

Abstract
Dilated Cardiomyopathy (DCM) is a disease of the myocardium characterized by left ventricular dilatation and diminished contractile function. In this report we describe a novel DCM mutation identified for the first time in the myosin regulatory light chain (RLC), replacing Aspartic Acid at position 94 with Alanine (D94A). The mutation was identified by exome sequencing of three adult first-degree relatives who met formal criteria for idiopathic DCM. To gain insight into the functional significance of this pathogenic MYL2 variant, we have cloned and purified the human ventricular RLC wild-type (WT) and D94A-mutant proteins and performed in vitro experiments using RLC-exchanged porcine cardiac preparations. The mutation was observed to induce a reduction in the α-helical content of the RLC and imposed intra-molecular rearrangements. The Ca2+-calmodulin-activated myosin light chain kinase phosphorylation of RLC was not affected by D94A. The mutation was seen to impair the binding of RLC to the MHC (myosin heavy chain), and its incorporation into the RLC-depleted porcine myosin. The actin-activated ATPase activity of mutant-reconstituted porcine cardiac myosin was significantly higher compared to ATPase of WT. No changes in myofibrillar ATPase-pCa relationship were observed in WT- or D94A-reconstituted preparations. Measurements of contractile force showed a slightly reduced maximal tension per cross-section of muscle with no change in calcium sensitivity of force in D94A-reconstituted skinned porcine papillary muscle strips compared with WT. Our data indicate that subtle structural rearrangements in the RLC molecule followed by its impaired interaction with the MHC may trigger functional abnormalities contributing to the DCM phenotype.
Keywords: Myosin Regulatory Light Chain (RLC), Secondary structure, Phosphorylation, ATPase activity, Muscle contraction, RLC-reconstituted β-myosin
INTRODUCTION
Dilated cardiomyopathy (DCM) is a disease of the myocardium characterized by left ventricular dilatation and diminished contractile function [1]. DCM presents with heart failure (HF), arrhythmia and sudden death [2], and is one of the leading causes of HF with high morbidity and mortality and a prevalence of 1/2500 individuals [3]. DCM can be caused by a variety of factors, including ischemia, alcohol toxicity, viral infections, as well as genetic factors [4]. Among these factors, it is estimated that 25% to 48% of all DCM cases are due to familial and genetic factors [3]. Parvari and Levitas have reported that ~35% of DCM is autosomal dominant, although some familial cases could be explained by autosomal recessive or X-linked recessive traits [5]. While clinically available molecular genetic testing can identify those at risk for genetic DCM, epigenetic and sentinel phenomic staging may help to identify those at highest risk in need for intervention [6]. Phenomic staging includes integrating clinical and imaging features, transcriptomic, higher order proteomic and metabolomic interactions, and epidemiological data. Combining family history with clinical screening of relatives, and emphasizing echocardiography to assess left ventricle (LV) size and function is essential to identify familial DCM [6]. More than 30 genes encoding essentially sarcomeric, cytoskeletal, and nuclear proteins are associated with DCM [7]. To date, mutations in at least eight genes encoding for sarcomeric proteins were found to result in DCM: β-MHC (myosin heavy chain), α-MHC, MyBP-C, titin, cardiac actin, α-Tm and troponins [6].
In this report we describe a novel sarcomeric protein mutation in the MYL2 gene encoding for the ventricular regulatory light chain (RLC) of myosin that was identified by exome sequencing in a pedigree with familial dilated cardiomyopathy. The missense variant occurred at amino acid position 94, replacing Aspartic acid by Alanine (D94A), and was not observed in 5400 Exome Variant Server control DNAs. The cardiomyopathy-linked mutations in the MYL2 gene that have been identified to date are mostly associated with hypertrophic cardiomyopathy (HCM) (for review see [8]), and this newly discovered MYL2 variant is the first RLC mutation ever associated with DCM.
To address the mechanisms responsible for triggering cardiac abnormalities due to the D94A mutation in myosin RLC, we have cloned, expressed and purified recombinant human ventricular RLC proteins, D94A mutant and control RLC wild-type (WT) (NCBI accession: NP_000423.2). The proteins were reconstituted into the RLC-depleted porcine cardiac β-myosin, myofibrils and papillary muscle strips and investigated for the structural and functional effects. The ultimate goal was to gain insight into this pathogenic MYL2 variant and provide evidence of its relevance for DCM. Our data indicate that mutation-induced structural rearrangements in the RLC molecule and decreased α-helical content are sufficient to modify the interaction of the RLC with its immediate binding partner, the myosin heavy chain (MHC), and the mutant myosin with actin. These changes when placed in vivo are expected to affect sarcomeric structure and function contributing to the DCM phenotype.
RESULTS
Identification of D94A-MYL2 variant in DCM patients
Twenty two variants, meeting our established criteria for exome sequencing analysis [9, 10], were identified in the three affected family members of the pedigree (Fig. 1). Of these 22, only one variant, missense mutation in TTN (Trp7406Cys), was identified in a gene previously associated with DCM. This variant was not prioritized due to the uncertain relevance of TTN missense mutations in DCM given their relatively high prevalence in control samples. A PKP2 variant (Val587Ile) was also identified; however, its pathogenicity is uncertain given a 0.46% allele frequency among control chromosomes of European ancestry [11]. The MYL2 mutation, on the other hand, is absent in the exome variant server database and had a GERP score of 5.07. Therefore, we hypothesized that this variant may explain DCM in this family [11]. None of the genes harboring the remaining 22 variants were associated with known cardiovascular disorders. No additional MYL2 variants were identified in the 16 other families. Sanger sequencing confirmed the MYL2 variant was present in all three affected family members (I.2, II.1, and II.2).
Figure 1.

Pedigree with DCM, solid symbols denote DCM, open symbols denote unaffected, “+” denotes heterozygous for D94A mutation, arrow points to the proband. Individuals who underwent exome sequencing are denoted “exome”.
Clinical characteristics of family members with the MYL2 mutation are provided in Table 1. The proband (II.2) and his sister (II.1) were diagnosed at ages 24 and 29 after preventive cardiovascular screening revealed mild global left ventricular systolic dysfunction. Medical therapy was initiated upon evidence of decreased ejection fraction. Their mother (I.2) was diagnosed with peripartum cardiomyopathy (PPCM) at age 21. Septal wall thickness measurements were within normal range in all affected family members.
Table 1.
Clinical information of pedigree with D94A-DCM
| Pedigree position |
Age of diagnosis (years) |
ECG / Arrhythmia |
LVEDD, mm (Z- score) |
LVEDD Framingham percentile |
EF (%) |
PWd (mm) |
SWd (mm) |
Comment |
|---|---|---|---|---|---|---|---|---|
| I.1 | NA | none | NA | NA | NA | NA | NA | Normal ECG at age 56 |
| I.2 | 21 | 1 AVB | 56 (3) | > 99th | 33 | 11 | 10 | PPCM |
| II.1 | 29 | NSSTT, ectopic atrial rhythm, PVCs, PACs |
47.2 (0.44) | < 95th | 40 | 7.9 | 5.9 | DCM assignment by systolic dysfunction only. Medical therapy initiated at earliest evidence of decreased ejection fraction with ongoing periodic clinical screening of at-risk relatives. |
| II.2 | 24 | PACs | 55 (1.3) | < 95th | 49 | NA | 7 | |
| II.3 | NA | NSR | 49 (−0.9) | < 95th | 58 | 9 | 9 | Normal echo and ECG at age 31 |
LVEDD = left ventricular end diastolic dimension; EF = ejection fraction; PWd = posterior wall diastole; SWd = septal wall diastole, 1AVB = first degree atrioventricular block; PPCM = peripartum cardiomyopathy; NSSTT = non-specific ST-T wave abnormality; PVCs = premature ventricular contractions; PACs = premature atrial contractions; NSR = normal sinus rhythm.
Effect of D94A on the secondary structure of the human cardiac RLC
Far-UV CD spectroscopy was used to analyze the effect of the D94A mutation on the secondary structure of the RLC. As shown in Fig. 2A, the mutation significantly changed the CD spectrum of the RLC, and the mean residue ellipticity [θ]MRE at 222 nm was [θ]MRE = −6358±140 (n=12) for the D94A mutant versus [θ]MRE = −7424±44 (n=11) for RLC-WT. The difference between the groups was statistically significant (P<0.001). The calculated α-helical content at wavelength 222 nm at 22°C (Eq.1) was 13.3±0.5% in the mutant versus 16.8±0.1% for WT (P<0.001). The latter value for the WT-RLC agrees with the previously determined [θ]MRE at 222 nm [12, 13]. This small but significant decrease in the α-helical content (Fig. 2B) due to the Aspartic acid to Alanine substitution implies that the mutation is able to impose some conformational changes in the RLC structure sufficient to affect the inter-domain organization within the RLC molecule and perhaps its interaction with the MHC.
Figure 2.


A. Effect of the D94A RLC mutation on the CD spectra of RLC. Far-UV CD was performed utilizing a 1-mm path quartz cell in a Jasco J-720 spectropolarimeter. Spectra were recorded at 190–250 nm with a bandwidth of 1 nm. B. The [θ]222nm value was used to calculate the α-helical content (fH) using the following equation: [θ]222 = −30,300 fH − 2,340. Note significant reduction in α-helical content in D94A mutant compared to WT.
Using I-TASSER software, we have analyzed the five lowest energy secondary structures of the human ventricular RLC. The structures were computed using protein templates extracted from the PDB library. The following PDB structures of high similarity to the structure of RLC were used: 3jvtB (Chain B, calcium-bound Scallop Myosin Regulatory Domain (Lever Arm) with reconstituted complete Light Chains), 1prwA (Chain A, crystal structure of Bovine Brain Ca2+ Calmodulin in a compact form), 4ik1A (Chain A, high resolution structure of Gcampj at pH 8.5), 2mysA (Chain A, Myosin Subfragment-1, alpha carbon coordinates only for the two Light Chains), 4i2yA (Chain A, crystal structure of the Calcium Indicator Rgeco1) and 2w4aB (Chain B, isometrically contracting Insect Asynchronous Flight Muscle). The resulting model of the D94A mutant structure (C-score=0.01) is presented in Fig. 3. Fig. 3A demonstrates the WT structure with the Aspartic acid at position 94, and Fig. 3B shows the D94A mutant with the Alanine residue inserted at amino acid position 94. The superimposed structures are pictured in Fig. 3C. Interestingly, the position of the D94A mutation is predicted to be in close proximity to the N-terminal α-helical region of the RLC containing the MLCK-specific phosphorylation site at Ser15. D94A renders a decrease in the apparent distance between the site of the mutation and Ser15 (Fig. 3B). The overlapped structures of WT and the mutant reveal significant intramolecular rearrangements in the RLC molecule imposed by the D94A mutation.
Figure 3.



Modeled structure of human ventricular RLCs, WT (A), D94A (B) and superimposed structures of WT (blue) and D94A (red) (C), using I-TASSER. Note that the Ala94 site is positioned closer to Ser15 than Asp94. The distance between the C-α of Ser15 and C-α of Asp94 is 21.9 Å while that of Ala94 is 13.8 Å. The predicted structures of RLCs were based on 3jvtB, 1prwA, 4ik1A, 2mysA, 4i2yA and 2w4aB PDB structures.
Assessment of time-dependent MLCK phosphorylation of WT and D94A RLCs
To investigate the effect of D94A on RLC function, the time-dependent phosphorylation by exogenously added smooth muscle myosin light chain kinase (smMLCK) [14] (generously provided by Dr. Zenon Grabarek, Boston Biomedical Research Institute, Boston, MA) was conducted on WT and D94A RLCs and the data analyzed with Eq. 2 and 3 (Fig. 4). Near 100% of RLC phosphorylation could be achieved for both WT (n=3) and D94A mutant (n=3) following the treatment with smMLCK (Fig. 4A). The calculated rates of phosphorylation were 1.08±0.05 min−1 in D94A (n=3) and 1.09±0.06 min−1 WT (n=3) (Fig. 4B). As shown in Fig. 3, the D94A mutation was predicted to slightly decrease the distance between the site of the mutation and phosphorylatable Ser15, but this change was found to have no effect on the RLC phosphorylation in vitro.
Figure 4.
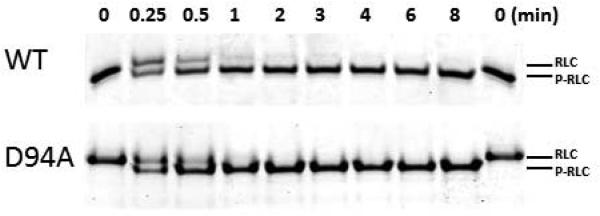

A. Representative Urea-gel picture of the time course of phosphorylation of WT and D94A RLCs. Note a charge based separation of the phosphorylated (P-RLC) and non-phosphorylated RLC bands. B. Time course of MLCK-dependent phosphorylation of WT and D94A RLCs (0 to 8 minutes). Data points were fitted to Eq. 3 to generate phosphorylation kinetics rates (k). Note that both WT and D94A were phosphorylated with the rate of 1.1 min−1 (n=3 independent experiments) and reached the same maximal level of RLC phosphorylation.
Effect of D94A on the RLC - MHC interaction
To gain insight into the effect of D94A on the assembly of the RLC into the lever arm domain of MHC, we have studied the binding profiles of the WT and D94A proteins to the RLC-depleted porcine cardiac myosin. The CDTA/Triton-based treatment yielded ~85% RLC-free myosin, which was then titrated with increasing concentrations of human recombinant WT or D94A RLCs. The degree of RLC reconstitution was calculated based on the ELC/RLC band ratio of reconstituted versus native (untreated) myosin obtained from SDS-PAGE images (Fig. 5A). There was a significant difference in the maximal level of reconstitution between D94A and WT groups incubated with RLC-depleted myosin at ~1.5 µM of RLC per 1 µM of MHC binding sites, with 57±3% saturation for D94A and 70±5% for WT and (P<0.05). The fit of binding isotherms (10 for each group) to Eq.4 yielded a slightly lower binding affinity for the D94A mutant (Kd=4.9±1.4 µM) compared with WT (Kd=3.2±1.4 µM) (not significant), and m1 = 0.15±0.02 and 0.16±0.04; m2 = 0.78±0.28 and 0.90±0.31, and n (apparent stoichiometry) = 0.90±0.30 and 1.26±0.38 for D94A and WT, respectively (Fig. 5B). These results imply that subtle mutation-induced changes in the secondary structure of the RLC (Figs. 2 and 3) are most likely responsible for the inability of the mutant to stoichiometrically bind to RLC-depleted myosin and saturate the MHC binding sites to the level observed for WT.
Figure 5.


The interaction of WT and D94A with the myosin heavy chain. A. Representative 15% SDS-PAGE gel picture of titration experiments of RLC-depleted porcine cardiac myosin with increasing concentrations of WT or D94A RLCs. ELC that remains intact during the depletion-reconstitution procedures was used as a loading control. B. Binding isotherms of WT or D94A to RLC-depleted porcine myosin. The data points were fitted into the nonlinear binding model (Eq. 4 and 5) yielding the apparent dissociation constant (Kd) and stoichiometry (n). Compared to WT, the D94A mutation significantly decreased the maximal level of RLC reconstitution.
Actin activated WT- and D94A-myosin ATPase activity
To further investigate the effect of D94A on the RLC function, WT and D94A RLCs were reconstituted into RLC-depleted porcine cardiac myosin and subjected to actin activated myosin ATPase activity assays. The ATPase activity was plotted as a function of F-actin concentrations (in μM) with data points expressed as average ± SEM of n=13 experiments. The data were fitted to the Michaelis-Menten equation yielding the Vmax and Km parameters. The Vmax represents the maximal rate of ATP turnover by the acto-myosin complex and depicts the transition from the weakly (A·M·ATP↔ A·M·ADP·Pi) to strongly (A·M·ADP↔A·M) attached states [15]. The Km determines the concentration of actin needed to produce half of the activation and stands for the binding affinity of actin to myosin. Fig. 6A shows a significant (P<0.01) increase in Vmax for D94A myosin (0.231±0.009 s−1, n=13) compared to WT myosin (0.195±0.009 s−1, n=10). The Km values were: 0.98±0.03 μM (n=10) for WT and 1.23±0.28 μM (n=13) for D94A. These results demonstrate that while the D94A mutation does not change the affinity of binding (no change in Km), it produces the quicker ATPase cycle (increase in Vmax) compared to WT (Fig. 6A) and predisposes the myosin cross-bridges to undergo a faster transition from the weakly and strongly attached states.
Figure 6.


ATPase activity of WT and D94A reconstituted porcine cardiac myosin and myofibrils.
A. Actin activated myosin ATPase activity. Data points were fitted to the Michaelis-Menten equation to generate Vmax and Km. Note that the D94A mutation significantly increased the maximal level of ATPase activity with no change in Km compared with WT. B. Myofibrillar ATPase activity assay and the ATPase-pCa relationship in WT and D94A reconstituted porcine cardiac myofibrils. Small, but not significant, decreases in the Ca2+ sensitivity and nH were observed for the mutant compared with WT reconstituted myofibrils.
WT- and D94A-reconstituted myofibrillar ATPase activity
We also examined the Ca2+ regulation of acto-myosin interaction in the unloaded myofibrillar ATPase activity assays performed on RLC-depleted and then WT- and D94A- reconstituted porcine cardiac myofibrils. Fig. 6B shows the myofibrillar ATPase activity as a function of increasing Ca2+ concentrations. There was a mutation induced decrease in the Ca2+ sensitivity of myofibrillar ATPase and a decrease in the Hill coefficient compared to WT myofibrils, but the changes were not statistically significant. The midpoint pCa50=5.90±0.04 (n=9) for D94A and pCa50=5.95±0.06 (n=13) for WT and the cooperativity coefficient nH=1.87±0.21 for D94A and nH=2.46±0.30 for WT (Fig 6B). It was quite surprising to observe very little effect of the D94A mutation on the myofibrillar ATPase activity, whereas the mutant significantly enhanced the actin-activated myosin ATPase activity (Fig. 6A). These combine results suggest that while in the acto-myosin complex the D94A mutation predisposes myosin cross-bridges to undergo a faster transition from the weakly to strongly attached states, in the myofibrils this transition is closely regulated by the Ca2+-tropomyosin-troponin complex. The cross-bridge cycling rates in the D94A mutant-reconstituted myofibrils were tuned by the regulatory system to the level observed for WT myofibrils.
Steady state force and force-pCa relationship in WT- and D94A- reconstituted porcine papillary muscle strips
To further study the effect of D94A at the sarcomeric level, WT- and D94A-RLCs were reconstituted into the RLC-free skinned porcine papillary muscle strips and the steady-state force and force-pCa relationship were measured. As shown in Fig. 7, a slight decrease in maximal force generation was observed in D94A-reconstituted strips compared with WT. The values of tension per cross sectional area of muscle strip (in kN/m2) were: D94A: 35.7±1.1 (n=25) versus WT: 37.1±1.1 (n=25) (Fig. 7A). There were no significant alterations in pCa50 of the force-pCa dependence: pCa50=5.67±0.01 (n=25) observed for D94A compared with 5.69±0.01 (n=25) for WT (Fig. 7B). No changes in the Hill coefficient were noted, nH=2.99±0.10 (n=25) for D94A and 2.93±0.10 (n=25) for WT (Fig. 7B). These results indicate that the D94A mutation imposes only small (statistically insignificant) alterations at the level of the sarcomere under unloaded (myofibrillar ATPase) or loaded (force generation) conditions. It is worth mentioning that the myofibrillar ATPase-pCa dependence of the mutant versus WT matches that of the force-pCa relationship observed in skinned papillary muscle fibers.
Figure 7.



Maximal force generation (A) and the force-pCa relationship (B) in skinned papillary muscle strips reconstituted with WT and D94A RLCs (C). A. The difference in maximal tension per cross section of muscle strip was ~3 kN/m2 (P>0.05). B. The force-pCa curve of D94A was overlapping that of WT, indicating no changes in the calcium sensitivity or Hill coefficient. C. Representative force-pCa traces in WT-reconstituted porcine papillary muscle strips.
Effect of RLC phosphorylation on steady state force in WT- and D94A-reconstituted porcine papillary muscle strips
To investigate the effect of RLC phosphorylation on steady-state force, D94A- and WT-reconstituted fibers were treated with smMLCK as described in Materials and Methods (Fig. 8). Measurements of maximal steady-state force showed approximately 1.1-fold increase in maximal tension following the RLC-phosphorylation in both D94A- and WT-reconstituted papillary muscle fibers (Fig. 8A). The P-RLC/RLCtot ratios determined in D94A- versus WT-reconstituted fibers were similar indicating the same level of RLC phosphorylation achieved in both group of reconstituted muscle strips (Fig. 8B).
Figure 8.


The effect of RLC phosphorylation on maximal steady state force in WT- and D94A-reconstituted porcine papillary muscle strips. A. RLC phosphorylation increased maximal force generation by ~1.1-fold in both WT- and D94A-reconstituted fibers. B. Representative Western blots of WT- and D94A-reconstituted fibers before and after the treatment with MLCK.
DISCUSSION
Dilated cardiomyopathy is one of the leading causes for cardiac transplantations and accounts for about 1/3 of all heart failure cases. DCM is characterized by enlarged ventricular dimensions and impaired systolic and diastolic function [16]. In contrast to HCM, DCM is more genetically heterogeneous, with mutations in sarcomeric, nucleoskeletal, mitochondrial, and calcium-handling proteins [16]. Rare variants in over 30 genes, some also involved in other cardiomyopathies, perturb a diverse set of important myocardial proteins to produce a final DCM phenotype [2]. The D94A mutation in MYL2 was identified in a Caucasian family with DCM. Three family members were heterozygous for the D94A mutation. Patient I.2 was diagnosed at the age of 21 with a typical DCM phenotype with increased LVEDD and reduced EF. Patients II.1 and II.2 (proband) displayed abnormal ECG findings and systolic dysfunction as manifested by reduced EF. However, the LV and septal wall thickness were within the normal range (Fig. 1 and Table 1). More than 10 single point mutations in MYL2 have been associated with another form of cardiomyopathy, namely HCM. Interestingly, two of those, D166A [17] and P95A [18] resulted in the alanine for aspartic acid or proline substitution similar to the D94A mutation. The former had also a negatively charged Aspartic acid replaced by Valine (D166V) [19, 20], and this particular MYL2 variant has been extensively studied in our laboratory. The phenotype associated with the D166V mutation closely recapitulated the human HCM phenotype when investigated in transgenic mice expressing human cardiac D166V-RLC [21-23]. One can speculate that at the molecular level, the reduction in charge and potential interruption of the D94-mediated interaction with neighboring amino acids in the RLC may trigger a series of abnormal protein-protein interactions resulting in a DCM phenotype. Therefore, in order to investigate the structure-function relationship as an underlying mechanism, one has to execute a thorough characterization of the structural and functional consequences of the D94A mutation in the RLC molecule.
The regulatory light chain of myosin is a major regulatory subunit of striated-muscle myosin and a modulator of the troponin and Ca2+-controlled regulation of cardiac muscle contraction [24]. RLC is localized at the head-rod junction of the MHC and, together with the ELC, stabilizes the α-helical neck region of the myosin head also called the lever arm [25-27]. The N-terminal domain of RLC contains a divalent cation binding site and the MLCK-specific phosphorylatable Ser15 in the human ventricular RLC sequence. The MLCK-dependent phosphorylation of Ser15 has been shown to be important for the normal function of the heart in health and in disease [28-31]. A significant role of RLC phosphorylation was shown in cardiogenesis [32] and proper myofibrillogenesis [33]. Depressed levels of RLC phosphorylation were observed in patients with heart failure [34, 35] and the inability of RLC to become phosphorylated was shown to result in a DCM phenotype in mice [36]. Interestingly, our studies with HCM causing mutations in the myosin RLC suggest a correlation between the severity of cardiomyopathy phenotype and the level of RLC phosphorylation in vivo [21, 23, 37]. The lack of effects on RLC phosphorylation in the D94A mutant compared to WT observed in this study suggests that RLC phosphorylation may have an important physiological role in the D94A-myocardium to alleviate the consequences of the DCM phenotype. The question exists pertaining to the molecular trigger of DCM in the mutated D94A hearts. It is likely that a mutation induced subtle reduction in the α-helical content of RLC affects the incorporation of the mutant RLC into the lever arm domain and impose changes on the interaction of RLC with the MHC and possibly with the myosin ELC modulating myosin unitary step-size [38]. RLC phosphorylation was shown to be a significant modulator of the myosin activation mechanism facilitating the movement of actin filaments with the largest 8 nm step-size compared to non-phosphorylated 5 nm step-size translation [39]. One can speculate that the ability of D94A to become as phosphorylated as the WT protein in vivo offsets the molecular defects associated with the mutation, e.g. abnormal D94A-MHC interaction. The I-TASSER predicted 3D structure of the RLC showed that the D94A mutation lies in close proximity to the α-helical N-terminal region of RLC that contains the Ser15 site of phosphorylation, and is also in close proximity to the RLC binding site on the MHC. Therefore, it is very likely that the charge changing D94A mutation affects the structure of its N-terminus, disturbs the MHC-RLC and/or ELC-RLC interactions which are offset by MLCK-dependent phosphorylation of D94A mutant.
Given the critical role of the lever arm in regulating the properties of the thick and thin filament interaction, mutations in the RLC molecule are expected to alter the force production and cardiac muscle contraction. However, functional studies utilizing skinned porcine cardiac muscle preparations reconstituted with human recombinant D94A and WT showed no significant changes in force generation or myofilament Ca2+-sensitivity elicited by D94A. Both of these parameters have been reported to be diminished in DCM hearts [40]. One can speculate that the incorporation of RLC-D94A into the MHC has no effect on the steady-state function (ATPase, force, tension-pCa, and force after RLC phosphorylation), but may render the effect on the kinetics of force development, shortening, relaxation, etc., the contractile parameters that were not measured in this study. On the other hand, the experimental system used in our investigation, where papillary muscle strips have to be RLC-depleted and reconstituted with the mutant, might not be sensitive enough to detect subtle differences between the phenotypes. The fiber experiment for RLC-extraction and mutant-reconstitution is quite long in-time and this may affect the ability of the reconstituted muscle strip to develop force to the level of native fibers. The observed irreversible fall in isometric force could be the reason that the system is not sensitive enough to the mutant-elicited functional changes. Perhaps studying the D94A-induced pathogenesis and the effect on heart contractility in transgenic mice in vivo would reveal more physiologically relevant differences. On the other hand, it is possible that this particular DCM causing mutation in the myosin RLC does not recapitulate the Ca2+ dependent changes in force generation as observed for mutations in the regulatory troponins [41, 42].
In summary, we report here a novel single point mutation in the myosin RLC shown to be associated with familial DCM. The D94A mutation is the first disease causing mutation in MYL2 that is linked to the DCM phenotype. The mutation significantly alters the N-terminal α-helical region of the RLC and triggers intramolecular rearrangements in the RLC molecule that ultimately result in alterations in the RLC function. We propose that this is a result of changes in myosin-based chemo-mechanical coupling secondary to alterations in molecular interactions of the RLC with myosin heavy chain, which elicits the DCM phenotype.
MATERIALS AND METHODS
Exome sequencing and characterization of DCM patients
Written informed consent was obtained and study methods were conducted in accordance with the Declaration of Helsinki (1964). In addition, the study was approved by the Institutional Review Boards of Oregon Health and Science University and the University of Miami, Miami FL. All participants provided blood samples for research. Medical records were used to confirm diagnoses. DCM was defined by LV enlargement and systolic dysfunction, with all other detectable causes excluded, including coronary artery disease. Genomic DNA was extracted using standard procedures. Exome sequencing was performed on 3 family members (Fig. 1) at the University of Washington Genome Sciences with capture, sequencing, alignment, and data analysis as previously reported [9, 10]. Variants selected for analysis met the following criteria: ≤0.5% allele frequency in 5400 exomes from the Exome Variant Server (EVS); predicted to alter an amino acid, affect splicing, or lead to coding insertion-deletions; read depth ≥5 and quality scores ≥40; Phastcons score >0.4 or a Genomic Evolutionary Rate Profiling (GERP) score >2; and shared among exomed samples from the same family.
Cloning, expression and purification of WT and D94A Mutant
The cDNA for wild-type (WT) human cardiac RLC (hcRLC) was cloned by reverse transcription-PCR using primers based on the published cDNA sequence (GenBankTM accession number AF020768) and standard methods [13]. The D94A-RLC mutant was generated using overlapping sequential polymerase chain reaction as described earlier [13]. The cDNAs of WT and D94A-RLC were transformed into BL21 expression host cells, and proteins expressed in 16-liter large cultures. Expressed proteins were purified using S-Sepharose column followed by Q-Sepharose column. S-Sepharose column was equilibrated with 2 M urea, 20mM Citrate, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), 0.02% NaN3, pH 6.0. For Q-Sepharose purification the following buffer was used: 2 M urea, 25 mM Tris-HCl, pH 7.5, 0.1 mM PMSF, 1 mM DTT and 0.02% NaN3. The proteins were eluted with a 1000-ml salt gradient of 0–450 mM KCl. The final purity of the proteins was tested using 15% SDS-PAGE. They were stored in Q-Sepharose buffer in −80°C.
CD measurements
Far-UV circular dichroism (CD) spectra were obtained using a 1-mm-path quartz cell in a Jasco J-720 Spectropolarimeter. Spectra were recorded at 190–250 nm with a bandwidth of 1nm at a speed of 50 nm/min and a resolution of 0.5 nm as described in Szczesna et al. [13]. The measurements were performed at 22 °C in 30 mM NaCl, 0.3 mM EGTA, 0.7 mM MgCl2, and 3 mM Tris-HCl buffer at pH 7.4. Analysis and processing of data were done using the Jasco system software (Windows standard analysis, version 1.20). Ten scans were averaged, base lines subtracted, and no numerical smoothing applied. Spectra were presented as mean residue ellipticity and the values of mean residue ellipticity ([θ]MRE, in degree*cm2/dmol) were calculated as shown previously [13]. The α-helical content was calculated using [θ]222:
| (Eq. 1) |
where fH is the fraction of α-helix (in 0-1 scale). fH*100 indicated the percent of α-helical content in WT and D94A proteins.
Secondary structure prediction
The secondary structure prediction was conducted with I-TASSER (online server from Zhanglab, University of Michigan): http://zhanglab.ccmb.med.umich.edu/I-TASSER/. In brief, the amino acid sequence of D94A-RLC was compared against template proteins selected from the PDB library of similar structures. The full length protein was assembled from the excised fragments and simulated into the lowest energy model using specific algorithms. The confidence of each predicted model structure was presented as C-score, ranging from −5 to 2. The quality of prediction was proportional to the value of C-score [43-45]. The predicted structures were then modeled using PyMOL molecular visualization system allowing for the C-α distance determination (in Angstroms, Å) of neighboring amino acid residues.
MLCK phosphorylation of WT and D94A- RLCs
WT and D94A proteins were first dialyzed into phosphorylation buffer containing 30 mM KCl, 20 mM PO4, pH 8.0 and the concentration adjusted to 2 mg/ml. The proteins (1mg/ml) were mixed with 0.5 µM of smooth muscle MLCK, 5 µM calmodulin (CaM), 0.1mM CaCl2 and 12.5 mM MgCl2. The reaction was initiated with the addition of 5mM ATP and carried out at room temperature for different time intervals (in minutes): 0.25, 0.5, 1, 2, 3, 4, 6 and 8. To analyze the level of phosphorylation, 100 µl samples were mixed with 70 mg of ultrapure urea, 10 µl β-ME (β-mercaptoethanol) and 5 µl of Bromophenol Blue and run on 12% Urea gels at 120 Volts for 90 minutes. The bands of non-phosphorylated and phosphorylated (P) RLCs, separated due to charge differences, were scanned using ImageJ software. The level of RLC phosphorylation was calculated as:
| (Eq. 2) |
The time-dependent phosphorylation isotherms for WT and D94A were fitted to the following equation (exponential rise to maximum):
| (Eq. 3) |
where “a” is a constant and “b” is a phosphorylation rate in min−1.
Depletion of native RLC from porcine cardiac myosin and reconstitution with WT and D94A RLCs
For all experiments using porcine cardiac muscle preparations, pig hearts were obtained postmortem from a slaughterhouse. Porcine cardiac myosin was purified as described in Pant et al. [46]. Endogenous RLC was depleted from porcine myosin by incubation of myosin with 1% Triton X-100 and 5 mM CDTA (1, 2-cyclohexylenedinitrilotetraacetic acid), pH 8.5 for 90 minutes on ice. The Triton/CDTA-treated myosin was precipitated in 1 mM ice cold dithiothreitol (DTT) and collected by centrifugation at 750xg for 10 minutes at 4°C. Myosin depleted of endogenous RLC was then re-suspended in reconstitution buffer (0.4 M KCl, 50 mM MOPS, pH 7.0, 2 mM MgCl2, and 1 mM DTT) to 2.6 µM concentration, and was titrated with increasing concentrations of WT or D94A (from 0.3-13 µM). The molar ratio of RLC to depleted myosin ranged from 0.1 to 5.0. Titrations were performed in the presence of BSA to prevent nonspecific RLC binding. The mixtures were incubated for 30 minutes at room temperature and left to precipitate in 13 volumes of ice-cold 1mM DTT for 30 minutes. Protein complexes were then pelleted by centrifugation (8,000xg for 10 minutes) at 4°C, dissolved in small volumes of 3 M KCl (~20 µl) and clarified by ultracentrifugation at 200,000xg for 45 minutes at 4°C. The supernatant was mixed 1:1 ratio with SDS-PAGE sample buffer with ¼ volume of β-ME, heated at 95°C for two minutes and loaded into SDS-PAGE. Coomassie stained gels were scanned and the bands were quantified using the Odyssey Infrared Imaging System. The essential light chain of myosin (ELC), not extracted under the experimental conditions used [46], served as a loading control and the ELC/RLC band ratios between the reconstituted and native (untreated) myosin preparations were used to determine the degree of reconstitution with D94A or WT proteins. The binding isotherms were fitted to the nonlinear binding model:
| (Eq. 4) |
where m1 is the band intensity at 0 µM RLC, m2 is the intensity of the signal at >13 µM RLC and ν is the fractional saturation of binding sites and is a root of the quadratic equation:
| (Eq. 5) |
where “a” and “b” are the total concentrations of RLC-depleted myosin (a=2.6 µM) and WT/D94A RLC, respectively; “n” is the apparent stoichiometry and Kd is the apparent equilibrium dissociation constant [15].
Actin activated myosin ATPase activity assay
Rabbit skeletal F-actin was prepared according to Kazmierczak et al. [47]. The RLC-depleted porcine myosin, obtained as described above, was incubated with a 3 molar excess of human recombinant ventricular WT or D94A and the mixtures were dialyzed for 2 hours against the reconstitution buffer (composition as above). The protein complexes were dialyzed overnight in the cold room against 5 mM DTT. The reconstituted myosins were collected by centrifugation at 8,000xg for 10 minutes and the pellets resuspended in the ATPase buffer (0.4 mM KCl, 10 mM MOPS, pH 7.0, 1 mM DTT). This procedure yielded fully reconstituted D94A- and WT-myosins.
Actin-activated myosin ATPase activity was measured as a function of actin concentration and the data analyzed as described in detail in [47]. Briefly, myosin at ~2.5 µM was added to the 96-well microplate and then mixed with increasing concentrations of F-actin (in μM): 0.1, 2.5, 5, 7.5, 10, 15, 20 and 25. The assay was performed in a 120 μl reaction volume in 25 mM Imidazole, pH 7.0, 4 mM MgCl2, 1 mM EGTA, and 1 mM DTT. The final KCl concentration was 107 mM. Protein mixtures were first incubated on ice for 10 minutes and then for another 10 minutes at 30°C. The reactions (run in triplicate) were initiated with the addition of 2.5 mM ATP with mixing in a Jitterbug incubator shaker (Boekel), allowed to proceed for 20 minutes at 30°C and then terminated by the addition of 5% trichloroacetic acid. Precipitated proteins were cleared by centrifugation and the inorganic phosphate was determined using the Fiske Subbarow method [48]. Data were analyzed using the Michaelis–Menten equation yielding the Vmax and Km parameters [15].
Depletion of native RLC from porcine cardiac myofibrils and reconstitution with WT and D94A
Cardiac myofibrils (CMF) were prepared from the left ventricular walls and papillary muscles of porcine hearts from healthy young animals according to Solaro et al. [49]. CMF were stored at ~20 mg/ml in the CMF buffer containing 30 mM Imidazole, 60 mM KCl, 2 mM MgCl2, pH 7, and 50% glycerol at −20 °C. Before experiments, CMF were removed from 50% glycerol stock, diluted with an equal volume of CMF buffer containing 1 mM DTT, and pelleted at 750xg for 7 minutes. They were re-suspended in CMF buffer and tested for protein concentration (by Coomassie assay). CMF were then diluted to 2.5 mg/ml in the a solution containing 40 mM Tris-HCl, 50 mM KCl,1mM DTT, pH 8.4. The RLC extraction was performed in the buffer containing 5 mM CDTA, 50 mM KCl, 1% Triton X-100, 40 mM Tris-HCl, pH 8.4, for 30 minutes at room temperature. CMF were then washed three times with CMF buffer, assayed for protein concentration, and diluted to 3 mg/ml. The RLC-depleted CMF were then reconstituted with 40 µM WT and/or D94A. Due to potential extraction of troponin C (TnC), ~8 µM porcine cardiac TnC was added to the reconstitution mixture. Complexes were incubated for 1.5 hours on ice. RLC/TnC reconstituted CMF were then washed twice with CMF buffer, assayed for protein concentration, and diluted to ~5 mg/ml in CMF buffer for experiments.
Myofibrillar ATPase assays
Reconstituted and control (not extracted) CMF at ~5 mg/ml were added to the 96-well microplate (~50 µg/well) and myofibrillar ATPase activity assays were performed in a buffer containing 20 mM MOPS, 40 mM KCl, 2.5 mM MgCl2, 2 mM EGTA, 1 mM DTT, pH 7.0, and different concentrations of Ca2+, from pCa 8 to 4.5 (reaction volume 120 μl). After a 5 minute-incubation at 25°C, the reaction was initiated with 2.5 mM MgATP and terminated after 15 minutes with 6.25% trichloroacetic acid. Released inorganic phosphate was measured according to Fiske and Subbarow [48]. The ATPase-pCa curves were analyzed using the Hill equation [50], where "[Ca2+]50 or pCa50" is the free Ca2+ concentration which produces 50% of the maximal ATPase and nH is the Hill coefficient.
Depletion of native RLC from porcine cardiac muscle strips and reconstitution with WT and D94A
Freshly isolated porcine hearts were placed in oxygenated physiological salt solution of 140 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 1.8 mM NaH2PO4, 5.5 mM glucose, and 50 mM HEPES buffer, pH 7.4. The papillary muscles of the left ventricles were isolated, dissected into muscle bundles of about 20 × 3 mm, and chemically skinned in a 50% glycerol, 50% pCa 8 solution (10−8 M [Ca2+], 1 mM free [Mg2+] (total MgPr (propionate) =3.88 mM), 7 mM EGTA, 2.5 mM [Mg-ATP2−], 20 mM MOPS, pH 7.0, 15 mM creatine phosphate and 15 units/ml of phosphocreatine kinase, ionic strength =150 mM adjusted with KPr) containing 1% Triton X-100 for 24 h at 4 °C. Then the strips were transferred to the same solution without Triton X-100 and stored at −20 °C for about 2 months.
Depletion of endogenous RLC from porcine cardiac muscle preparations was achieved in 5 mM CDTA, 40 mM Tris, 50 mM KCl, 1 µg/ml pepstatin A, 0.6 mM NaN3, 0.2 mM PMSF, and 1% Triton X-100, pH 8.4. Muscle strips were incubated in this solution for 5 minutes at room temperature and then transferred to the fresh solution of the same composition for another 30 minutes. The extent of RLC extraction was tested by SDS-PAGE.
Reconstitution of the RLC-depleted muscle strips with WT or D94A mutant (and porcine cardiac TnC) was performed in pCa 8 solution containing 40 µM RLC and 15 µM TnC. The solution of TnC was included in the reconstitution protein mixture during the first 30 minutes of fiber incubation followed by a 30 minute incubation with freshly added 40 µM RLC at room temperature. The addition of cTnC was to assure that the strips were not deficient of TnC, because its partial extraction could affect the Ca2+ sensitivity of force development. Reconstituted strips were then washed in pCa 8 solution and subjected to force measurements.
Steady-state force development
Small muscle strips of approximately 1.4 mm in length and 100 μm in diameter were attached by tweezer clips to a force transducer. The strips were placed in a 1 ml cuvette and freshly skinned in 1% Triton X-100 dissolved in pCa 8 buffer for 30 minutes. They were rinsed 3 times × 5 minutes in pCa 8 buffer and their length adjusted to remove the slack. This procedure resulted in sarcomere length of ~2.2 μm as judged by the first order optical diffraction pattern as described in [51]. Then the strips were tested for maximal steady state force development in pCa 4 solution (composition is the same as pCa 8 buffer except the [Ca2+] =10−4 M). Maximal tension readings at pCa 4 were taken before and after the force-pCa curve, averaged and expressed in kN/m2. The cross sectional area of the muscle strip was assumed to be circular.
The Ca2+ dependence of force development
After the initial steady state force was determined, muscle strips were relaxed in pCa 8 buffer and exposed to solutions of increasing Ca2+ concentrations from pCa 8 to pCa 4. The level of force was measured in each "pCa" solution. Data were analyzed using the Hill equation, where "[Ca2+]50 or pCa50" is the free Ca2+ concentration which produces 50% of the maximal force and nH is the Hill coefficient. The pCa50 represents the measure of Ca2+ sensitivity of force and the nH is the measure of myofilament cooperativity.
Effect of RLC phosphorylation on maximal steady-state force development
After reconstitution of the RLC-depleted papillary muscle strips with WT or D94A mutant (performed on the force transducer - see above), the fibers were subsequently incubated with 1μM smMLCK and 5 μM CaM dissolved in the pCa 6 buffer (composition is the same as pCa 8 buffer except the [Ca2+] =10−6 M) for 30 minutes at room temperature. Then the strips were washed twice in pCa 8 solution and maximal force was measured. After experiments, the RLC-phosphorylated strips were dissolved in SDS-PAGE sample buffer and loaded on 15% SDS-PAGE. The level of RLC phosphorylation was determined with phospho-specific RLC antibody (produced in this laboratory) [52] and compared to the total RLC content assessed with a rabbit polyclonal RLC CT-1 antibody recognizing total RLC protein. Band intensities were measured using Image J software and the +P-RLC/RLCtotal ratios were determined in D94A- versus WT-reconstituted fibers. Total ELC probed with the anti-ELC antibody was used as a loading control.
Statistical Analysis
All values are shown as means ±SEM. Statistically significant differences between two groups (WT and D94A) were determined using an unpaired Student’s t-test (Sigma Plot 11; Systat Software, San Jose, CA), with significance defined as P<0.05.
ACKNOWLEDGEMENTS
The authors thank Dr. Zenon Grabarek for the preparation of smooth muscle myosin light chain kinase. This work was supported by NIH grants R01 HL-071778 and HL-108343 to DSC, R01 HL-58626 to REH, and AHA-12PRE12030412 to WH.
ABBREVIATIONS
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- PPCM
peripartum cardiomyopathy
- ELC
essential light chain of myosin
- RLC
regulatory light chain of myosin
- MHC
myosin heavy chain
- MYL2
gene encoding human ventricular RLC
- TTN
gene encoding human titin
- PKP2
gene encoding human plakophilin
- LVEDD
left ventricular end diastolic dimension
- EF
ejection fraction
- PWd
posterior wall diastole
- WT
wild-type
REFERENCES
- 1.Schonberger J, Seidman CE. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet. 2001;69:249–60. doi: 10.1086/321978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nature reviews Cardiology. 2013;10:531–47. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 3.Millat G, Bouvagnet P, Chevalier P, Sebbag L, Dulac A, Dauphin C, Jouk PS, Delrue MA, Thambo JB, Le Metayer P, Seronde MF, Faivre L, Eicher JC, Rousson R. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Genet. 2011;54:e570–5. doi: 10.1016/j.ejmg.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Szczesna-Cordary D, Morimoto S, Gomes AV, Moore JR. Cardiomyopathies: classification, clinical characterization, and functional phenotypes. Biochem Res Int. 2012;2012:870942. doi: 10.1155/2012/870942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parvari R, Levitas A. The mutations associated with dilated cardiomyopathy. Biochem Res Int. 2012;2012:639250. doi: 10.1155/2012/639250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piran S, Liu P, Morales A, Hershberger RE. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. Journal of the American College of Cardiology. 2012;60:283–9. doi: 10.1016/j.jacc.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Current opinion in cardiology. 2011;25:198–204. doi: 10.1097/HCO.0b013e328337ba52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muthu P, Huang W, Kazmierczak K, Szczesna-Cordary D. Functional Consequences of Mutations in the Myosin Regulatory Light Chain Associated with Hypertrophic Cardiomyopathy. In: Veselka J, editor. Cardiomyopathies – From Basic Research to Clinical Management. InTech; Croatia: 2012. pp. 383–408. Ch. 17. [Google Scholar]
- 9.Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S, Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA, Hershberger RE. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–53. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA, Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. American journal of human genetics. 2011;88:273–82. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norton N, Robertson PD, Rieder MJ, Zuchner S, Rampersaud E, Martin E, Li D, Nickerson DA, Hershberger RE, National Heart L, Blood Institute, G. O. E. S. P. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet. 2012;5:167–74. doi: 10.1161/CIRCGENETICS.111.961805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang W, Wilson GJ, Brown LJ, Lam H, Hambly BD. EPR and CD spectroscopy of fast myosin light chain conformation during binding of trifluoperazine. Eur J Biochem. 1998;257:457–65. doi: 10.1046/j.1432-1327.1998.2570457.x. [DOI] [PubMed] [Google Scholar]
- 13.Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, Zhi G, Stull JT, Potter JD. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. The Journal of biological chemistry. 2001;276:7086–92. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- 14.Mabuchi Y, Mabuchi K, Stafford WF, Grabarek Z. Modular structure of smooth muscle Myosin light chain kinase: hydrodynamic modeling and functional implications. Biochemistry. 2010;49:2903–17. doi: 10.1021/bi901963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazmierczak K, Muthu P, Huang W, Jones M, Wang Y, Szczesna-Cordary D. Myosin Regulatory Light Chain Mutation Found In Hypertrophic Cardiomyopathy Patients Increases Isometric Force Production in Transgenic Mice. Biochem J. 2012;442:95–103. doi: 10.1042/BJ20111145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. The Journal of clinical investigation. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez-Acosta L, Mazzanti A, Fernández X, Ortí M, Barriales-Villa R, García D, Maneiro E, Rebolo P, Álvarez E, Monserrat L. Regulatory Light Chain (MYL2) Mutations in Familial Hypertrophic Cardiomyopathy. Journal of Cardiovascular Disease. 2014;2 [Google Scholar]
- 18.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–9. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 19.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet J-P, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M, EUROGENE Heart Failure Project Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 20.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet J-P, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M, EUROGENE Heart Failure Project Correction to: "Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy". Circulation. 2004;109:3258. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 21.Kerrick WGL, Kazmierczak K, Xu Y, Wang Y, Szczesna-Cordary D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2009;23:855–865. doi: 10.1096/fj.08-118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muthu P, Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski Z, Szczesna-Cordary D, Borejdo J. Single molecule kinetics in the familial hypertrophic cardiomyopathy D166V mutant mouse heart. Journal of molecular and cellular cardiology. 2010;48:989–998. doi: 10.1016/j.yjmcc.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muthu P, Kazmierczak K, Jones M, Szczesna-Cordary D. The effect of myosin RLC phosphorylation in normal and cardiomyopathic mouse hearts. Journal of cellular and molecular medicine. 2012;16:911–919. doi: 10.1111/j.1582-4934.2011.01371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szczesna D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:187–97. doi: 10.2174/1568006033481474. [DOI] [PubMed] [Google Scholar]
- 25.Burghardt TP, Sikkink LA. Regulatory light chain mutants linked to heart disease modify the cardiac myosin lever arm. Biochemistry. 2013;52:1249–59. doi: 10.1021/bi301500d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geeves MA. Molecular motors: Stretching the lever-arm theory. Nature. 2002;415:129–131. doi: 10.1038/415129a. [DOI] [PubMed] [Google Scholar]
- 27.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–8. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 28.Toepfer C, Caorsi V, Kampourakis T, Sikkel MB, West TG, Leung MC, Al-Saud SA, MacLeod KT, Lyon AR, Marston SB, Sellers JR, Ferenczi MA. Myosin regulatory light chain (RLC) phosphorylation change as a modulator of cardiac muscle contraction in disease. The Journal of biological chemistry. 2013;288:13446–54. doi: 10.1074/jbc.M113.455444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM, Solaro RJ. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. The Journal of biological chemistry. 2009;284:5097–106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis JS, Satorius CL, Epstein ND. Kinetic effects of myosin regulatory light chain phosphorylation on skeletal muscle contraction. Biophys J. 2002;83:359–370. doi: 10.1016/S0006-3495(02)75175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. The American journal of physiology. 1993;264:C1085–95. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 32.Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, Shintani Y, Wakeno M, Minamino T, Kondo H, Furukawa H, Nakamaru K, Naito A, Takahashi T, Ohtsuka T, Kawakami K, Isomura T, Kitamura S, Tomoike H, Mochizuki N, Kitakaze M. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest. 2007;117:2812–2824. doi: 10.1172/JCI30804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terry M, Walker DD, Ferrari MB. Protein phosphatase activity is necessary for myofibrillogenesis. Cell Biochem Biophys. 2006;45:265–78. doi: 10.1385/CBB:45:3:265. [DOI] [PubMed] [Google Scholar]
- 34.van der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PML, Hasenfuss G, Stienen GJM. The effect of myosin light chain 2 dephosphorylation on Ca2+-sensitivity of force is enhanced in failing human hearts. Cardiovascular research. 2003;57:505–514. doi: 10.1016/s0008-6363(02)00662-4. [DOI] [PubMed] [Google Scholar]
- 35.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PBJ, Goldmann P, Jaquet K, Stienen GJM. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovascular research. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 36.Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, Tangney J, Hidalgo CG, Chung CS, Cheng H, Dalton ND, Gu Y, Kasahara H, Ghassemian M, Omens JH, Peterson KL, Granzier HL, Moss RL, McCulloch AD, Chen J. Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest. 2012;122:1209–21. doi: 10.1172/JCI61134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang W, Liang J, Kazmierczak K, Muthu P, Duggal D, Farman GP, Sorensen L, Pozios I, Abraham T, Moore JR, Borejdo J, Szczesna-Cordary D. Hypertrophic Cardiomyopathy Associated Lys104Glu Mutation in the Myosin Regulatory Light Chain Causes Diastolic Disturbance in Mice. Journal of molecular and cellular cardiology. 2014;74:318–329. doi: 10.1016/j.yjmcc.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Ajtai K, Burghardt TP. The Qdot-labeled actin super-resolution motility assay measures low-duty cycle muscle myosin step size. Biochemistry. 2013;52:1611–21. doi: 10.1021/bi301702p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Ajtai K, Burghardt TP. Ventricular myosin modifies in vitro step-size when phosphorylated. Journal of molecular and cellular cardiology. 2014;72:231–7. doi: 10.1016/j.yjmcc.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–49. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatraman G, Harada K, Gomes AV, Kerrick WG, Potter JD. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. The Journal of biological chemistry. 2003;278:41670–6. doi: 10.1074/jbc.M302148200. [DOI] [PubMed] [Google Scholar]
- 42.Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS, Potter JD. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2009;2:306–13. doi: 10.1161/CIRCGENETICS.108.846733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic acids research. 2012;40:W471–7. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pant K, Watt J, Greenberg M, Jones M, Szczesna-Cordary D, Moore JR. Removal of the cardiac myosin regulatory light chain increases isometric force production. FASEB J. 2009;23:3571–3580. doi: 10.1096/fj.08-126672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kazmierczak K, Xu Y, Jones M, Guzman G, Hernandez OM, Kerrick WGL, Szczesna-Cordary D. The Role of the N-Terminus of the Myosin Essential Light Chain in Cardiac Muscle Contraction. J Mol Biol. 2009;387:706–725. doi: 10.1016/j.jmb.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. The Journal of biological chemistry. 1925;66:375–400. [Google Scholar]
- 49.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochimica et biophysica acta. 1971;245:259–62. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 50.Hill TL, Einsenberg E, Greene LE. Theoretical model for the cooperative equilibrium binding of myosin subfragment-1 to the actin-troponin-tropomyosin complex. Proc Natl Acad Sci. 1980;77:3186–3190. doi: 10.1073/pnas.77.6.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Muthu P, Szczesna-Cordary D, Kawai M. Characterizations of myosin essential light chain's N-terminal truncation mutant Delta43 in transgenic mouse papillary muscles by using tension transients in response to sinusoidal length alterations. J Muscle Res Cell Motil. 2013;34:93–105. doi: 10.1007/s10974-013-9337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muthu P, Wang L, Yuan CC, Kazmierczak K, Huang W, Hernandez OM, Kawai M, Irving TC, Szczesna-Cordary D. Structural and functional aspects of the myosin essential light chain in cardiac muscle contraction. FASEB J. 2011;25:4394–4405. doi: 10.1096/fj.11-191973. [DOI] [PMC free article] [PubMed] [Google Scholar]