

Abstract
Activation of the immune system in the central nervous system and oxidative stress play important roles in traumatic brain injury (TBI)-induced cognitive impairment. Rhamnetin possesses anti-inflammatory and anti-oxidative properties. This study aimed to detect the possible effects of rhamnetin on cognitive deficit, hippocampal inflammatory factors, and oxidative stress in rats with TBI. In this study, we established the traumatic brain injury model in rats. Rats respectively received vehicle saline or rhamnetin for 21 days. Cognitive functions were evaluated by assessing the acquisition of spatial learning and memory retention in Morris Water Maze test from day 15 to 19 post TBI. Levels of interleukin (IL)-1β, IL-6, IL-8, tumor necrosis factor a (TNF-a), IL-10, and nuclear factor κB (NF-κB) in hippocampal homogenate were measured using ELISA. Oxidative stress was analysed by investigating the activities of MDA, H2O2, SOD, and GSH-Px. We found that rhamnetin significantly improved cognitive impairment in rats with TBI, and inhibited the inflammatory response and oxidative stress in the hippocampus. The results suggested that rhamnetin could enhance the recovery of cognitive deficits induced by TBI, and that its mechanism might be associated with the inhibition of inflammation and oxidative stress in the hippocampus.
Keywords: rhamnetin, cognitive deficit, TBI, inflammation, oxidative stress
Introduction
Traumatic brain injury (TBI) is a serious public health issue that affects millions of people worldwide (more than 1.7 million new cases each year within the United States alone), and severe TBI is a leading cause of death and disability in industrialised countries [1]. Besides the initial trauma (the direct impact or penetrating injury), the secondary injury to the brain, resulting from delayed biochemical and cellular changes induced by TBI, contributes to the occurrence of the disability. Cognitive deficit is one of the most significant disabilities after TBI, with an incidence of approximately 65% in moderate to severe TBI cases [2, 3]. However, few agents are available to effectively control the cognitive deficit caused by TBI.
It has been increasingly recognised that activation of the immune system in the central nervous system is a key component of the normal aging process, but also of the pathological development and progression of many neurological disorders, including TBI. Traumatic brain injury often triggers a series of chronic neuroinflammatory cascades in the brain [4, 5]. The neuroinflammatory response develops within the first week and persists for several months after traumatic brain injury, but returns to control levels after several years [7]. The secondary post-traumatic neuroinflammation can lead to neuronal injury that may contribute to the occurrence of cognitive deficit, especially neuroinflammation and neurodegeneration in the hippocampus [4, 8, 9]. The roles of inflammatory cytokines tumor necrosis factor α (TNF-α), interleukin (IL)-1β, IL-6, and IL-8 are well known in neurodegeneration and cognitive deficit. Recently, Su reported that levels of inflammatory cytokine high-sensitivity C-reactive protein (hs-CRP) are associated with cognitive impairment after TBI [10]. Furthermore, some drugs or exercises that counteracted the inflammation (including hippocampal inflammation) induced by TBI, for example reducing the expression of IL-1β, IL-6, and TNF-α, also improved the cognitive recovery [11–13]. Thus, it seems that anti-inflammation may be a potential method to manage the cognitive deficit induced by TBI.
Evidence showed that TBI also results in an excess of oxidative stress [14] that may further exacerbate the inflammation or directly induce neuronal injury, which is associated with cognitive impairment after TBI. Elevated levels of H2O2 were found in subjects with TBI. The overproduction of H2O2 can activate nuclear factor κB (NF-κB), which can promote the production of IL-1β, IL-6, and TNF-α, and ultimately exacerbates the inflammatory response. Overproduction of malondialdehyde (MDA), a highly reactive electrophilic aldehyde, in the brain is also frequently reported in TBI. Some studies have shown levels of antioxidant enzymes SOD, and GPx increase in animal models of TBI, but some other studies have reported the opposite results [15]. Nonetheless, some drugs possessing anti-oxidative activity improved the cognitive impairment in animal model [16]. Thus, anti-oxidative stress seems potentially useful in the management of cognitive deficit caused by TBI. The relationship between TBI and oxidative stress has generated interest in the development of antioxidant therapies for TBI management.
Many natural phenolic compounds have been reported for their anti-inflammatory and anti-oxidative activities. Rhamnetin, a phenolic flavonoid compound, has been used to manage some disorders in animal models. Recently, it was shown to have anti-inflammatory and anti-oxidative activities in some cell and animal models, such as in B16 cells, mouse macrophage-derived RAW264.7 cells, and in rats with paw oedema [17–19]. However, its activity in animal models of TBI has not been investigated, and its effect on cognitive deficit caused by TBI is unknown to date.
Thus, in this study we aimed to investigate the effects of rhamnetin on cognitive deficit as well as hippocampal inflammatory markers and oxidative stress in rats with TBI.
Material and methods
Animals
All procedures and protocols used in the present study were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, and they were approved by the Committee on the Ethics of Animal Experiments of Yidu Central Hospital. Male Sprague-Dawley (SD) rats, weighing 300-350 grams (Animal Centre of LUKANG Company), were randomly assigned to 6 groups (10 rats in each group): control group, sham group, TBI group, rhamnetin low-dose group (TBI + L), rhamnetin middle-dose group (TBI + M), and rhamnetin high-dose group (TBI + H). The rats were housed in a temperature-controlled (20-24°C) and humidity-controlled (60 ±3%) room with a 12: 12 hour light/dark cycle. The rats were fed with free access to food and water (5 rats in each cage).
Traumatic brain injury model
Traumatic brain injury was induced in rats according to the methods previously described elsewhere [20]. Briefly, prior to the surgical procedures, rats were anesthetised using chloral hydrate. Following a midline incision, the rats underwent a 6-mm diameter bilateral frontal craniotomy (the lesion site was set 3 mm anterior and lateral to the bregma). Subsequently, the animal was connected to a pneumatic cortical contusion device and a 3.0-mmdeep contusion was produced using an impact velocity of 2.25 m/s for 500 ms. The sham animals only received midline incision without craniotomy and brain injury. The control animals received no surgical procedures. The incision was immediately sutured after the TBI procedure.
Rhamnetin treatment
Rhamnetin dissolved in saline was given daily to rats in the TBI + L group, TBI + M group, and TBI + H group, respectively, at the dose of 50, 100, and 200 mg/kg/day by intragastric administration for 21 consecutive days, starting from the day TBI was performed (day 1). The control group and sham group were administrated with vehicle saline. The range of doses was based on our pilot test.
Cognitive assessment
For cognitive assessment, spatial learning and memory were examined using the Morris water maze (MWM) test, as previously described [20, 21]. Briefly, a large circular tank (180 cm in diameter, 60 cm in height) was filled with tap water (30 cm in depth) (26 ±1°C) and surrounded by curtains to eliminate spatial cues from surroundings in the testing room. A platform (10 cm in diameter) was placed 2 cm below the water surface in the southwest quadrant. Spatial learning consisted of four trials per day with an intertribal of 4 minutes for five consecutive days starting from day 15. In the spatial learning testing, the rat was randomly placed in each of the 4 start locations (north, east, south, and west) to climb onto the platform, where the rats remained for 30 seconds before being removed from the apparatus. Animals unable to find the hidden platform within 2 minutes were gently guided to it by the experimenter. The time and swim distance of the four trials of each animal were recorded and averaged. Visible platform trials with a platform 2 cm above the water surface were conducted on day 20, rats unable to find the visible platform were removed from all data sets. Memory retention was evaluated by a single probe trial 24 hours after the visible platform trials, with the platform removed. Briefly, each rat was placed into the maze at the most distal point from where the platform was previously set and allowed to swim freely for 30 seconds. The time spent in the target quadrant and the swim speed of each animal were recorded.
Tissue homogenate preparation
Animals were sacrificed on day 21 under deep anesthetisation with chloral hydrate. The rat hippocampus was immediately removed from the brain and homogenised in iced saline (ratio: 0.1 g hippocampus tissue: 1 ml saline). Supernatant of the tissue homogenate was collected, sub-packaged, and stored (at –80°C) for the following detections.
Cytokines and nuclear factor κB analysis
Concentrations of TNF-α, IL-1β, IL-6, IL-8, IL-10, hs-CRP, and NF-κB in supernatant of the hippocampus homogenate were measured using ELISA assay kits strictly according to the manufacturer's instructions (Bioss Company, Beijng, China).
Oxidative stress analysis
Contents of MDA, H2O2, SOD, and GSH-Px in supernatant of the hippocampus homogenate were detected using commercially available assay kits (Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer's instruction.
Statistical analysis
Statistical analysis was performed using one-way ANOVA with subsequent Students-Newman-Keuls (SNK). Data were reported as mean ± SD. Differences were considered statistically significant if p < 0.05.
Results
Cognitive function: acquisition of spatial learning
Rats in the TBI group required longer times and travelled farther distances to locate the escape platform than did the control group and the sham group on day 15, 16, 17, 18, and 19 (all p < 0.05), which suggests significant cognitive impairment in TBI rats. Rats in the TBI + M group and TBI + + H group required less time and travelled shorter distances to locate the platform than did the rats in the TBI group on day 15, 16, 17, 18, and 19 (all p < 0.05), but the TBI + L group did not (all p > 0.05). No significant difference in the performance of the control group and the sham group was observed on day 15, 16, 17, 18, and 19 (all p > 0.05). The results indicated that rhamnetin could partially reverse the cognitive impairments induced by TBI. Shown in Tables 1 and 2.
Table 1.
Effects of rhamnetin on the time spent in locating the escape platform
| Day 15 (s) | Day 16 (s) | Day 17 (s) | Day 18 (s) | Day 19 (s) | |
|---|---|---|---|---|---|
| Control | 60.2 ±5.1 | 50.3 ±4.8 | 43.6 ±3.8 | 30.5 ±3.9 | 20.1 ±3.3 |
| Sham | 62.1 ±6.0 | 53.2 ±5.5 | 44.1 ±3.1 | 33.4 ±4.0 | 23.6 ±3.7 |
| TBI | 103.9 ±8.6a,b | 99.4 ±8.6a,b | 93.5 ±7.6a,b | 82.2 ±5.9a,b | 75.9 ±7.0a,b |
| TBI + L | 95.6 ±8.4a,b | 90.4 ±9.5a,b | 82.6 ±8.0a,b | 70.3 ±6.2a,b | 65.1 ±8.2a,b |
| TBI + M | 86.8 ±6.9a,b,c | 79.7 ±7.1a,b,c | 70.5 ±5.7a,b,c | 58.1 ±6.3a,b,c | 39.6 ±4.9a,b,c |
| TBI + H | 79.2 ±6.1a,b,c | 75.3 ±8.0a,b,c | 62.8 ±4.9a,b,c | 51.7 ±4.5a,b,c | 35.8 ±4.1a,b,c |
Control – control group; Sham – sham group; TBI – TBI group; TBI + L = rhamnetin low-dose group; TBI + M = rhamnetin middle-dose group; TBI + H = = rhamnetin high-dose group. Spatial learning consisted of four repeated trials per day with an intertribal of 4 minutes. The time of the four trials of each animal were averaged. Data were expressed as mean ± SD (N = 10 per group) and analysed using one-way ANOVA with subsequent SNK test.
p < 0.05, vs. the control group
p < 0.05, vs. the sham group
p < 0.05, vs. the TBI group
Table 2.
Effects of rhamnetin on the path length to locate the escape platform
| Day 15 (cm) | Day 16 (cm) | Day 17 (cm) | Day 18 (cm) | Day 19 (cm) | |
|---|---|---|---|---|---|
| Control | 1630 ±136 | 1390 ±140 | 1106 ±96 | 826 ±79 | 570 ±66 |
| Sham | 1707 ±146 | 1406 ±149 | 1195 ±101 | 900 ±114 | 631 ±71 |
| TBI | 2884 ±297a,b | 2605 ±302a,b | 2401 ±260a,b | 2139 ±249a,b | 1956 ±335a,b |
| TBI + L | 2655 ±200a,b | 2349 ±186a,b | 2235 ±201a,b | 1761 ±267a,b | 1503 ±250a,b |
| TBI + M | 2305 ±198a,b,c | 2018 ±198a,b,c | 1890 ±153a,b,c | 1618 ±182a,b,c | 999 ±116a,b,c |
| TBI + H | 2235 ±207a,b,c | 1965 ±171a,b,c | 1629 ±184a,b,c | 1406 ±135a,b,c | 963 ±91a,b,c |
Control – control group; Sham – sham group; TBI – TBI group; TBI + L – rhamnetin low-dose group; TBI + M – rhamnetin middle-dose group; TBI + H – rhamnetin high-dose group. Spatial learning consisted of four repeated trials per day with an intertribal of 4 minutes. The swim distances of the four trials of each animal were averaged. Data were expressed as mean ± SD (N = 10 per group) and were analysed using one-way ANOVA with subsequent SNK test.
p < 0.05, vs. the control group
p < 0.05, vs. the sham group
p < 0.05, vs. the TBI group
Cognitive function: probe trial (memory retention)
Rats in the TBI group spent a lower percentage of the allotted time (30 seconds) in the target quadrant than did the control group and sham group (both p < 0.05). Rats in the TBI + M group and TBI + H group spent greater percentages of time in the target quadrant than did the rats of the TBI group (both p < 0.05), but the TBI + L group did not (p > 0.05); however, their performance was still worse than the control group and the sham group (both p < 0.05). No significant difference in the performance between the control group and the sham group was observed (p > 0.05). The results indicate that rhamnetin could improve the memory retention in the TBI rats. Shown in Table 3.
Table 3.
The effects of rhamnetin on the percentage of time spent in the target quadrant and on swim speed
| Percentage (%) | Swim speed (cm/s) | |
|---|---|---|
| Control | 40.6 ±5.1 | 28.3 ±2.2 |
| Sham | 43.9 ±4.0 | 27.6 ±2.0 |
| TBI | 23.2 ±3.0a,b | 27.3 ±2.5 |
| TBI + L | 26.0 ±2.3a,b | 27.6 ±2.1 |
| TBI + M | 31.5 ±2.8a,b,c | 28.1 ±3.0 |
| TBI + H | 35.6 ±4.1a,b,c | 28.0 ±3.2 |
Control – control group; Sham – sham group; TBI – TBI group; TBI + L – rhamnetin low-dose group; TBI + M – rhamnetin middle-dose group; TBI + H – rhamnetin high-dose group. The probe trial consisted of a single 30-second trial. Data were expressed as mean ± SD (N = 10 per group) and were analysed using one-way ANOVA with subsequent SNK test.
p < 0.05, vs. the control group
p < 0.05, vs. the sham group
p < 0.05, vs. the TBI group
Cognitive: swim speed
There were no significant differences in swim speed among any of the groups during the last day's testing (all p > 0.05). The result excluded the possible false effects of the performance resulting from the motor impairments in the Morris water maze test. Shown in Table 3.
Cytokines and nuclear factor κB
We found increased hippocampal levels of TNF-α, IL-1β, IL-6, IL-8, hs-CRP, and NF-κB, as well as decreased levels of IL-10, in the rats of the TBI group compared to the control group and the sham group (all p < 0.05). The changes induced by TBI were significantly reversed by rhamnetin in the TBI + M group and the TBI + H group, but not in the TBI + L group, compared to the TBI group. No differences were observed between the control group and the sham group (all p > 0.05). Shown in Figures 1 and 2.
Fig. 1.
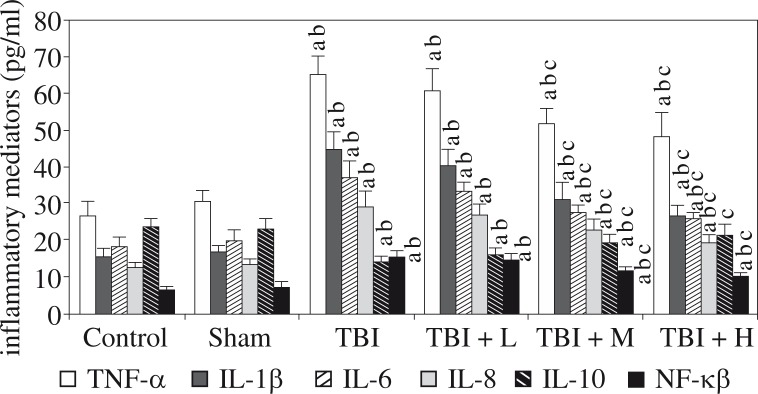
Effects of rhamnetin on inflammatory mediators Control – control group; Sham – sham group; TBI – TBI group; TBI + L – rhamnetin low-dose group; TBI + M – rhamnetin middle-dose group; TBI + H – rhamnetin high-dose group. The investigations were repeated 3 times and the 3 values of each animal were averaged. Data were expressed as mean ± SD (N =10 per group) and analyzed using one-way ANOVA with subsequent SNK test. ap < 0.05, vs. the control group; bp < 0.05, vs. the sham group; cp < 0.05, vs. the TBI group
Fig. 2.

Effects of rhamnetin on hs-CRP Control – control group; Sham – sham group; TBI – TBI group; TBI + L – rhamnetin low-dose group; TBI + M – rhamnetin middle-dose group; TBI + H – rhamnetin high-dose group. The investigations were repeated 3 times and the 3 values of each animal were averaged. Data were expressed as mean ± SD (N = 10 per group) and analyzed using oneway ANOVA with subsequent SNK test. ap < 0.05, vs. the control group; bp < 0.05, vs. the sham group; cp < 0.05, vs. the TBI group
Oxidative stress
Elevated hippocampal levels of MDA and H2O2 as well as reduced levels of SOD and GSH-Px were observed in the TBI group compared to the control group and the sham group. Rhamnetin significantly reversed the changes in the TBI + M group and TBI + H group, but not in the TBI + L group, compared to the TBI group; these levels in the control group and the sham group were similar (all p > 0.05). Shown in Figures 3 and 4.
Fig. 3.

Effects of rhamnetin on oxidative markers Control – control group; Sham – sham group; TBI – TBI group; TBI + L – rhamnetin low dose group; TBI + M – rhamnetin middle dose group; TBI + H – rhamnetin high dose group. The investigations were repeated 3 times and the 3 values of each animal were averaged. Data were expressed as mean ± SD (N = 10 per group) and analyzed using one-way ANOVA with subsequent SNK test. ap < 0.05, vs. the control group; bp < 0.05, vs. the sham group; cp < 0.05, vs. the TBI group
Fig. 4.

Effects of rhamnetin on anti-oxidative markers Control – control group; Sham – sham group; TBI – TBI group; TBI + L – rhamnetin low dose group; TBI + M – rhamnetin middle dose group; TBI + H – rhamnetin high dose group. The investigations were repeated 3 times and the 3 values of each animal were averaged. Data were expressed as mean ± SD (N = 10 per group) and analyzed using one-way ANOVA with subsequent SNK test. ap < 0.05, vs. the control group; bp < 0.05, vs. the sham group; cp < 0.05, vs. the TBI group
Discussion
Our work showed that administration of rhamnetin improved the cognitive deficit and inhibited hippocampal inflammatory response and oxidative stress in rats with TBI.
Many patients with TBI experience a short-term or long-term cognitive deficit, which reduces the quality of life of TBI survivors. Nonetheless, limited effective methods in treating the cognitive deficit induced by TBI are available in clinics. Rhamnetin is a phenolic flavonoid compound possessing various pharmacological activities, such as cardioprotective, anti-inflammatory, anti-oxidative, and anti-apoptotic effects [17–19, 22]. In order to detect the possible effects of rhamnetin on the cognitive deficit induced by TBI, we established a TBI rat model using a pneumatic cortical contusion device as described previously, the use of which can be seen extensively in literature [20]. Then, a Morris water maze apparatus system was employed to examine the cognitive impairment of the TBI rats. Acquisition of spatial learning and memory in the Morris water maze test is sensitive in monitoring the cognitive function of animals and its use can be seen widely in literature [20, 21]. We found that the rats in the TBI group spent more time in locating the escape platform and travelled longer path length in the spatial learning test and spent less time in target quadrant in the probe trial than the rats in the control group and the sham group. The results suggest that the experimental TBI induced significant cognitive impairment in rats. However, rhamnetin treatment significantly improved the performance of the rats in the TBI + M group and TBI + H group, but not in the TBI + L group, compared to the TBI group. The improved performance in the Morris water maze test indicated that rhamnetin had protective effects on cognitive function in rats with TBI. However, it is thought that the possible motor impairments may have an influence on the performance of animals in the Morris water maze test. In this study, we observed the swim speed of the rats and found no significant differences in swim speed among any of the groups. The result excluded the possible false effects of the performance resulting from the motor impairments during the Morris water maze test.
It has been increasingly recognised that activation of the immune system in the central nervous system is a key component of the normal aging process, but also of the pathological development and progression of many neurological disorders, including TBI [23]. Studies have confirmed the chronic neuroinflammatory cascade in the brain after TBI [4, 5]. Immune dysfunction, especially inflammation, has been well demonstrated to play a role in the pathogenesis of cognitive deficit [9, 16, 24, 25]. Anti-inflammatory management seems effective on cognitive impairment. Consistently, our results showed the levels of inflammatory factors TNF-α, IL-1β, IL-6, IL-8, and hs-CRP in the hippocampus increased and the anti-inflammatory cytokine IL-10 in the hippocampus decreased, in the TBI group, compared to the control group and the sham group. This confirmed the neuroinflammation in the hippocampus of the TBI rats. However, rhamnetin treatment significantly reduced the levels of these inflammatory factors and elevated IL-10 levels in the TBI + M group and TBI + H group, but not in the TBI + L group, compared to the TBI group. The results indicate that rhamnetin could inhibit the hippocampal inflammatory response in TBI rats, and that the effect was dose dependent. The findings were consistent with previous studies that reported the anti-inflammatory effects of rhamnetin [17–19]. The inhibition of the inflammatory response by rhamnetin should play a role in the improvement of the cognitive function in TBI rats.
In TBI there is a considerable increase in the production of free radicals and a decrease in the production of antioxidant enzymes, supporting the theory that oxidative stress plays an important role in the secondary injury [14–16, 26]. Consistently, our results showed elevated hippocampal levels of oxidative stress markers MDA and H2O2 as well as reduced levels of antioxidant enzymes SOD and GSH-Px in the TBI group, compared to the control group and the sham group, indicating an excess of oxidative stress in hippocampus. Besides its direct neurotoxicity, oxidative stress can promote an inflammatory response. H2O2 is a factor that can activate NF-κB, which promotes the production of inflammatory cytokines IL-1β, IL-6, and TNF-α. Consistently, we found increased levels of hippocampal NF-κB in rats of the TBI group in the present study. However, rhamnetin treatment significantly reduced the oxidative stress and NF-κB production in the TBI + M group and TBI + H group, but not in the TBI + L group, compared to the TBI group. The inhibition of oxidative stress and NF-κB should contribute to the attenuation of hippocampal inflammatory response and to the improvement of cognitive function in TBI rats.
The mechanisms underlying the secondary injury of TBI induced by neuroinflammation and oxidative stress are complex. It has been well demonstrated that the activation of microglia is one of the contributors [27]. In some studies, microglial activation has been found to persist for years after the initial brain trauma, particularly in moderate-to-severe TBI [27]. The microglial activation may occur in many regions of the brain, including the hippocampus [28]. Upon activation, microglia transforms from a resting state to an activated state. The activated microglia can promote the secretion of many factors, including inflammatory mediators such as TNF-α, IL-1β, IL-6, and NF-κB [29], leading to neuroinflammation. In addition, the highly activated microglia at the margins of the expanding lesion can express NADPH oxidase, which leads to the overproduction of reactive oxygen species (ROS), ultimately resulting in excessive oxidative stress in the brain [27]. However, the excessive ROS in turn can activate NF-κB, which promotes the production of cytokines TNF-α, IL-1β, and IL-6 [30–32]. Thus, microglial activation can induce an inflammatory-oxidative cascade in the body, and NF-κB plays an important role in the process. Subsequently, the microglial-mediated neuroinflammation and oxidative stress produce chronic and progressive neurodegenerative changes leading to late neurological dysfunction, including cognitive deficit. Consistently, as mentioned above, our findings in the present study showed rhamnetin both improved the cognitive deficit induced by TBI and inhibited overproduction of hippocampal inflammatory mediators, including NF-κB and oxidative stress markers, which might be associated with microglial activation. Furthermore, interestingly, a recent study by Lutz revealed that rhamnetin could inhibit the lipopolysaccharide-induced release of inflammatory mediators from BV2 microglia [33], suggesting the microglial inhibition activity of rhamnetin. Taken together, we suppose that the microglia-mediated neuroinflammation and oxidative stress in the hippocampus might be the target of rhamnetin in our study.
In summary, rhamnetin can improve the cognitive deficit induced by TBI and may inhibit hippocampal inflammatory response and oxidative stress, and the effects are dose dependent. However, the enhancement in cognitive performance should be further evaluated and replicated in other models of TBI. More other possible mechanisms that mediate the beneficial effects should be further investigated.
The authors declare no conflicts of interest.
References
- 1.Robertson CS, Garcia R, Gaddam SS, et al. Treatment of mild traumatic brain injury with an erythropoietin-mimetic peptide. J Neurotrauma. 2013;30:765–774. doi: 10.1089/neu.2012.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dikmen SS, Corrigan JD, Levin HS, et al. Cognitive outcome following traumatic brain injury. J Head Trauma Rehabil. 2009;24:430–438. doi: 10.1097/HTR.0b013e3181c133e9. [DOI] [PubMed] [Google Scholar]
- 3.Walker KR, Tesco G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front Aging Neurosci. 2013;5:29. doi: 10.3389/fnagi.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cordaro M, Impellizzeri D, Paterniti I, et al. Neuroprotective effects of Co-ultraPEALut on secondary inflammatory process and autophagy involved in traumatic brain injury. J Neurotrauma. 2014 doi: 10.1089/neu.2014.3460. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 5.Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics. 2015;12:143–150. doi: 10.1007/s13311-014-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith C, Gentleman SM, Leclercq PD, et al. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol. 2013;39:654–666. doi: 10.1111/nan.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aungst SL, Kabadi SV, Thompson SM, et al. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab. 2014;34:1223–1232. doi: 10.1038/jcbfm.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gentleman SM, Leclercq PD, Moyes L, et al. Longterm intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 9.Tompkins P, Tesiram Y, Lerner M, et al. Brain injury: neuro-inflammation, cognitive deficit, and magnetic resonance imaging in a model of blast induced traumatic brain injury. J Neurotrauma. 2013;30:1888–1897. doi: 10.1089/neu.2012.2674. [DOI] [PubMed] [Google Scholar]
- 10.Su SH, Xu W, Li M, et al. Elevated C-reactive protein levels may be a predictor of persistent unfavourable symptoms in patients with mild traumatic brain injury: a preliminary study. Brain Behav Immun. 2014;38:111–117. doi: 10.1016/j.bbi.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Wei J, Pan X, Pei Z, et al. The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J Trauma Acute Care Surg. 2012;73:654–660. doi: 10.1097/TA.0b013e31825133c0. [DOI] [PubMed] [Google Scholar]
- 12.Sauerbeck A, Gao J, Readnower R, et al. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol. 2011;227:128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Si D, Wang H, Wang Q, et al. Progesterone treatment improves cognitive outcome following experimental traumatic brain injury in rats. Neurosci Lett. 2013;553:18–23. doi: 10.1016/j.neulet.2013.07.052. [DOI] [PubMed] [Google Scholar]
- 14.Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. Biomed Res Int. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva LFA, Hoffmann MS, Gerbatin RDR, et al. Treadmill exercise protects against pentylenetetrazol-induced seizures and oxidative stress after traumatic brain injury. J Neurotrauma. 2013;30:1278–1287. doi: 10.1089/neu.2012.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tiwari V, Chopra K. Attenuation of oxidative stress, neuroinflammation, and apoptosis by curcumin prevents cognitive deficits in rats postnatally exposed to ethanol. Psychopharmacology (Berl) 2012;224:519–535. doi: 10.1007/s00213-012-2779-9. [DOI] [PubMed] [Google Scholar]
- 17.Jnawali HN, Lee E, Jeong KW, et al. Anti-inflammatory activity of rhamnetin and a model of its binding to c-Jun NH2-terminal kinase 1 and p38 MAPK. J Nat Prod. 2014;77:258–263. doi: 10.1021/np400803n. [DOI] [PubMed] [Google Scholar]
- 18.Kim YJ. Rhamnetin attenuates melanogenesis by suppressing oxidative stress and pro-inflammatory mediators. Biol Pharm Bull. 2013;36:1341–1347. doi: 10.1248/bpb.b13-00276. [DOI] [PubMed] [Google Scholar]
- 19.Mondal A, Rajalingam D, Kumar Maity T. Anti-inflammatory effect of O-methylated flavonol 2-(3,4-dihydroxy-phenyl)-3,5-dihydroxy-7-methoxy-chromen-4-one obtained from Cassia sophera Linn in rats. J Ethnopharmacol. 2013;147:525–529. doi: 10.1016/j.jep.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Cope EC, Morris DR, Scrimgeour AG, Levenson CW. Use of zinc as a treatment for traumatic brain injury in the rat: effects on cognitive and behavioral outcomes. Neurorehabil Neural Repair. 2012;26:907–913. doi: 10.1177/1545968311435337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaw KE, Bondi CO, Light SH, et al. Donepezil is ineffective in promoting motor and cognitive benefits after controlled cortical impact injury in male rats. J Neurotrauma. 2013;30:557–564. doi: 10.1089/neu.2012.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park ES, Kang JC, Jang YC, et al. Cardioprotective effects of rhamnetin in H9c2 cardiomyoblast cells under H2O2-induced apoptosis. J Ethnopharmacol. 2014;153:552–560. doi: 10.1016/j.jep.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Marks W, Golabek-Dropiewska K, Bryl E, et al. Immunomonitoring in patients with early moderate and severe head trauma. Centr Eur J Immunol. 2013;38:494–499. [Google Scholar]
- 24.Martín-Moreno AM, Brera B, Spuch C, et al. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers (-amyloid levels and improvescognitive performance in Tg APP 2576 mice. J Neuroinflammation. 2012;9:8. doi: 10.1186/1742-2094-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwalm MT, Pasquali M, Miguel SP, et al. Acute brain inflammation and oxidative damage are related to longterm cognitive deficits and markers of neurodegeneration in sepsis-survivor rats. Mol Neurobiol. 2014;49:380–385. doi: 10.1007/s12035-013-8526-3. [DOI] [PubMed] [Google Scholar]
- 26.Ji X, Liu W, Xie K, et al. Beneficial effects of hydrogen gas in rat model of traumatic brain injury via reducing oxidative stress. Brain Res. 2010;1354:196–205. doi: 10.1016/j.brainres.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 27.Loane DJ, Kumar A, Stoica BA, et al. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bedi SS, Hetz R, Thomas C, et al. Intravenous multipotent adult progenitor cell therapy attenuates activated microglial/macrophage response and improves spatial learning after traumatic brain injury. Stem Cells Transl Med. 2013;2:953–960. doi: 10.5966/sctm.2013-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Min JS, Kim B, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response inmicroglia cells by regulating MAPK and NF-(B pathways. Neurosci Lett. 2015;584:191–196. doi: 10.1016/j.neulet.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Li J, Yue Y, et al. Genistein suppresses tumor necrosis factor α-induced inflammation via modulating reactive oxygen species/Akt/nuclear factor κB and adenosine monophosphate-activated protein kinase signal pathways in human synoviocyte MH7A cells. Drug Des Devel Ther. 2014;8:315–323. doi: 10.2147/DDDT.S52354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Luo F, Zhao H. Paraquat-induced reactive oxygen species inhibit neutrophil apoptosis via a p38 MAPK/NF-κB-IL-6/TNF-α positive-feedback circuit. PLoS One. 2014;9:e93837. doi: 10.1371/journal.pone.0093837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Wang L, Li NL, et al. Subanesthetic isoflurane reduces zymosan-induced inflammation in murine Kupffer cells by inhibiting ROS-activated p38 MAPK/NF-(B signaling. Oxid Med Cell Longev. 2014;2014:851692. doi: 10.1155/2014/851692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lutz JA, Kulshrestha M, Rogers DT, et al. A nicotinic receptor-mediated anti-inflammatory effect of the flavonoid rhamnetin in BV2 microglia. Fitoterapia. 2014;98:11–21. doi: 10.1016/j.fitote.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]