

Abstract
Background
Repolarization-delay is a common clinical problem which can promote ventricular arrhythmias. In myocytes, abnormal sarcoplasmic reticulum Ca2+-release is proposed as the mechanism that causes early afterdepolarizations, the cellular equivalent of ectopic-activity in drug-induced long QT syndrome. A crucial missing link is how such a stochastic process can overcome the source-sink mismatch to depolarize sufficient ventricular tissue to initiate arrhythmias.
Methods and Results
Optical maps of action potentials (APs) and Ca2+-transients (CaT) from Langendorff rabbit hearts were measured at low (150×150 μm2/pixel) and high (1.5×1.5 μm2/pixel) resolution before and during arrhythmias. Drug-induced long QT type 2, elicited with dofetilide inhibition, produced spontaneous Ca2+-elevations during diastole and systole, before the onset of arrhythmias. Diastolic Ca2+− waves appeared randomly, propagated within individual myocytes, were out-of-phase with adjacent myocytes and often died-out. Systolic secondary Ca2+− elevations were synchronous within individual myocytes, appeared 188±30ms after the AP-upstroke, occurred during high cytosolic-Ca2+ (40–60% of peak-CaT), appeared first in small islands (0.5×0.5 mm2) that enlarged and spread throughout the epicardium. Synchronous systolic Ca2+-elevations preceded voltage-depolarizations (9.2±5ms, n=5) and produced pronounced Spatial Heterogeneities of CaT-durations and AP-durations. Early afterdepolarizations originating from sites with the steepest gradients of membrane-potential propagated and initiated arrhythmias. Interestingly, more complex subcellular Ca2+-dynamics (multiple chaotic Ca2+-waves) occurred during arrhythmias. K201, a ryanodine receptor stabilizer, eliminated Ca2+-elevations and arrhythmias.
Conclusions
The results indicate that systolic and diastolic Ca2+-elevations emanate from sarcoplasmic reticulum Ca2+-release and systolic Ca2+-elevations are synchronous because of high cytosolic and luminal-sarcoplasmic reticulum Ca2+ which overcomes source-sink mismatch to trigger arrhythmias in intact hearts.
Keywords: calcium transients, electrophysiology, long QT syndrome, optical mapping, arrhythmia (mechanisms), early afterdepolarization, sarcoplasmic reticulum, ryanodine receptor stabilizer, K201
Introduction
Delayed ventricular repolarization is frequently encountered in clinical practice. It is manifested by prolongation of QT interval on surface ECG and a propensity to potentially lethal polymorphic ventricular tachycardia.1, 2 Occasionally, the condition is congenital, caused by mutation(s) in one of the genes encoding cardiac ion channel subunits or auxiliary proteins.3–7 More commonly, it is acquired,8–10 caused by exposure to certain medications, electrolyte abnormalities or coronary ischemia. The mechanism linking action potential (AP) prolongation to ventricular arrhythmias remains incompletely understood.
At the single-cell level, early afterdepolarizations are observed as a result of AP prolongation.11 It is believed that triggered activity caused by afterdepolarizations, along with increased dispersion of AP duration (APD), result in functional reentry and ventricular arrhythmias.12 The precise sequence of events leading to early afterdepolarizations remains disputed. It was suggested that early afterdepolarizations are caused by the “window-current” of L-type Ca2+-channels during phase 3 of the AP, and the spontaneous reactivation of ICa,L.13, 14 However, subsequent compelling evidence showed that early afterdepolarizations are caused by Ca2+-release from overloaded sarcoplasmic reticulum,15 resulting in an elevation of intracellular Ca2+ (Cai) and subsequent augmentation of the Cai-dependent depolarizing current carried by the forward mode of the sodium-calcium exchanger.16, 17 Another related issue is whether early afterdepolarizations caused by sarcoplasmic reticulum Ca2+-overload and spontaneous Ca2+-release can overcome the source-sink mismatch to successfully propagate and initiate an arrhythmia in intact hearts. Synchronization of afterdepolarizations was experimentally demonstrated by a local injection of a high concentration of norepinephrine which elicited Ca2+-overload and a focal arrhythmia.18 Similar mechanisms cause delayed afterdepolarizations in digoxin toxicity or catecholaminergic polymorphic ventricular tachycardia.19, 20
We have recently reported that development of secondary systolic Ca2+-elevations (SSCEs) precedes the development of ventricular arrhythmias by several minutes in a rabbit Langendorff heart with drug-induced repolarization-delay.21 In this model, SSCEs preceded membrane depolarizations at sites of ectopic foci. The temporal relationship between Cai and Vm and pharmacological manipulation supported sarcoplasmic reticulum Ca2+-release as the primary mechanism of early afterdepolarizations. However, the limited spatial resolution did not allow the detection of subcellular Ca2+ phenomena such as Ca2+-waves, which have been described in excitable22 and non-excitable23 cells. Extensive studies have established that cytosolic Ca2+-elevation can elicit voltage depolarization by reverse Cai to voltage coupling and may contribute to arrhythmogenesis under specific circumstances. A critical challenge remains to explain how a stochastic process such as sarcoplasmic reticulum Ca2+-release can depolarize the cellular membrane sufficiently to produce a propagating early afterdepolarization and thereby initiate an arrhythmia. Here, we investigate Ca2+-dynamics at subcellular and the whole-heart resolution in perfused hearts (rather than dissociated cells) to gain new insights on the role of Ca2+-handling in eliciting afterdepolarizations and ventricular arrhythmias in rabbit model of drug-induced long QT type-2 (LQT2).
Methods
Langendorff preparation
Adult New Zealand White rabbits (females, 60 to 120 days old) were anesthetized with pentobarbital sodium (75 mg/kg, intravenously) and anticoagulated with heparin (200 U/kg intravenously). The heart was rapidly isolated, cannulated and perfused with Tyrode’s solution containing (in mM) 130 NaCl, 24 NaHCO3, 1.0 MgCl2, 4 KCl, 1.2 NaH2PO4, 50 dextrose, and 1.25 CaCl2 (at pH 7.2–7.4), gassed with 95% O2 plus 5% CO2. Temperature was continuously monitored and regulated by a feedback system at 37 ± 2°C. Epicardial bipolar pseudo-ECG was continuously monitored.24 The atrioventricular node was ablated by electrocautery to allow full control of heart rate by pacing.
This investigation conformed to the current Guide for Care and Use of Laboratory Animals, published by the National Institutes of Health, and was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Optical mapping
Dual optical mapping setups for macroscopic and microscopic measurements of CaTs and Vm in Langendorff perfused whole hearts were previously described.25 Briefly, the isolated heart was perfused with blebbistatin (5–10 μM; Sigma, St Louis, MO) for 5–10 min to minimize motion artifact, immobilized in a chamber and stained with a voltage-sensitive dye (PGH1 or RH237: 200 μl of 1 mg/ml in dimethyl sulfoxide) and loaded with a Ca2+ indicator (Rhod-2 AM: 200 μl of 1 mg/ml in dimethyl sulfoxide). The anterior surface of the heart was illuminated with a 520 ± 30-nm excitation beam, and the fluorescence emitted by Rhod-2 and PGH1 was separated by a dichroic mirror (660 nm) and was focused on two complementary metal-oxide-semi-conductor cameras.25 For macroscopic imaging, each camera viewed 1.5 × 1.5 cm2 from the anterior surface of the heart using a Nikon camera lens (50 mm, 1:1.2). For subcellular imaging, the heart was perfused horizontally in a chamber on the stage of an upright microscope (Olympus BX61W1) using a ×40 water immersion objective (Olympus, ×40/LUMPLFL).26 The sampling frequency was 500 Hz for macroscopic and 200–500 Hz for microscopic imaging.
Study protocol
The tolerance for motion artifact is much lower for high magnification compared to low-magnification imaging. To address this issue, we used a custom-designed plastic Lucite which has a heart-shaped cavity in the bottom, carved out of sylgard. Here, the heart is perfused and imaged in a horizontal position and is further stabilized by a plastic plate suspended over the heart by 4 metal bars fastened to the rim of the chamber. The pressure on the heart by the plate is adjustable by 4 spring-loaded screws. The epicardium is imaged from the top with an upright microscope where the 40× water-immersion objective accesses the heart through an oval opening on the compression plate allowing for the short working distance between the objective and epicardial surface. For these measurements, the heart was perfused continuously with a high concentration of blebbistatin (15 μM).
Simultaneous optical measurement of CaT and AP was performed at baseline and during drug-induced LQT2 conditions. Isolated whole hearts were paced with an epicardial unipolar electrode placed on the lateral wall of the right ventricle, approximately halfway between the apex and base. Hearts were paced at a cycle length of 0.5 sec (120 beats/minute), and subsequently slowed to 1.2 seconds (50 beats/minute; profound bradycardia for rabbit hearts). After baseline recordings, LQT2 was induced by perfusion of Tyrode’s solution containing dofetilide (500 nM, Pfizer, New York, NY), a selective IKr blocker, and by lowering K+ and Mg2+ concentrations by 50%.21, 26 In some experiments, 3-(4-benzylcyclohexyl)-1-(7-methoxy-2,3-dihydrobenzo[f][1,4]thiazepin-4(5H)-yl)propan-1-one (K201, 1 μM) was added to the perfusate before or after the induction of long QT type 2. K201 was a kind gift from Dr. Jonathan J. Abramson and Dr. Robert Strongin from Portland State University, Portland OR.
Data analysis
The amplitude of optically measured CaT and AP in each pixel was normalized between 0 and 1, and activation time at each site was calculated from maximum first derivative of the fluorescent signal [(dF/dt)max] of the local AP or CaT upstroke. APD and CaT duration (CaTD) at each site were calculated from the interval between (dF/dt)max and the recovery of Vm and CaT traces to 20% of baseline (APD80 or CaTD80), respectively. Automatic measurement of APD80 and CaTD80 from all pixels (100 × 100 pixels) was used to calculate mean APD80 and CaTD80. The dispersion of APD80 was defined as the standard deviation of APD80 measured from all pixels. The amplitude of the Vm gradient vector was defined as
The gradients were calculated from discrete data (100 × 100 pixels) and spatial step of 3 pixel size was used to approximate the partial derivatives in the formula above. Standard deviation of Vm and CaT amplitude was considered a measure of spatial heterogeneity (SH) of the respective signals. It was calculated from all 100 × 100 pixels at each time and then averaged over repolarization time-interval (from 100 ms after action potential upstroke to the longest APD80). SH values are reported as a natural logarithm of the time-averaged standard deviation. Phase angles of CaT and AP at each pixel were calculated based on modified phase analysis and used to identify the rise-time of CaT and AP. Briefly, after local maxima and local minima were defined at a given pixel, individual segments between neighboring local minima and maxima were normalized between 0 and π (rising phase) or –π and 0 (falling phase). Phase maps were used to visualize the spatiotemporal patterns of CaT/AP oscillation. Zero phase angles were used to identify the rise-times (take-off times) of Cai and Vm elevation during AP plateau. Correlation coefficient (r) between amplitudes of normalized CaT/AP at a given time or APD/CaTD was calculated by using MATLAB software. In microscopic imaging, changes in CaT and AP were analyzed by converting our two-dimensional data to stacked line scans along the individual cardiomyocyte as previously described. Wilcoxon signed-rank test (XLSTAT, Addinsoft) was used for statistical analysis and box-whisker diagrams displaying data range, interquartile range and median were used to visualize the distribution of the data.
Cell Isolation
Ventricular myocytes were isolated from female New Zealand white rabbits (10–12 weeks) with a standard protocol. Briefly, rabbits were euthanized as described above, the heart was excised and Langendorff-perfused with a Ca2+ containing Tyrode’s solution (containing (in mM): 140 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, 5.5 Glucose, at pH 7.4) for 5 min, then with a Ca2+-free Tyrode’s solution supplemented with 0.02% BSA for 10 min, after which collagenase type II (Worthington, 0.6 mg/ml) was added for a 15 min digestion at 37°C, followed by Ca2+-free Tyrode for 5 min. Hearts were removed and placed in a high K+, Kraft-Bruhe (KB) solution containing (in mM): 110 potassium glutamate, 25 KCl, 10 KH2PO4, 2 MgSO4, 20 taurine, 5 creatine, 0.5 EGTA, 5 HEPES, 20 glucose, pH 7.4 with KOH. The sub-epicardium at the base of the left ventricle was excised, minced and myocytes were obtained by filtering through 200 μm nylon mesh. Cells were allowed to settle to form a pellet and washed with KB solution, then re-suspended in an incubation medium: M199 (free of phenol red) with 5% FBS (fetal bovine serum) and 100 μg/ml primocin (InvivoGen).27–29
Electrophysiology
ICa,L was measured with whole-cell configuration of the patch-clamp technique. The pipettes were filled with (in mM): 130 CsCl, 20 tetraethylammonium chloride, 5 EGTA, 5 HEPES, 5 Mg-ATP, 0.1 Tris-GTP, 0.1 cAMP, 10 Creatine phosphate (pH 7.2 with CsOH). The external solution contained (in mM): 140 NaCl, 5.4 CsCl, 2.5 CaCl2, 0.5 MgCl2, 11 glucose, and 5.5 HEPES (pH 7.4 with NaOH). Currents were measured with an Axopatch 1D amplifier. ICa,L was measured from a holding potential of −80 mV and peak ICa-L was recorded during 100 ms voltage-clamp steps from −30 to +60 mV in 10 mV increments following a 40 ms pre-pulse at −30 mV which served to inactivate INa. Capacitance measurements were obtained from membrane test parameters. The current density (pA/pF) for each cell was normalized in relation to its capacitance.27, 28
Results
Seven low-resolution experiments and a separate set of eight high-resolution experiments were analyzed. Ventricular tachycardia, either sustained or in the form of recurrent brief runs, eventually developed in all hearts under LQT2 conditions.
Diastolic and systolic Ca2+-dynamics before the onset of LQT2-related arrhythmias
During 2 Hz pacing, subcellular Ca2+-transients (CaTs) were smooth, monophasic and spatially homogenous (Fig. 1A, Movie A). Under LQT2 conditions, abnormal Cai elevations appeared in all hearts in systole and diastole during paced beats (Fig. 1B–C, Movies B–C), before the development of ectopy (Fig. 1D, Movie D). At the subcellular level, diastolic Ca2+-elevations corresponded to Ca2+-waves, propagating typically along the long axis of myocytes with a velocity of ~100–500 μm/s (n=8 hearts), and were not synchronous among neighboring cells. In a given cell, a single Ca2+-wave was typically observed per diastolic interval; occasionally 2–3 waves occurred consecutively and less frequently, more than one wave occurred simultaneously.
Figure 1.
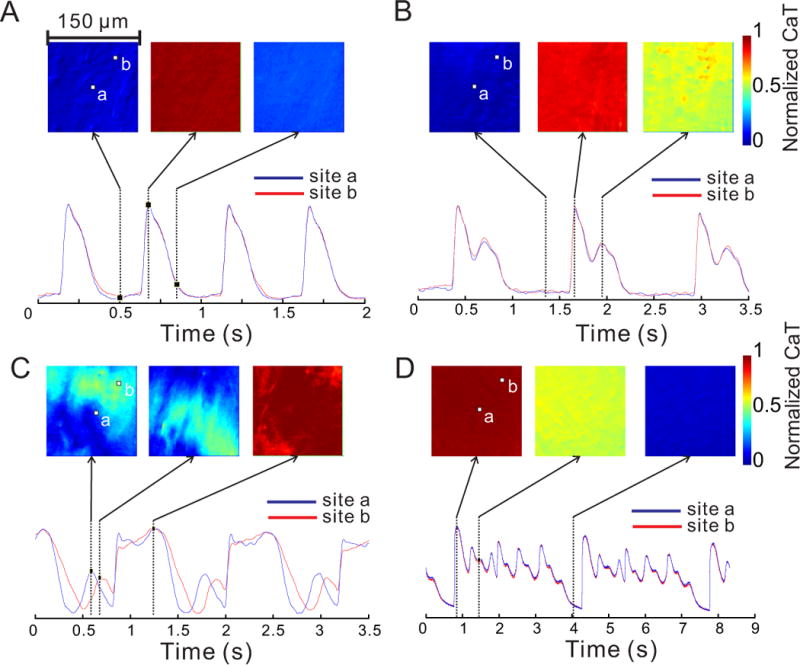
Optical imaging of cytoplasmic Ca2+ at high resolution. Each of the square images displays color-coded intensity of Ca2+ signal from an epicardial region of ventricular myocardium 150×150 μm2 in size. A: During baseline paced rhythm at 2 Hz, Ca2+ concentration is spatially homogenous and CaT tracings from two distinct pixels (a and b) are nearly superimposable. B: In paced rhythm under LQT2 conditions, a secondary systolic Ca2+ elevation occurs. The Ca2+ concentration remains homogenous within the field of view. C: Another example of slow pacing in LQT2 illustrates the contrast between the propagating diastolic Ca2+ wave (left and middle panels) and the synchronous systolic Ca2+ elevation (right). D: Multiple systolic peaks of CaT in LQT2 during arrhythmia are also spatially homogenous. See also Movies A–D.
In contrast to diastolic Ca2+-waves, SSCEs were cell-synchronous within myocytes when observed at subcellular-resolution and had similar amplitudes in neighboring myocytes. Most significant, SSCEs occurred at about the same time in myocytes in the field-of-view or 188±30 ms after the AP upstroke (n=5 hearts). Systolic and diastolic Cai events are more easily visualized in pseudo line-scan format (Fig. 2).
Figure 2.
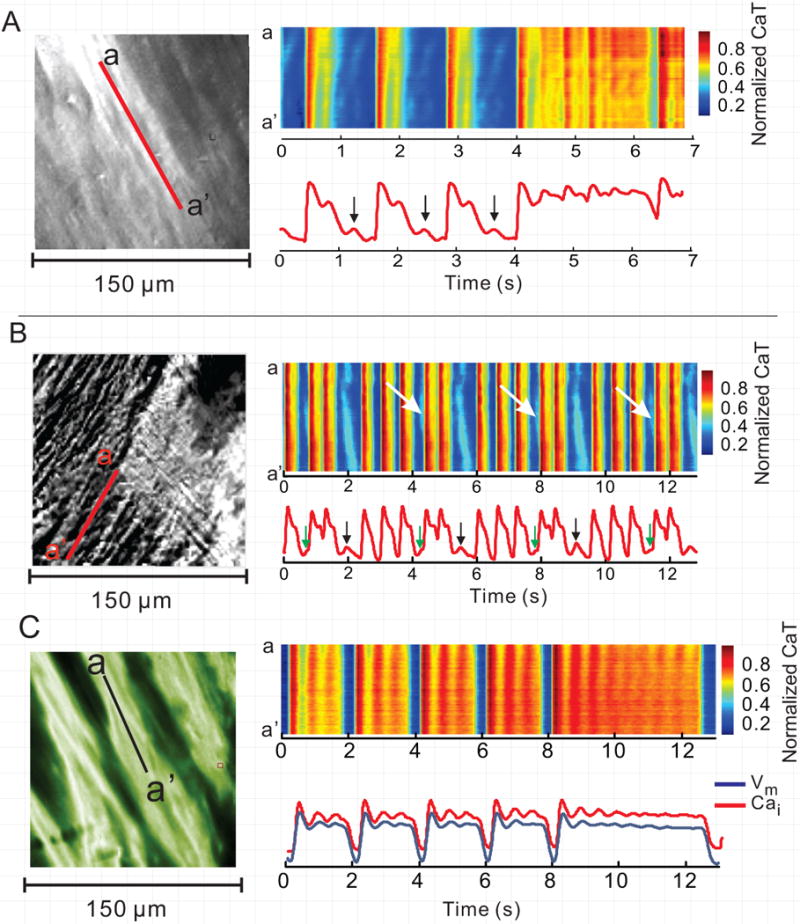
High-resolution Ca2+ imaging displayed as pseudo line-scans in LQT2. A and B: The scan lines are identified by the red lines in the subcellular images in left most panels (field-of-view 150×150 μm). The diastolic Cai elevations (arrows) correspond to propagating waves, as evidenced by the oblique orientation of the corresponding pseudo line-scan feature. The systolic Cai oscillations are cell-synchronous, with vertical pattern on pseudo line-scan. C: An example of simultaneous subcellular Cai and Vm imaging. The pseudo line-scan again shows cell-synchronous systolic Ca2+ oscillations during the long CaTs. The simultaneous Cai and Vm tracings show that the corresponding Vm oscillations are less pronounced than CaT oscillations.
Spatial heterogeneity of CaT and APD in LQT2
In LQT2, SSCE amplitudes varied in different regions of epicardium when viewed at low-resolution (15×15 mm2) (Fig. 3A). Spatial variations of CaTs during paced beats (before ventricular arrhythmias) led to an increase in SH of CaT during phase 2 and 3 of APs compared to control (−4.56±0.67 vs. −3.28±0.45; p<0.05). SH of APDs also increased in LQT2 (−4.40±0.67 vs. −3.55±0.41; p<0.05), although to a significantly smaller degree (n=7, p<0.05) (Fig. 3B&E). SH of CaT and AP were low and similar at baseline, increased in LQT2 and the increase was higher for CaTD than for APD (AP: 0.85±0.71; CaT: 1.28±0.76; p<0.05) (Fig. 3E).
Figure 3.
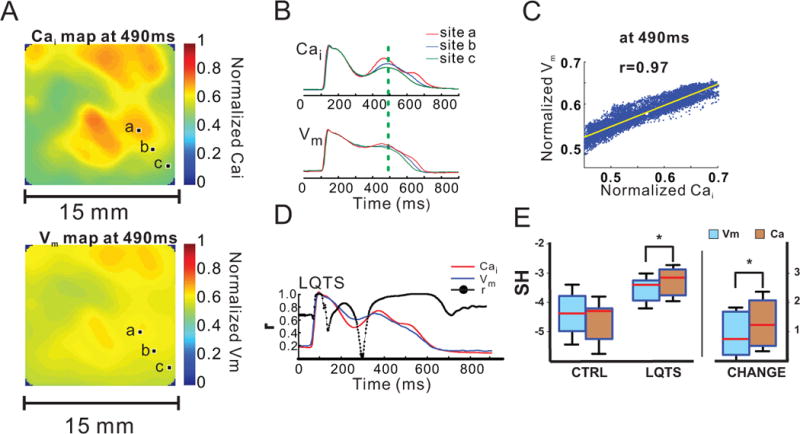
Relationship between Ca2+ and Vm dynamics in LQT2 at low resolution. A: In paced rhythm under LQT2 conditions, the Cai and Vm maps (15×15 mm2) are similar, but the Vm map is substantially “smoother”. The images are taken 490 ms after pacing during regular paced rhythm. B: Superimposed CaT and AP from the 3 pixels (a–c) labeled in A. C: The scatterplot of Cai and Vm from different pixels at 490 ms shows that the signals are highly correlated. D: During AP phase 2 and 3, Vm and Cai signals remain well correlated except at the onset of SSCEs precedes the Vm rise during the AP plateau. See Movie E for another example. E: During baseline slow pacing (CTRL), Spatial Heterogeneity (SH) of both AP and CaT is low and similar. During LQT2 (paced rhythm), SH increases for both signals, but CaT heterogeneity is higher and its increase from baseline is more pronounced than for AP. * indicates p<0.05.
In LQT2, areas with elevated CaT correlated well with areas of depolarized Vm (membrane potential) at the time of SSCE (r=0.95±0.03, n=5; Fig. 3B, C–D; Movie E). The durations of AP (APD80) and CaT (CaTD80) were tightly correlated (r=0.96±0.01 at baseline, r=0.99±0.01 in LQT2; Fig. 1S). Areas of SSCE were irregular and did not correspond to obvious anatomical features. With appropriate contrast setting, initial Cai rise could be resolved in small “islands” of tissue (<1-mm) (Fig. 4D), which spread and fused during later plateau phase. SSCEs occurred before ectopy and their appearance was constant on beat-to-beat basis over a few seconds.
Figure 4.
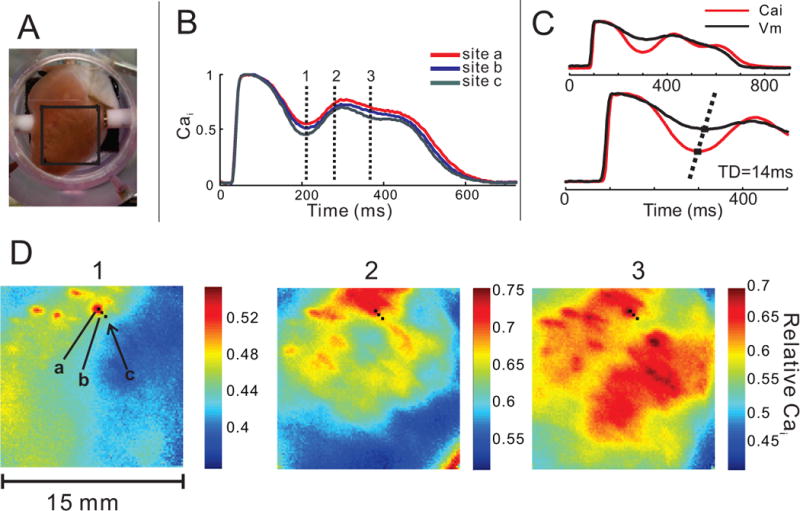
Ca2+ dynamics in LQT2 at low resolution during paced rhythm. A: Field-of-view (15×15 mm) is selected on the anterior surface of the ventricles. B: Spatial heterogeneity (SH) of Ca2+ signal in LQT2 is evidenced by a different course of CaT in nearby pixels (a–c). The vertical lines (1–3) indicate the timing of the snapshots D1–D3. C: Superposition of AP and CaT from the same pixel show that the onset of the SSCE precedes the rise in membrane potential; the time delay, TD=14 ms in this example. D: SSCEs start to rise in several small (~ 0.5–1 mm) distinct areas (D1) that form isolated “islands” of Ca2+ elevation that spread and enlarge (panels D 2–3). The fluorescence intensity scale differs in these panels and in the corresponding Movie E to enhance the contrast of spatial heterogeneities of CaT on a millimeter-scale.
Propagated early afterdepolarizations arise in regions of steep Vm gradients
When the earliest ectopic depolarization occurred in the field-of-view, it typically corresponded to an early afterdepolarizations. Interestingly, the sites of earliest ectopy occurred in regions of steep spatial Vm gradients. At the origin of ectopic beats, Vm gradient was at 94.9±3.2 percentile (median 95.7, interquartile range 95.0–96.7; n=6 hearts) immediately before the ectopic beat (Fig. 2S). Mean gradient of CaT calculated from all pixels was steeper than mean gradient of Vm (0.28±0.05 vs. 0.20±0.04; p<0.05, n=6) immediately before the onset of ectopic activity. At the origins of early afterdepolarizations, SSCEs upstrokes preceded Vm upstroke by median 12.2 ms (interquartile range 2.8–14.6 ms).
Effect of RyR2 stabilization on SSCEs, SH of CaT, and ventricular arrhythmia
Addition of K201 (1 μΜ) to the perfusate either before or after switching to LQT2 blocked SSCEs, markedly reduced spatial heterogeneities of CaT and APs, and abolished ectopy (n=4/4) (Fig. 5A,B,C). Addition of K201 to dofetilide-induced LQT2 suppressed Torsade de Pointes (Fig. 5C) and in additional subcellular imaging experiments, K201 blocked SSCEs and diastolic Ca2+-waves, as well as the development of ventricular arrhythmia (n=4/5 hearts). In LQT2, K201 (1 μM) also caused a marked shortening of APDs (from 538.5±53 to 294.5±12, p<0.05) and CaTD (from 559±6 to 307.7±16.7, p<0.05). Besides its ability to stabilize RyR2, K201 was reported to have off-target effects and to inhibit ICa,L, IKr and SERCA pumps in a concentration dependent manner.30 Voltage-clamp experiments showed that K201 at 1μM does not inhibit the L-type Ca2+ current (Fig. 5 D–E, n=5), whereas at 10 μM K201 suppressed ICa,L by > 20% (not shown) in agreement with previous reports.30 To safeguard against possible off-target effects, additional pilot experiments were repeated with 0.5 μM K201 and showed that the lower concentration was equally effective at suppressing early afterdepolarization and Torsade de Pointes, n=3/3 (Figure 3S). Hence, the suppression of SSCEs by 1μM K201 was primarily attributed to inhibition of SR Ca2+ release since at that concentration K201 did not alter ICa,L, possible suppression of IKr would have prolonged rather than reduced action potential durations and inhibition of SERCA would reduce SR Ca2+ content and like RyR2 stabilization would suppress SSCEs.
Figure 5.
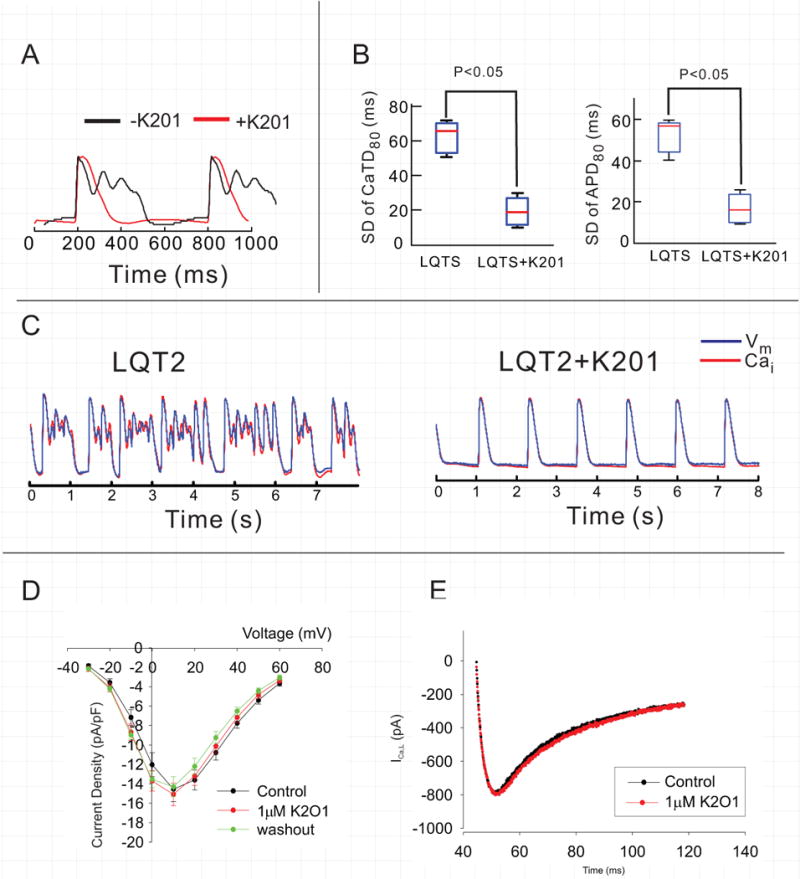
Effect of K201 on ICa,L. K201 suppresses systolic Ca2+ oscillations (A), dispersion of APD and CaTD (B) and ventricular arrhythmia (C). The data shown in panels A–C are from a low-resolution experiment. D&E: Rabbit ventricular myocytes were isolated from the base of the epicardium to measure ICa,L before and after 1μM K201. Current-to-voltage plots show that K201 did not alter ICa,L.
Ectopic beats due to delayed afterdepolarizations in LQT2
Diastolic and systolic Ca2+-elevations were observed in low-resolution experiments, and occurred in the same regions of the ventricles during paced beats (Fig. 4S). During ventricular arrhythmias, propagated delayed and early afterdepolarizations were observed in most experiments (Fig. 5S, traces). At the origin of delayed afterdepolarizations (black arrow-heads), Cai rose before Vm (Fig. 5S, green arrow-heads). This is supported by the phase-zero maps of Ca2+ and Vm signals, where phase zero of the signal corresponds to the onset of the signal upstroke. The timing is consistently earlier for Ca2+ than for Vm (n=5).
Patterns of subcellular Ca2+ handling during ventricular arrhythmias
In addition to stochastic diastolic waves and cell-synchronous SSCEs, more complex patterns of Ca2+-handling abnormalities were observed in ventricular arrhythmias by high-resolution imaging:
Multiple CaT spikes. During runs of non-sustained ventricular tachycardia, the first CaT rise did not fully recover to baseline, but was followed by a series of CaT spikes with sharp upstrokes. Similar to systolic Ca2+-elevations seen during paced rhythm, they were spatially synchronous in myocytes within the field-of-view (Fig. 1D, Movie D).
Ca2+ “ripples” superimposed on the plateau of long CaTs. We use the term “Ca2+ ripples” to denote the presence of multiple brief, small-amplitude Ca2+ elevations during prolonged elevated “plateau-like” CaTs, occurring in cell-asynchronous manner (as seen in Movie F). During episodes of non-sustained ventricular tachycardia, rectangular shaped CaTs lasting several seconds were often observed, with small amplitude Ca2+ ripples superimposed on the CaT (Fig. 6S, Movie F). Ca2+ ripples were not synchronized among adjacent myocytes and appeared to propagate within individual cells. Ripples can only be observed with high-resolution imaging, since these subcellular, regional signals will cancel themselves out if a pixel contains more than 1 cell. The phenomenon is mechanistically important as it represent an instability of sarcoplasmic reticulum Ca2+ uptake and release from multiple Ca2+ release units, since it is very difficult to imagine that ICa,L reactivation could drive ripples which occur out-of-phase in different regions of the same cell.
“Calcium fibrillation”. Chaotic Ca2+ dynamics was occasionally observed in a single cell or in all cells in the field-of-view during ventricular arrhythmias. These high frequency subcellular Cai oscillations (Fig. 6, Movie G) appeared as a persistent generation and annihilation of Ca2+ waves reminiscent of reentrant arrhythmias, though the scale and mechanism is obviously different.
Figure 6.
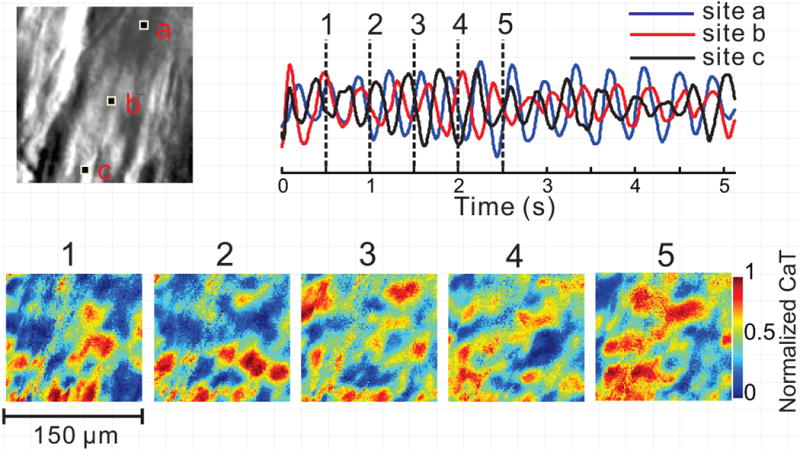
Random Subcellular Ca2+ oscillations during ventricular arrhythmia. Disorganized subcellular Ca2+ dynamics is occasionally observed under LQT2 conditions, with multiple Ca2+ wavefronts which collide, break and annihilate each other (bottom, 1–5). Ca2+ signal tracings (top right) from 3 pixels indicated in high-resolution image (top left, 150×150 μm2 image size) illustrate the lack of spatial synchrony. Stable Ca2+ baseline is not reached in any cell in the field-of-view, and the signals from different regions of the same cell are poorly correlated. This may reflect an extreme degree of cellular Ca2+ overload. See also Movie G.
Discussion
The mechanism of arrhythmogenesis in LQT2 is disputed. One hypothesis posits that the decrease in repolarization rate allows the L-type Ca2+ channel to occupy a state where neither the activation nor the inactivation gates are completely closed, enhancing the ICaL “window current”, which overwhelms the repolarizing currents and gives rise to early afterdepolarizations.14 In support of this mechanism, Marban et al.,31 reported that the chelation of cytosolic free Ca2+ with BAPTA and the depletion of sarcoplasmic reticulum Ca2+ with ryanodine failed to prevent early afterdepolarizations, but they were prevented by an L-type Ca2+ channel blocker. These data obtained in papillary muscles bathed in a cesium containing Tyrode’s solution as a model of LQT2 suggested that early afterdepolarizations are elicited by the ICaL window current and are independent of cytosolic Ca2+ handling (assuming BAPTA chelates Cai in all myocytes) and sarcoplasmic reticulum Ca2+ overload.
DeFerrari et al.,32 loaded isolated cardiac myocytes with a Ca2+ indicator dye to investigate the mechanisms that induce delayed and early afterdepolarizations during β-adrenergic stimulation. Despite low temporal resolution, cytosolic Ca2+ waves were found to correspond to delayed afterdepolarizations and secondary Ca2+ elevations to early afterdepolarizations. The latter appeared to be synchronous within the cell rather than a random Ca2+ release from sarcoplasmic reticulum Ca2+-release units, consistent with a process driven by a voltage-dependent sarcolemmal channel such as ICaL. Similar systolic secondary CaT elevations were recently described in mouse ventricular myocytes with increased sarcoplasmic reticulum load; in contrast to diastolic waves, they could not be easily suppressed with verapamil.33
In contrast, Volders et al.,16 argued that both delayed and early afterdepolarizations are caused by spontaneous intracellular Ca2+ release in isolated canine myocytes exposed to bradycardia and β-adrenergic stimulation. This interpretation was based on the timing of myocyte contractions and Ca2+ release that preceded the onset of early afterdepolarizations, and on dissociation of Ca2+ release from early afterdepolarizations by sodium-calcium exchange blockade. A similar finding was obtained in isolated ventricular rabbit myocytes, where we elicited early afterdepolarization by IKr blockade which were suppressed by sodium-calcium exchange inhibition,29 implicating SR Ca2+ release as the underlying mechanism. Choi and Salama24 proposed that sarcoplasmic reticulum Ca2+ release elicited early afterdepolarizations induced by IKr blockade in isolated rabbit hearts because the upstroke of CaTs preceded the upstroke of early afterdepolarizations at the sites of early afterdepolarization foci. More recently, we reported that in LQT2, abnormal Ca2+ handling and secondary CaT elevations precede development of early afterdepolarizations and voltage-instabilities during the AP plateau by minutes, and that multiple Ca2+ re-elevations occurred during regular paced rhythm even before the onset of polymorphic ventricular tachycardia. In contrast to previous reports, depletion of sarcoplasmic reticulum Ca2+ with ryanodine and thapsigargin prevented secondary CaT oscillations, early afterdepolarizations and polymorphic ventricular tachycardia, in perfused rabbit hearts.21 In acute bradycardia without IKr block, the repolarization delay prolonged CaT durations resulting in SSCEs of sufficient magnitude to elicit early afterdepolarizations.26
Finally, Horvath et al.,34 observed SSCEs in isolated ventricular myocytes treated with ATX-II (LQT3 model). Both SSCEs and early afterdepolarizations were suppressed by chelation of cytosolic Ca2+ with BAPTA, supporting the role of SSCE in the generation of early afterdepolarizations in that model.
The data reported here demonstrate that in rabbit heart perfused at physiological temperature, the secondary Ca2+ rise corresponding to early afterdepolarization is indeed cell-synchronous. However, Ca2+ release from overloaded sarcoplasmic reticulum rather than ICa,L reactivation is responsible, since SSCEs are suppressed by K201 (1 μΜ). At this concentration, K201 has no effect on ICa,L.30 Given the importance of the latter result, we repeated the voltage-clamp measurements of ICa,L from rabbit base myocytes and demonstrate that K201 at 1 μΜ, does not alter ICa,L. Hence, K201 abolished early afterdepolarizations by stabilizing ryanodine receptors and not by inhibiting ICa,L. In addition, the rise of CaT consistently precedes the rise of Vm at the site of the ectopic focus and multiple Ca2+-elevations are frequently observed during a single AP. In contrast to SSCEs, diastolic Ca2+ waves in LQT2 were not synchronous within or between adjacent cells. The diastolic waves and were typically of lower amplitude than SSCEs and more difficult to detect at low resolution. Nevertheless, K201 eliminated both SSCEs and diastolic Ca2+ waves, suggesting a common mechanism of SR Ca2+ release in both phenomena.
Different experimental setting may explain why others reached a different conclusion regarding mechanism of early afterdepolarizations. We performed experiments in perfused heart at physiological temperature, as opposed to single-cells or superfused tissue. We used a highly selective IKr inhibitor to elicit early afterdepolarization at a high concentration, dictated by the need to reliably induce Torsade de Pointes within minutes due to the life-time of perfused hearts (3–4 hours). We acknowledge that the details of arrhythmogenesis may differ in patients, who may experience a single Torsade episode in their lifetime. Non-confocal microscopy was used to improve light throughput and temporal resolution. The resulting decrease in depth resolution to about 5 μm improves the detection of Ca2+ waves and is still considerably less than the myocyte thickness.
Diastolic Ca2+ waves and delayed afterdepolarizations in LQT2
Our data indicate that diastolic Ca2+ waves classically associated with sarcoplasmic reticulum Ca2+ overload and delayed afterdepolarizations also occur in LQT2 and may contribute to the initiation of arrhythmias. Ectopic beats occurring during diastole are preceded by a rise of Ca2+ (Figure 4S). Epicardial regions exhibiting diastolic Ca2+ waves and SSCEs are well correlated (Fig. 3S), suggesting that sarcoplasmic reticulum Ca2+ overload triggers both SSCEs and diastolic waves. Visualization of diastolic Ca2+ waves in LQT2 requires high spatial and temporal resolution imaging; their role in LQT2 arrhythmogenesis may thus have been underappreciated.
Synchronization of seemingly random diastolic Ca2+ waves in individual cells is required to overcome the source-sink mismatch and to elicit an ectopic beat. A mechanism of synchronization was demonstrated during a pause following rapid pacing in a rat heart perfused with elevated extracellular Ca2+ at room temperature.35 Despite questions regarding the clinical relevance of the model, the mechanism proposed by Wasserstrom, i.e. increasing degree of temporal overlap among diastolic waves in adjacent cells with increasing sarcoplasmic reticulum Ca2+ load, may well apply to LQT2.35
K201 effect on APD
The precise mechanism for the striking shortening of CaTD and APD by K201 in LQT2 is uncertain. Our published data suggest that in the absence of dofetilide, K201 has little effect on APD during 2 Hz pacing in rabbit hearts, but partially prevents APD prolongation in right ventricular base.26 Given the lack of effect of K201 on ICaL in the rabbit heart at this concentration,30 and lack of effect on late sodium current,36 it is likely that K201-dependent shortening of APD in LQT2 could be mediated by RyR2 stabilization. It is possible that a complete IKr block initially prolongs APD only to a moderate degree, which is nevertheless sufficient to increase myocyte Ca2+ load and SSCEs. Higher cytosolic Ca2+ during phase 3 of the AP caused by SSCE would augment INCX, slow the AP downstroke and increase Ca2+ influx via ICaL. Under certain conditions, the increase in Ca2+ influx during phase 3 might exceed the increase in Ca2+ efflux via NCX. In such a case, APD would increase gradually with increasing Ca2+ load, even during full IKr block. Hence, it is possible that K201 reduces APDs in LQT2 by inhibiting SSCEs. Additional experiments will be required to address this hypothesis directly.
Mechanism of arrhythmogenesis in LQT2
Based on the above considerations, we propose the following sequence of events to explain the role of Ca2+ dynamics in LQT2-related arrhythmias:
Prolonged APD results in increased Ca2+ entry into myocyte because ICa,L remains partially activated during prolonged plateau phases and because Ca2+ efflux via sodium-calcium exchange is impaired due to shorter diastolic intervals.
The initial CaT upstroke is caused by Ca2+-induced Ca2+-release, and empties the junctional sarcoplasmic reticulum. Ca2+ is subsequently pumped to the non-junctional sarcoplasmic reticulum by SERCA, and diffuses towards the junctional sarcoplasmic reticulum, increasing the Ca2+ load in junctional sarcoplasmic reticulum and eliciting a second reopening of ryanodine receptors during the AP plateau phase.
The open probability of cardiac ryanodine receptor is known to increase with both higher cytoplasmic and luminal Ca2+ concentrations.37–39 The high cytosolic Ca2+ during the AP plateau in LQT2 may explain why systolic, but not diastolic sarcoplasmic reticulum Ca2+-release is cell-synchronous. We hypothesize that when junctional sarcoplasmic reticulum is reloaded before cytoplasmic Ca2+ recovers to baseline, the threshold of luminal Ca2+ needed to re-open cardiac ryanodine receptors is low. Reloading the terminal cisternae may occur in most junctions at approximately the same time. In contrast, the luminal threshold for release would be higher during diastole, and reached in only one or a few regions of a myocyte.
The amplitude of SSCEs differs in different regions, resulting in marked spatial heterogeneity of CaT. The reasons for this large-scale spatial heterogeneity are uncertain and likely include spatial variations in expression of Ca2+ handling proteins.26
Spatial heterogeneity of CaT drives the spatial heterogeneity of AP by means of the electrogenic sodium-calcium exchanger, i.e. regions of higher cytoplasmic Ca2+ during AP phases 2 and 3 experience higher inward sodium-calcium exchange current.
Steep spatial gradients of Vm cause cell membrane depolarization and trigger propagated early afterdepolarizations,40 perhaps through ICaL reactivation.
Chaotic intracellular Ca2+ dynamics may reflect a marked degree of myocyte Ca2+ overload, with failure of cytoplasmic Ca2+ to reach a low concentration in the entire cell even transiently. The myocyte or ventricular region with such dynamics would be expected to remain at least partially depolarized and serve as a source of depolarizing current for adjacent cells. Clearly, ICaL window current cannot account for this form of Ca2+ dynamics.
In summary, we describe Ca2+ dynamics in LQT2 at whole-heart and subcellular resolutions. The abnormal Ca2+ handling appears to involve store overload-induced Ca2+ release and directly participates in the generation of triggered activity. Compared to acute bradycardia without IKr block, well-developed secondary CaT peaks were observed in all LQT2 experiments and triggered early afterdepolarizations, likely reflecting a higher degree of sarcoplasmic reticulum load.
Improved understanding of arrhythmogenesis due to delayed repolarization may lead to novel therapeutic and diagnostic strategies. For example, it is possible that targeting SSCEs may be protective with respect to polymorphic ventricular tachycardia in the clinical setting. Spatial heterogeneity of CaT is the likely mechanism of regional heterogeneity in ventricular contraction timing detected in LQT2 patients with echocardiography.41 More generally, our findings underscore the role of store overload-induced Ca2+ release in generation of triggered activity, which is not limited to delayed afterdepolarizations.
Supplementary Material
Acknowledgments
Funding Sources: Supported in part by NHLBI HL-70722 and HL-093074 to GS and AHA to JK
Footnotes
Journal Subject Codes: [5] Arrhythmias, clinical electrophysiology, drugs, [106] Electrophysiology, [132] Arrhythmias – basic studies
Conflict of Interest Disclosures: None
References
- 1.Dessertenne F. ventricular tachycardia with 2 variable opposing foci. Arch Mal Coeur Vaiss. 1966;59:263–272. [PubMed] [Google Scholar]
- 2.Morita H, Wu J, Zipes DP. The qt syndromes: Long and short. Lancet. 2008;372:750–763. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 3.Roden DM. Clinical practice. Long-qt syndrome. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 4.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of k(v)lqt1 and mink (isk) proteins to form cardiac i(ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 5.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: Herg encodes the ikr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 6.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: Kvlqt1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. Scn5a mutations associated with an inherited cardiac arrhythmia, long qt syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 8.Roden DM, Viswanathan PC. Genetics of acquired long qt syndrome. J Clin Invest. 2005;115:2025–2032. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Digby GC, Perez Riera AR, Barbosa Barros R, Simpson CS, Redfearn DP, Methot M, Femenia F, Baranchuk A. Acquired long qt interval: A case series of multifactorial qt prolongation. Clin Cardiol. 2011;34:577–582. doi: 10.1002/clc.20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monnig G, Kobe J, Loher A, Wasmer K, Milberg P, Zellerhoff S, Pott C, Zumhagen S, Radu R, Scheld HH, Haverkamp W, Schulze-Bahr E, Eckardt L. Role of implantable cardioverter defibrillator therapy in patients with acquired long qt syndrome: A long-term follow-up. Europace. 2012;14:396–401. doi: 10.1093/europace/eur316. [DOI] [PubMed] [Google Scholar]
- 11.Bailie DS, Inoue H, Kaseda S, Ben-David J, Zipes DP. Magnesium suppression of early afterdepolarizations and ventricular tachyarrhythmias induced by cesium in dogs. Circulation. 1988;77:1395–1402. doi: 10.1161/01.cir.77.6.1395. [DOI] [PubMed] [Google Scholar]
- 12.el-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long qt syndrome. Tridimensional mapping of activation and recovery patterns. Circ Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- 13.January CT, Riddle JM. Early afterdepolarizations: Mechanism of induction and block. A role for l-type ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 14.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: Mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. Ryr2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced ca2+ release (soicr) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc Res. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 17.Milberg P, Pott C, Fink M, Frommeyer G, Matsuda T, Baba A, Osada N, Breithardt G, Noble D, Eckardt L. Inhibition of the na+/ca2+ exchanger suppresses torsades de pointes in an intact heart model of long qt syndrome-2 and long qt syndrome-3. Heart Rhythm. 2008;5:1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 18.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local beta-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubalova Z, Gyorke I, Terentyeva R, Viatchenko-Karpinski S, Terentyev D, Williams SC, Gyorke S. Modulation of cytosolic and intra-sarcoplasmic reticulum calcium waves by calsequestrin in rat cardiac myocytes. J Physiol. 2004;561:515–524. doi: 10.1113/jphysiol.2004.073940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in purkinje fibres. J Physiol. 1976;263:73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemec J, Kim JJ, Gabris B, Salama G. Calcium oscillations and t-wave lability precede ventricular arrhythmias in acquired long qt type 2. Heart Rhythm. 2010;7:1686–1694. doi: 10.1016/j.hrthm.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fabiato A. Spontaneous versus triggered contractions of “calcium-tolerant” cardiac cells from the adult rat ventricle. Basic Res Cardiol. 1985;80(Suppl 2):83–87. [PubMed] [Google Scholar]
- 23.Lechleiter J, Girard S, Peralta E, Clapham D. Spiral calcium wave propagation and annihilation in xenopus laevis oocytes. Science. 1991;252:123–126. doi: 10.1126/science.2011747. [DOI] [PubMed] [Google Scholar]
- 24.Choi BR, Burton F, Salama G. Cytosolic ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long qt syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salama G, Hwang SM. Simultaneous optical mapping of intracellular free calcium and action potentials from langendorff perfused hearts. Curr Protoc Cytom. 2009;Chapter 12(Unit 12):17. doi: 10.1002/0471142956.cy1217s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JJ, Nemec J, Papp R, Strongin R, Abramson JJ, Salama G. Bradycardia alters ca(2+) dynamics enhancing dispersion of repolarization and arrhythmia risk. Am J Physiol Heart Circ Physiol. 2013;304:H848–860. doi: 10.1152/ajpheart.00787.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G. Sex, age, and regional differences in l-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long qt type 2. Circ Res. 2008;102:e86–100. doi: 10.1161/CIRCRESAHA.108.173740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Chen G, Papp R, Defranco DB, Zeng F, Salama G. Oestrogen upregulates l-type ca(2)(+) channels via oestrogen-receptor- by a regional genomic mechanism in female rabbit hearts. J Physiol. 2012;590:493–508. doi: 10.1113/jphysiol.2011.219501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen G, Yang X, Alber S, Shusterman V, Salama G. Regional genomic regulation of cardiac sodium-calcium exchanger by oestrogen. J Physiol. 2011;589:1061–1080. doi: 10.1113/jphysiol.2010.203398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loughrey CM, Otani N, Seidler T, Craig MA, Matsuda R, Kaneko N, Smith GL. K201 modulates excitation-contraction coupling and spontaneous ca2+ release in normal adult rabbit ventricular cardiomyocytes. Cardiovasc Res. 2007;76:236–246. doi: 10.1016/j.cardiores.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early after depolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Ferrari GM, Viola MC, D’Amato E, Antolini R, Forti S. Distinct patterns of calcium transients during early and delayed afterdepolarizations induced by isoproterenol in ventricular myocytes. Circulation. 1995;91:2510–2515. doi: 10.1161/01.cir.91.10.2510. [DOI] [PubMed] [Google Scholar]
- 33.Stokke MK, Tovsrud N, Louch WE, Oyehaug L, Hougen K, Sejersted OM, Swift F, Sjaastad I. I(cal) inhibition prevents arrhythmogenic ca(2+) waves caused by abnormal ca(2+) sensitivity of ryr or sr ca(2+) accumulation. Cardiovasc Res. 2013;98:315–325. doi: 10.1093/cvr/cvt037. [DOI] [PubMed] [Google Scholar]
- 34.Horvath B, Banyasz T, Jian Z, Hegyi B, Kistamas K, Nanasi PP, Izu LT, Chen-Izu Y. Dynamics of the late na(+) current during cardiac action potential and its contribution to afterdepolarizations. J Mol Cell Cardiol. 2013;64:59–68. doi: 10.1016/j.yjmcc.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wasserstrom JA, Shiferaw Y, Chen W, Ramakrishna S, Patel H, Kelly JE, O’Toole MJ, Pappas A, Chirayil N, Bassi N, Akintilo L, Wu M, Arora R, Aistrup GL. Variability in timing of spontaneous calcium release in the intact rat heart is determined by the time course of sarcoplasmic reticulum calcium load. Circ Res. 2010;107:1117–1126. doi: 10.1161/CIRCRESAHA.110.229294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura J, Kawahara M, Sakai E, Yatabe J, Nakanishi H. Effects of a novel cardioprotective drug, jtv-519, on membrane currents of guinea pig ventricular myocytes. Jpn J Pharmacol. 1999;79:275–281. doi: 10.1254/jjp.79.275. [DOI] [PubMed] [Google Scholar]
- 37.Laver DR. Ca2+ stores regulate ryanodine receptor ca2+ release channels via luminal and cytosolic ca2+ sites. Biophys J. 2007;92:3541–3555. doi: 10.1529/biophysj.106.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal ca2+ involves luminal ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, Shinohara T, Shen MJ, Qu Z, Weiss JN, Chen PS. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-qt syndrome. Circ Arrhythm Electrophysiol. 2011;4:103–111. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haugaa KH, Amlie JP, Berge KE, Leren TP, Smiseth OA, Edvardsen T. Transmural differences in myocardial contraction in long-qt syndrome: Mechanical consequences of ion channel dysfunction. Circulation. 2010;122:1355–1363. doi: 10.1161/CIRCULATIONAHA.110.960377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.