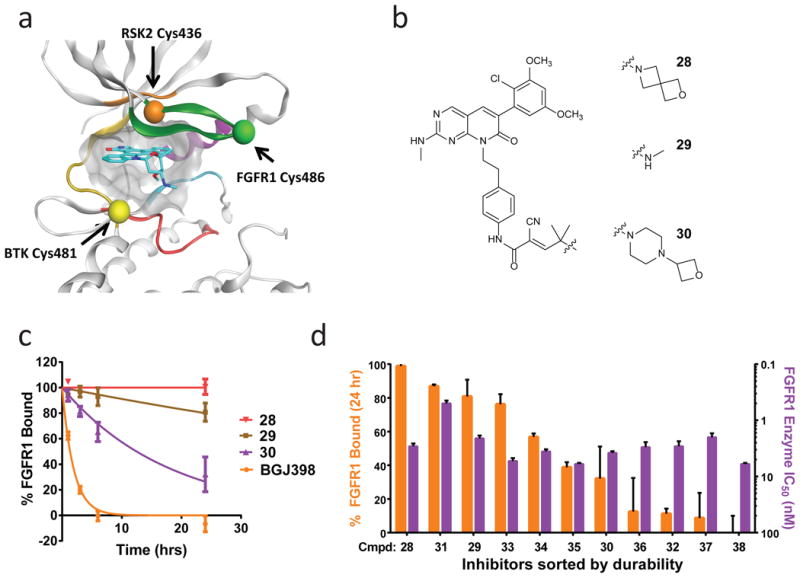
(a) Model of the kinase ATP binding site demonstrating the disparate locations of cysteine residues targeted by cyanoacrylamide-based RSK2, BTK, and FGFR inhibitors. Structural elements are colored as follows: green:glycine rich loop; purple: αC-helix; cyan:DFG loop; yellow:hinge region; red: phosphate binding region. (b) Inverted cyanoacrylamides linked to a pyridopyrimidinone scaffold were synthesized with a variety of different capping groups. (c) Dissociation curves of FGFR inhibitors. Shown is the percent FGFR1 with inhibitor bound (% FGFR1 Bound) versus time (hrs) for a 24-hour dissociation experiment. Residence times, τ, were determined as follows: 28: >150 hrs; 29: 110 ± 41 hrs; 30: 18 ± 5 hrs; BGJ398: 1.9 ± 0.4 hrs. Data from representative experiments are shown (mean ± SD, n = 2). (d) Prolonged and tunable residence time of inverted cyanoacrylamide FGFR inhibitors. FGFR inhibitors 28 through 38 containing a variety of electrophile capping groups were characterized (See compounds and durability data in Supplementary Table 4). Inhibitors were found to display a continuum of residence times as evaluated by the % FGFR1 occupancy 24 hours after addition of a fluorescent competitive tracer (orange bars) (mean ± SD, n ≥ 2). Residence time was not predicted by potency in a FGFR1 enzymatic inhibition assay (purple bars) since IC50 potency for all compounds clustered in the range between 0.5 ± 0.05 nM and 6.0 ± 0.3 nM.
Discussion
By exploiting the intrinsic reversibility and facile tunability of inverted cyanoacrylamide-based inhibitors, we demonstrated that BTK inhibitor residence time can be modulated in a discrete, stepwise manner. The strategy focused specifically on maximizing BTK residence time rather than binding affinity, and the biochemical and cellular assays reported in this work are applicable to other therapeutic targets for which optimization of residence time is crucial. Fine-tuning of the cyanoacrylamide capping group and linker attached to a kinase-recognition scaffold culminated in the discovery of 9 (Fig. 2a), a reversible BTK inhibitor that binds to the intact kinase with a residence time of 7 days, while dissociating rapidly and quantitatively upon BTK proteolysis. Cyanoacrylamide 9 has good aqueous solubility (126 μM at pH 7), is orally bioavailable, and provides sustained BTK engagement in vivo, lasting 18 hours after clearance from the circulation. The apparent increased dissociation rate in cells relative to the biochemical assay likely reflects the increased temperature (37 °C vs. 23 °C) and a more dynamic intracellular environment, including BTK turnover, re-synthesis, and interaction with cellular binding partners. Despite these differences, results from the biochemical assay correlated with durability in cells (See Fig. 2b & Fig. 3).
A breakthrough came with the discovery that certain branched-alkyl capping groups with polar functionality can be attached to the cyanoacrylamide β-carbon to modulate BTK residence time. Such cyanoacrylamides have little precedent in the chemical literature, and prior to this work, it was unclear whether they would even be stable under physiological conditions. When certain capping groups and linkers are combined, noncovalent interactions with the protein (exemplified by the BTK/3 crystal structure), as well as conformational restriction proximal to the new covalent bond, increase the kinetic barrier to cysteine elimination and thereby stabilize the complex. Such capped cyanoacrylamides could potentially be used to modulate inhibitor residence time for many other cysteine-containing targets, and we demonstrated here generalizability beyond BTK by showing that Cys486 of FGFR1 could also be targeted with inverted cyanoacrylamide-based inhibitors (Fig. 6). No estimate of the total number of drug targets that contain an accessible cysteine (the “druggable cysteinome”) has yet been reported. However, among kinases, it has been estimated that 39% (200 of 518) contain an approachable cysteine in the ATP-binding pocket15, suggesting that the overall percentage of drug targets amenable to cysteine-targeting is significant.
While targeting cysteines with inverted cyanoacrylamides likely has broad application, there are certain limitations to this approach. First, cyanoacrylamides with branched-alkyl capping groups have a high steric demand, potentially limiting their application to relatively unhindered and solvent-exposed cysteines. Second, although structure-based design is useful for identifying initial scaffolds and linkers that orient the cyanoacrylamide near the targeted cysteine, predicting which linker/capping group combinations will provide the desired residence time is not currently feasible. Nevertheless, because the optimization path involves late-stage installation of both the linker and the capping group onto a pre-assembled scaffold, fine-tuning inhibitor residence time is highly efficient. Through different combinations of linkers and capping groups, a wide range of residence times were covered by synthesizing 21 closely related compounds, all of which inhibit BTK enzyme activity in the low nanomolar range (Fig. 2d; Supplementary Table 1).
This study revealed that cysteine-targeted, reversible covalent inhibitors can exhibit residence times from minutes to days. For targets where prolonged residence time is desired, reversible covalent inhibitors may be able to mimic the long-lasting activity of irreversible inhibitors, with dissociation rates in vivo that approach the rate of target degradation and re-synthesis. However, molecules with a fast off-rate are ideal for certain therapeutic targets, due to mechanism-based toxicity associated with permanent on-target inhibition. Well-characterized examples include the cyclooxygenase inhibitor ibuprofen39,40, the NMDA receptor antagonist memantine41, and the D2 dopamine receptor antagonist clozapine42,43. For these targets, irreversible inhibition is not a clinically viable approach, but reversible covalent inhibition would still be appropriate. The ability to generate a series of ligands with residence times that span a wide temporal range, as demonstrated in this study, suggests it may be possible to match a ligand's residence time to the biological requirements of a drug target.
BTK has been clinically validated as a target for B cell malignancies based on pioneering studies with the acrylamide-based inhibitor ibrutinib24,25. In addition to inhibiting BTK and related Tec-family kinases, ibrutinib targets more distantly related kinases, including EGFR, MKK7, ITK, and JAK3, which have a cysteine equivalent to BTK Cys48117,28,44. Moreover, when added to cells at therapeutically relevant concentrations, acrylamide-based probes nearly identical to ibrutinib and a clinical EGFR inhibitor were shown to form irreversible adducts with dozens of other cysteine-containing proteins28. Irreversible adducts have historically been a concern within drug discovery due to the relationship between covalent drug binding and the potential for idiosyncratic drug toxicity10,12,45,46. Irreversible modification of off-target cysteines may be especially problematic in tissues exposed to high initial drug concentrations after oral administration (often in the micromolar range), such as the gastrointestinal tract and liver. By eliminating the possibility of irreversible binding to off-target proteins, a reversible covalent molecule may provide reduced risk of idiosyncratic drug reactions and therefore may be safer than an analogous irreversible inhibitor. Given the intrinsically reversible nature of the cyanoacrylamide/cysteine interaction, the probability of forming permanent adducts using a reversible covalent BTK inhibitor such as 9 is negligible. Moreover, 9 has outstanding kinome-wide selectivity (Fig. 4), avoiding all kinases that lack the conserved cysteine and many physiologically important kinases that have this cysteine, including EGFR, ERB-B2, ITK, JAK3, and MKK7.
In addition to their established roles in the treatment of B cell cancers, BTK inhibitors may have beneficial effects in autoimmune diseases. Diseases such as rheumatoid arthritis and lupus erythematosus, which are ameliorated by BTK inhibitors in animal models17,19,26,27, typically require chronic dosing and a high safety margin, favoring drugs that are highly selective with minimal off-target effects. Reversible covalent BTK inhibitors such as 9 appear well suited for potential use in autoimmune disorders and are currently being advanced for clinical studies. More broadly, inverted cyanoacrylamides with branched-alkyl capping groups represent a new platform for targeting noncatalytic cysteines. Wide application of this approach has the potential to improve small-molecule residence times across diverse targets and disease areas.
Online Methods
Chemical synthesis
The synthesis of compounds is described in the Supplementary Note.
BTK kinase domain cloning, expression and purification
Human BTK kinase domain (residues 382-659) was inserted into pFastBac-1 by InFusion cloning following the manufacturers protocol (Clontech). During PCR amplification a TEV-cleavable 6x-His tag was introduced on the BTK N-terminus. This plasmid was then used to transform DH10α MultiBac cells for preparation of the expression bacmid. Sf21 cells were transfected with the MultiBac bacmid using Fugene HD transfection reagent according to the manufacturer's instructions (Roche), and virus was propagated following published methods. The BTK kinase domain was subsequently expressed in Sf21 cells by infection of 2.0 L of cultured cells with 20 mL virus solution such that cell growth was halted immediately. The cells were collected by centrifugation (800 g × 15 min) and the pellets resuspended in 50 mL lysis buffer (50 mM Hepes, pH 7.5, 400 mM NaCl, 1.5 mM DTT) supplemented with 1 × protease inhibitor cocktail (Roche). The cells were lysed by 5 passes through a cell homogenizer. The cellular debris was pelleted by centrifugation (30k g × 30 min). The protein was bound in batch to Ni-NTA agarose beads in binding buffer (lysis buffer + 20 mM imidazole) for 4 h at 4 °C. The beads were washed with additional binding buffer (4 × 5 mL) and the protein was eluted with 3 × 0.5 mL portions of elution buffer (lysis buffer + 300 mM imidazole). The His tag was cleaved by addition of AcTev protease with concomitant dialysis overnight into cleavage buffer (50 mM Tris, pH 8.0, 0.5 mM EDTA, 1 mM DTT). The resulting soluble protein was purified further by gel filtration on an S75 column (gel filtration buffer: 20 mM Tris, pH 8.0, 50 mM NaCl, 3 mM DTT). The protein was then concentrated to 16.8 mg/mL, flash frozen in LN2 and stored at -80 °C. The final yield was 4 mg of protein.
Crystallization, data collection and structure solution
BTK kinase domain (6.0 mg/ml, 0.18 mM) was treated with compound 3 (1 mM) in 10% DMSO and the mixture was incubated on ice for 30 min, followed by ultracentrifugation in a TLA100 rotor for 30 min at 90,000 rpm at 4 °C. Crystals of BTK-KD/3 were grown by mixing 1 μL of protein with 1 μL of precipitant solution composed of 0.1 M MES pH 6.5, 0.2 M (NH4)SO4, and 30% (w/v) PEG 5000 MME, at 4 °C. The crystals grew as stacked plates that were crushed and individual crystals were cryoprotected in mother liquor supplemented with 15% ethylene glycol and frozen in a stream of liquid nitrogen at 100 K. The crystals belonged to the space group P21 with unit cell parameters a=43.5 Å; b=76.1 Å; c= 88.2 Å; α=90.0°; β=96.5°; γ=90.0° and contained two copies of the complex. All datasets were collected on a stream of liquid nitrogen at 100K at 1.000 Å wavelength on the 8.2.1 beamline of the Advanced Light Source at the Berkeley National Laboratory. Diffraction data were integrated and scaled with the program XDS47. The structure of the BTK-KD/3 complex was solved by molecular replacement using data to 2.2 Å and structure 3GEN as search model in program Phaser48, followed by several rounds of manual rebuilding and restrained refinement with programs Coot49 and Phenix50. The structure refinement converged to R and Rfree values of 17.4% and 20.8%, respectively with 96% favored and 0.2% Ramachanran outliers as validated with the Molprobity server51. See Supplementary Fig. 3 for data collection and refinement statistics.
Dissociation of compound 3 from BTK after guanidine-mediated unfolding
BTK (1.5 μM in 20 mM Tris, 50 mM NaCl, pH 8.0) or buffer alone was incubated with 1 μM compound 3 (1% DMSO v/v final, 10 μL total volume) for 30 min at room temperature. Guanidinium hydrochloride (6 M, 10 μL) was added to each solution and the mixtures were incubated for 30 min at 37 °C. Acetonitrile (40 μL) was added to each solution, followed by water with 0.1% formic acid (10 μL). The resulting solutions were analyzed by LC-MS/MS, and 3 was quantified using a MRM method and calibration curve (Waters Acquity UPLC/ESI-TQD instrument with a 2.1 × 50 mm Acquity UPLC BEH C18 column). Percent recovery of the BTK-containing sample was determined relative to the control sample lacking BTK.
Competition labeling assay in Ramos cells with PP-BODIPY
Ramos cells were grown in RPMI 1640 media supplemented with 10% FBS and pen/strep antibiotics under 5% CO2. Cells (1 mL, 1 – 1.5 × 106) in growth media were treated with drug (0.1% DMSO) for 1 h at 37 °C. PP-BODIPY was added to a final concentration of 1 μM and the cells were incubated for an additional hour at 37 °C. The cells were collected by centrifugation (650 g × 3 min) and washed with PBS. The cells were flash frozen in LN2 and stored at -80 °C. The cell pellets were resuspended in 50 μL CelLytic M (Sigma, #C2978) supplemented with 1 × Roche complete protease inhibitors and incubated 15 min at 4 °C. The cellular debris was removed by centrifugation (20k g × 10 min) and the supernatents were normalized for total protein concentration and subjected to SDS-PAGE analysis. The resulting gel was scanned for fluorescence and either probed by western blot for total BTK (α-BTK antibody – Cell Signaling #C82B8 or BD Biosciences Cat#611117, 1:500) and α-tubulin (anti-α-tubulin antibody – Sigma #T6199) or stained with Coomassie Brilliant Blue for total protein.
Cellular durability studies using the PP-BODIPY occupancy probe
Ramos B cells were seeded at a concentration 1 × 106 cells/well in a 12-well plate. Compounds were added to a final inhibitor concentration of 1 μM and the plates were incubated at 37 °C for 1 hr. Cells were washed 3 times to remove unbound inhibitor and returned to culture for the indicated time. Following the washout period, cells were incubated with 1 μM PP-BODIPY for 1 hr at 37 °C to evaluate inhibitor occupancy. Cells were pelleted, washed with PBS once, and 40 μL of lysis buffer (CelLytic M, Sigma #C2978) was added to each well. To evaluate the occupancy of BTK with PP-BODIPY and the total BTK present in each sample, gels were analyzed by fluorescent scanning and Western blot as described above. When the dependence of BTK occupancy on new protein synthesis was evaluated, 100 μL of cycloheximide (50 μg/ml) or culture media alone (10% FBS/RPM1 1600) was added to 900 μL of cells for 30 minutes at 37 °C to a final concentration of 5 μg/ml cycloheximide, followed by addition of BTK inhibitors.
Biochemical residence time using fluorescence competition
Using an assay buffer of 50 mM Hepes pH 7.5, 10 mM MgCl2, 0.01% Triton-X 100, and 1 mM EGTA, 1 μL of 15 μM compound was added to 9 μL of 0.5 μM BTK (Invitrogen PV3587) in a 96 well polypropylene plate. Following 30 minutes of incubation, the mixture was diluted in assay buffer. A dilution of 25-fold was utilized for time course studies with 4-9, and a 5-fold dilution was used for occupancy determinations measured at solely 1, 6, and 24 hours; varying the dilution in this range was demonstrated to have negligible effect on determination of BTK occupancy. 10 μL of diluted mixture was transferred to a Greiner 384 well black plate. Europium-coupled Anti-6XHis Ab (Perkin Elmer AD0205) and Tracer 178 (Invitrogen PV5593) were added to a final concentration of 15 nM and 0.75 μM, respectively, in 20 μL volume. Data was acquired using a Perkin Elmer Envision plate reader (Model 2101) containing LANCE TR-FRET compatible excitation and emission filters. Fluorescence at 665 nM and 615 nM wavelengths were collected at various times. In each experiment, a condition that provides the maximum signal (max) was acquired consisting of the signal from enzyme, Europium-coupled Anti-6XHis Ab, and Tracer 178 in the absence of test compound. A background signal (bkg) was also acquired where a 1 μM concentration of PP-ir was added to completely block Tracer 178 binding. Data for each test compound was reported as % BTK bound, which is calculated as 100*(1 -(cmpd - bkg)/(max – bkg)). In order to determine the koff and residence time τ, data were fit using a decreasing exponential function of the form % occupancy = (100 - Plateau)*exp(-koff*time) + Plateau using GraphPad Prism software. Residence time τ was determined using τ = 1/koff. Biochemical residence time studies using FGFR1 (Invitrogen PV4105) were performed using a similar procedure except that a 60 minute enzyme/compound incubation time was utilized, the dilution following pre-incubation was 100-fold, and a Cy5-labeled pyridopyrimidinone was utilized as a tracer.
Enzymatic IC50 assays
Inhibitor potency in a BTK enzymatic assay was determined using the microfluidic-based LabChip 3000 Drug Discovery System from Caliper Life Sciences that uses capillary electrophoresis to separate phosphorylated and non-phosphorylated peptides. Twelve concentrations of inhibitor were first pre-incubated with BTK for 15 minutes. Enzyme, inhibitor, peptide substrate, and cofactors (ATP and Mg2+) were then combined and incubated at 25 °C. The final buffer was 100 mM HEPES, pH 7.5, 0.1% BSA, 0.01% Triton X-100, 1 mM DTT, 10 mM MgCl2, 10 μM Sodium Orthovanadate, 10 μM Beta-Glycerophosphate, 16 μM ATP, and 1% DMSO. At the end of the incubation, the reaction was quenched by an EDTA-containing buffer. Negative controls (containing no inhibitor) and positive controls (acquired in the presence of 20 mM EDTA) were simultaneously evaluated to calculate percent inhibition at each compound concentration. The IC50 values were determined by fitting the inhibition curves using a 4 parameter sigmoidal dose-response model using XLfit 4 software (IBDS). To measure the effect of pre-incubation time on compound IC50, the protocol above was modified such that the pre-incubation time between compound and BTK enzyme was 0 minutes, 10 minutes, 30 minutes, 90 minutes, and 180 minutes. The determination of enzymatic IC50 values for BLK, BMX, EGFR, ERB-B2, ERB-B4, ITK, JAK3, MKK7, TEC, and TXK was performed as described above except that the concentration of kinase, the peptide substrate, and the ATP concentration were optimized for each kinase. Enzymatic IC50 data was acquired by Nanosyn, Inc. (Santa Clara, CA; www.nanosyn.com).
BTK progress curve analysis
Progress curves of BTK peptide phosphorylation were acquired at six concentrations. The real-time curves were obtained for a total of 5 hrs using the climate controlled Caliper LabChip instrument. The obtained curves were fit using XLfit4 software to the time dependent inhibition equation: [P] = Vs*t + ((Vi-Vs)/Kobs)*(1-exp(-Kobs*t)). In the equation: Vi is the initial velocity, Vs is the steady state velocity, and Kobs reflects the rate of inactivation. For rapid equilibrium compounds, a linear fit was used: [P]=Vi*t. For time dependent inhibitors, the obtained Kobs values were plotted against compound concentration using either a hyperbolic or a linear fit. From these plots, kinact and Ki were determined. Progress curve data was acquired by Nanosyn, Inc. (Santa Clara, CA; www.nanosyn.com).
Kinase selectivity panel
Kinase selectivity determination using a 254-kinase panel was performed by Nanosyn, Inc. (Santa Clara, CA; www.nanosyn.com).
Dissociation of compound 9 from BTK after trypsinization
A mixture of 2 μM BTK (Invitrogen PV3587) and either 9 (0.6 μM) or PP-ir (0.6 μM) was incubated at room temperature for 60 minutes in an assay buffer of 20 mM Hepes pH 7.5, 10 mM MgCl2, 150 mM NaCl, 5% Glycerol, 0.05% Triton-X 100, and 1 mM 2-mercaptoethanol. In parallel 9 (0.6 μM) and PP-ir (0.6 μM) were also incubated with the BTK enzyme storage buffer alone (50 mM Hepes pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.05% Triton X-100, 4 mM 2-mercaptoethanol, 50% Glycerol). A trypsin stock solution (Sigma T1426) was prepared to 1 mg/ml in assay buffer. 60 μL of both the enzyme/compound mixture and the storage buffer/compound mixture were added to 120 μL of 1 mg/ml trypsin. Liquid chromatography-mass spectrometry of the samples was performed by the analytical chemistry group of the Molecular Medicine Research Institute (www.mmrx.org). In order to calculate inhibitor reversibility, the average mass spectrometry peak area of the compound/BTK/trypsin samples was divided by the peak area for the samples containing compound/storage buffer/trypsin to get the % Reversibility.
BTK cell occupancy using Alphascreen
To treat Ramos B cells with compounds, 100 μL of 1 × 106 cells were cultured in a 96-well round bottom polypropylene plate. 10 μL of a 1:3 dilution series of compound prepared in media containing 1.1% DMSO was added in duplicate, and 10 μL of media with 1.1% DMSO alone was added to control wells. The plates were incubated at 37 °C for 1 hr. The PP-biotin probe was prepared at 15 μM from a 5 mM stock using media, and 2.5 μL of 15 μM PP-biotin was added to all wells except DMSO control wells to a final concentration of 330 nM. In DMSO control wells, 2.5 μL of media containing 0.3% DMSO was added. The plates were incubated for 1 hr at 37 °C. Cells were pelleted, washed with PBS once, and 60 μL of lysis buffer (Cell Lytic M, Sigma #C2978) was added to each well. The plate was incubated for 15-20 minutes on ice and centrifuged for 5 minutes at 2000 rpm at 4 °C. The lysate was transferred to a new polypropylene plate and either stored at -80 °C or used immediately. According to the Alphascreen manufacturer's protocol (Alpha technology Protein A detection kit, Perkin Elmer catalog 6760617C), 1X buffer, 200 μg/ml acceptor beads, 30 nM BTK antibody (BTK, BD Biosciences, Cat# 611117) and 200 μg/ml streptavidin donor beads were prepared. To each well, 5 μL of acceptor beads and BTK antibody were added. The plate was sealed and incubated at room temperature for 30 minutes. Then 10 μL of lysate was added, and the plate was incubated at room temperature for 30 minutes. 5 μL of streptavidin donor beads was added to each well in a dark room. The plate was sealed with a plate sealer and incubated at room temp on the shaker for 30 minutes. The plate was read using the Perkin Elmer Envision Model 2101 plate reader compatible with Alphascreen. % Occupancy was calculated from the control samples that were either untreated with test compound (maximum signal) or were not treated with PP-biotin (minimum signal). IC50 values were determined by fitting dose response curves to a 4 parameter sigmoidal function using GraphPad Prism software.
BTK cell durability using Alphascreen
To treat Ramos B cells with compounds, 1 ml of 1 × 106 cells per well were cultured in a 12-well polystyrene plate. 100 μL of test compound was added to cells to a concentration of 1 μM, and 100 μL of media with 1.1% DMSO was added to control wells. The plates were incubated at 37 °C for 1 hr. Cells were transferred to microfuge tubes, pelleted and washed three times using media, and transferred to a new 12-well plate. The plates were incubated for either 4 hr or 18 hr. The PP-biotin probe was prepared at 15 μM from a 5mM stock using media, and the probe was added to wells at a concentration of 330 nM. The plate was incubated for 1 hr at 37 °C. Cells were pelleted, washed with PBS once, and 60 μL of lysis buffer (Cell Lytic M, Sigma #C2978) was added to each well. Lysates were evaluated for BTK occupancy using Alphascreen as described above.
Human whole blood CD69 expression assay
Human whole blood collected at Stanford Blood Bank in sodium heparin was incubated for 1 hr at 37 °C/5% CO2 in a humidified incubator with a dilution series of compound or 0.5% DMSO alone as a control. Samples were then stimulated with 50 μg/ml of Goat F(ab')2 anti-human IgM (Southern Biotech Cat#2022-14) for 16-18 hrs in a 37 °C/5% CO2 humidified incubator. The blood was stained for CD69 antigen expression on human B cells with BD Biosciences antibodies CD20-FITC (Cat#555622), CD69-APC (Cat#5555633), and corresponding isotype controls according to manufacturer's recommendations. Red blood cells were lysed with 1X BD Biosciences Lysis buffer (Cat#555899) according to manufacturer's recommendations. Cells were washed and resuspended in 1% bovine serum albumin in PBS and analyzed via flow cytometry on a Cytex DxP11 instrument using FlowJo v7.6.4. The percent of CD69 positive cells was plotted as a function of inhibitor concentration and fit to a sigmoidal dose-response using GraphPad Prism software to evaluate inhibitor potency.
T cell receptor and EGFR pathway analysis
The T cell receptor/NFAT reporter assay and the EGFR receptor/AP-1 reporter assay were performed by Thermo Fisher Scientific as part of their SelectScreen® Pathway Profiling Service.
BTK and BTK[C481S] cell assay
The transient transfection assay comparing the activity of BTK wt and BTK(C481S) was performed by Cell Assay Innovations (Beverly, MA) using ClariCELL™ technology (www.cellassayinnov.com). HEK293 cells were transfected with a vector expressing sequence verified for full length human BTK or BTK[C481S], then dispensed into multi-well plates. Test compounds were added, and cells were incubated for 2 hours. The cells were lysed, and an ELISA was performed by capturing the BTK or BTK[C481S] and detecting autophosphorylation levels using a generic anti-phosphotyrosine antibody. BTK and BTK[C481S] dependent kinase activity in these assays were validated by using a kinase-deficient BTK[K430R] control.
In vivo analysis
Sprague Dawley outbred female rats (Harlan Laboratories) were used in these studies. Study animals were dosed orally at 40 mg/kg with 9 formulated as a suspension in 0.5% methylcellulose. Treatment and vehicle groups consisted of 3 and 4 animals per group, respectively. BTK occupancy was measured on individual animals in each group, and the data represented is the average of two independent studies. Blood was drawn via cardiac puncture, 4-5 ml, for PK/PD analysis. Blood plasma exposure from each animal was analyzed for the levels of test compound by LC/MS/MS analysis methods. Samples were injected on a Shimadzu LC20AD HLPC system connected to a Sciex API4000 QTrap mass spectrometer. A Phenomenex Gemini column (2.1 × 50 mm) was used at 40 °C utilizing a flow rate of 400 μL/minute. Mobile Phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. A linear gradient of 5 – 98% B over 2.5 minutes was used. The mass spectrometer was operated in positive ion electrospray mode with multiple reaction monitoring for maximum sensitivity. Sciex Analyst software (version 1.6) was used for LC/MS/MS instrument control and acquisition. Compound concentration was determined against a standard curve of internal standard versus compound peak area ratio. Rat PBMC were isolated with GE Ficoll-Paque Plus (Cat#17-1440-03) according to manufacturer's recommendations. Freshly isolated PBMC, at 10-20 × 106 cells, were incubated with 1 μM PP-BODIPY for 1 hr. Protein lysates were separated on a 4-12% Bis-Tris SDS-Page gel. The BTK-associated fluorescence signal was detected by scanning the gel on a Typhoon image scanner (GE Healthcare). Total BTK was subsequently detected via Western blotting with a BTK antibody (D3H5 clone, Cell Signaling Cat#8547) and an AlexaFluor647 anti-rabbit IgG (H+L) secondary antibody (Invitrogen, Cat#A31573). The signal was quantified using ImageQuantTLv7.0 software. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and had prior approval by Principia Biopharma's IACUC adhering to Protocol PRN-01-2011.
Alphascreen ERK phosphorylation
Human umbilical vein endothelial cells (HUVECs) were utilized to determine compound potency by measuring FGF-induced phospho-Erk1/2 phosphorylation. Approximately 30,000 cells were seeded per well in a 96-well cell culture plate at 37 °C overnight. Cells were incubated in recommended HUVECs media with 10% fetal bovine serum. After 24 hrs of incubation, cells were grown in serum free media for 1 hr prior to compound treatment. Compound dilutions were added to cells and incubated for 1 hr at 37 °C, starting at a concentration of 1 μM, and decreasing in tripling dilutions to a final concentration of 0.05 nM. After a 1 hour incubation period, cells were stimulated with 50 ng/ml of FGF2 for 10 mins. The reaction was stopped by adding 100 μl of ice cold PBS and washed three times. After washing, cells were lysed with 50 μL of 1× Lysis Buffer (pERK SureFire kit, PerkinElmer). Lysates were incubated in a pERK SureFire reaction mixture for a total of 4 hrs as recommended by the manufacturer's protocol. At the end of the incubation period, pERK activity was measure using an Envision multilabel reader (PerkinElmer). The raw signal for pERK activity was used to calculate an IC50 inhibition value as a function of log compound concentration for each compound using Prism software from GraphPad.
phosphoERK washout assay
HUVECs were prepared at a concentration of 0.5 × 106 cells/well in a 6 well plate seeded overnight. Cells were starved in RPMI for one hour prior to the addition of compound (at a concentration 10-fold greater than the compound IC50) or DMSO for 1 hour at 37 °C. Cells were then washed three times to remove unbound inhibitor and returned to culture for the indicated time. Following the washout period, cells were stimulated with 50 ng/ml basic FGF (R&D systems Cat#233-FB) for 10 minutes at 37 °C. Cells were pelleted, washed with cold PBS once, and 40 μl of lysis buffer (Cell Lytic M, Sigma #C2978) was added to each tube. The samples were incubated for 15-20 min on ice and centrifuged for 5 min at 2000 rpm at 4 °C. The lysate was transferred to a new Eppendorf tube and either stored at -80 °C or used immediately. Lysates were added to gel loading buffer and separated using gel electrophoresis. Gels were transferred onto a nitrocellulose membrane and probed for pERK (1:1000 dilution; Cell Signaling Cat#4370) and total ERK (1:2000 dilution; Cell Signaling Cat#9102).
Kd determination using β-mercaptoethanol
Solutions were prepared containing 0, 0.5, 1.5, 5, 15, 50, 150, and 500 mM β-mercaptoethanol (BME) in a 1:1 mixture of ethanol and phosphate buffered saline (pH 7.4, PBS). Aliquots of a 10 mM DMSO stock solution of test compound (4 μL) were separately added to 196 μL aliquots of the above-described ethanol/PBS solutions containing 0 to 500 mM BME. After these solutions had been allowed to stand at room temperature for one hour, they were analyzed using an Agilent 1200 LCMS system equipped with a 50 × 2 mm Phenomenex Luna 5μ C18 100A column. Samples were eluted using a gradient of acetonitrile and water, with both solvents containing 0.1% formic acid. Peaks corresponding to parent and BME adduct were identified by their masses and the percent parent in each sample was determined by measuring the area under the curve for these peaks in the 254 nm UV trace. Percent parent was plotted versus the log of the BME concentration using GraphPad Prism to determine a Kd for the reaction. For 9, parent and BME adduct were not well separated on the LCMS and percent parent was determined using the area under the curve of the appropriate extracted mass peaks from the positive ion trace.
Rate of β-mercaptoethanol reversibility
A 10 μL aliquot of a 10 mM DMSO solution of the test compound was added to a 90 μL aliquot of a 15 mM solution of BME in 1:1 PBS/ethanol. After the mixture had been allowed to equilibrate for about one hour at room temperature, it was analyzed using an Agilent 1200 LCMS system equipped with a 50 × 2 mm Phenomenex Luna 5μ C18 100A column. The sample was eluted using a gradient of acetonitrile and water, with both solvents containing 0.1% formic acid. Peaks corresponding to parent and BME adduct were identified by their masses and the percent adduct in the sample was determined by measuring the area under the curve for these peaks in the 254 nm UV trace. This value was used as the T=0 percent adduct. The sample was then diluted 10-fold by adding a 50 μL aliquot to a 450 μL aliquot of 1:1 PBS/ethanol. The diluted sample was then immediately injected on the LCMS using the same conditions used for the T=0 data point. LCMS injections were repeated as often as the LCMS method would allow (approximately every 6 minutes) for about one half hour. Percent adduct for each time point was determined as described above, then plotted and analyzed using GraphPad Prism. For 9, parent and BME adduct were not well separated on the LCMS and percent adduct was determined using the areas under the curve of the appropriate extracted mass peaks from the positive ion trace.
Structure coordinates
The coordinates of the BTK/3 crystal structure are deposited in the Protein Data Bank with accession code 4YHF.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (NIH) (GM071434 to J.T., F32GM087052 to J.M.M.), the UCSF Stephen & Nancy Grand Multiple Myeloma Translational Initiative (J.T.), the Academy of Finland (V.O.P.), and the Sigrid Juselius Foundation (V.O.P.). We acknowledge the University of California San Francisco (UCSF) Mass Spectrometry Facility (supported by NIH grant P41RR001614).
Footnotes
Author contributions: J.M.M., V.O.P., and J.T. designed experiments involving compounds 1-3. J.M.M. and V.O.P. performed experiments and analyzed data for these compounds. J.M.B., A.B., D.T., V.P., S.R., P.A.N., D.E.K., M.E.G., J.O.F., T.D.O., E.V., K.A.B., R.J.H., and D.M.G. designed and managed experiments involving compounds 4 and higher; J.M.B., A.B., D.T., V.P., S.R., D.F., J.S., V.P., T.T., X.L., and D.L. performed experiments and analyzed data for these compounds; J.M.B. and J.T. wrote the manuscript with feedback from other authors; all authors read and approved the manuscript.
Competing financial interests: The authors declare competing financial interests. J.T., J.M.M., and V.O.P. have filed patent applications on cyanoacrylamide kinase inhibitors (licensed to Principia Biopharma, of which J.T. is a co-founder). J.M.B., A.B., D.T., D.F., J.S., V.P., T.T., X.L., P.A.N., D.E.K., M.E.G., J.O.F., T.D.O., E.V., K.A.B., R.J.H., and D.M.G. are members of Principia Biopharma, which is interested in developing BTK inhibitors for therapeutic applications.
Additional information: Supplementary information is available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for for lead optimization. Nat Rev Drug Disc. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 2.Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Disc. 2010;5:305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- 3.Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol. 2010;14:467–474. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo D, Hillger JM, IJzerman AP, Heitman LH. Drug-target residence time- a case for G protein-coupled receptors. Med Res Rev. 2014;34:856–892. doi: 10.1002/med.21307. [DOI] [PubMed] [Google Scholar]
- 5.Swinney DC, et al. A study of the molecular mechanism of binding kinetics and long residence times of human CCR5 receptor small molecule allosteric ligands. Br J Pharmacol. 2014;171:3364–3375. doi: 10.1111/bph.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louvel J, et al. Agonists for the adenosine A1 receptor with tunable residence time. A case for nonribose 4-amino-6-aryl-5-cyano-2-thiopyrimidines. J Med Chem. 2014;57:3213–3222. doi: 10.1021/jm401643m. [DOI] [PubMed] [Google Scholar]
- 7.Vilums M, et al. Structure-kinetic relationships-an overlooked parameter in hit-to-lead optimization: a case of cyclopentylamines as chemokine receptor 2 antagonists. J Med Chem. 2013;56:7706–7714. doi: 10.1021/jm4011737. [DOI] [PubMed] [Google Scholar]
- 8.Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J Am Chem Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serafimova IM, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barf T, Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem. 2012;55:6243–6262. doi: 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]
- 11.Mah R, Thomas JR, Shafer CM. Drug discovery considerations in the design of covalent inhibitors. Bioorg Med Chem. 2014;24:33–39. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discov. 2012;7:561–581. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]
- 13.Weerpana E, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leproult E, Barluenga S, Moras D, Wurtz JM, Winssinger N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J Med Chem. 2011;54:1347–1355. doi: 10.1021/jm101396q. [DOI] [PubMed] [Google Scholar]
- 15.Liu Q, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh J, Petter RC, Kluge AF. Targeted covalent drugs of the kinase family. Curr Opin Chem Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- 17.Honigberg LA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans EK, et al. Inhibition of BTK with CC-292 provides early pharmacodynamics assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346:219–228. doi: 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- 19.Rankin AL, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191:4540–4550. doi: 10.4049/jimmunol.1301553. [DOI] [PubMed] [Google Scholar]
- 20.Benson MJ, et al. Modeling the clinical phenotype of BTK inhibition in the mature murine immune system. J Immunol. 2014;193:185–197. doi: 10.4049/jimmunol.1302570. [DOI] [PubMed] [Google Scholar]
- 21.Wu H, et al. Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem Biol. 2014;9:1086–1091. doi: 10.1021/cb4008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akinleye A, Chen Y, Mukhi N, Song Y, Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol. 2013;6:59. doi: 10.1186/1756-8722-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lou Y, Owens TD, Kuglstatter A, Kondru RK, Goldstein DM. Bruton's tyrosine kinase inhibitors: approaches to potent and selective inhibition, preclinical and clinical evaluation for inflammatory diseases and B cell malignancies. J Med Chem. 2012;55:4539–4550. doi: 10.1021/jm300035p. [DOI] [PubMed] [Google Scholar]
- 24.Byrd JC, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ML, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Paolo JA, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7:41–50. doi: 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- 27.Xu D, et al. RN486, a selective Bruton's tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther. 2012;341:90–103. doi: 10.1124/jpet.111.187740. [DOI] [PubMed] [Google Scholar]
- 28.Lanning BR, et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuglstatter A, et al. Insights into the conformational flexibility of Bruton's tyrosine kinase from multiple ligand complex structures. Protein Sci. 2011;20:428–436. doi: 10.1002/pro.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcotte DJ, et al. Structures of human Bruton's tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010;19:429–439. doi: 10.1002/pro.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans DA, Ennis MD, Le T, Mandel N, Mandel G. Asymmetric acylation reactions of chiral imide enolates. The first direct approach to the construction of chiral .beta.-dicarbonyl synthons. J Am Chem Soc. 1984;106:1154–1156. [Google Scholar]
- 32.Lebakken CS, et al. Development and validation of a broad-coverage, TR-FRET-based kinase binding assay platform. J Biomol Screening. 2009;14:924–935. doi: 10.1177/1087057109339207. [DOI] [PubMed] [Google Scholar]
- 33.Motulsky HS, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- 34.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. 2nd. John Wiley & Sons; Hoboken, New Jersey: 2013. [PubMed] [Google Scholar]
- 35.Hantschel O, et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci USA. 2007;104:13283–13288. doi: 10.1073/pnas.0702654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eglen RM, et al. The use of Alphascreen technology in HTS: current status. Curr Chem Genomics. 2008;1:2–10. doi: 10.2174/1875397300801010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W, et al. A structure-guided approach to creating covalent FGFR inhibitors. Chem Biol. 2010;17:285–295. doi: 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guagnano V, et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012;2:1118–1133. doi: 10.1158/2159-8290.CD-12-0210. [DOI] [PubMed] [Google Scholar]
- 39.Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40:5172–5180. doi: 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- 40.Swinney DC. Can Binding Kinetics Translate to a Clinically Differentiated Drug? From Theory to Practice. Lett Drug Des Disc. 2006;3:569–574. [Google Scholar]
- 41.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat Rev Drug Disc. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 42.Vauquelin G, Bostoen S, Vanderheyden P, Seeman P. Clozapine, atypical antipsychotics, and the benefits of fast-off D2 dopamine receptor antagonism. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:337–372. doi: 10.1007/s00210-012-0734-2. [DOI] [PubMed] [Google Scholar]
- 43.Kapur S, Seeman P. Antipyschotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J Psychiatry Neurosci. 2000;25:161–166. [PMC free article] [PubMed] [Google Scholar]
- 44.Dubovsky JA, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–2549. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakayama S, et al. A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab Disp. 2009;37:1970–1977. doi: 10.1124/dmd.109.027797. [DOI] [PubMed] [Google Scholar]
- 46.Takakusa H, et al. Covalent binding and tissue distribution/retention assessment of drugs associated with idiosyncratic drug toxicity. Drug Metab Disp. 2008;36:1770–1779. doi: 10.1124/dmd.108.021725. [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.