

Abstract
Drugs that act more promiscuously provide fewer routes for the emergence of resistant mutants. But this benefit often comes at the cost of serious off-target and dose-limiting toxicities. The classic example is the antifungal amphotericin B (AmB), which has evaded resistance for more than half a century. We report dramatically less toxic amphotericins that nevertheless evade resistance. They are scalably accessed in just three steps from the natural product, and bind their target (the fungal sterol, ergosterol) with far greater selectivity than AmB. Hence, they are less toxic and far more effective in a mouse model of systemic candidiasis. Surprisingly, exhaustive efforts to select for mutants resistant to these more selective compounds revealed that they are just as impervious to resistance as AmB. Thus, highly selective cytocidal action and the evasion of resistance are not mutually exclusive, suggesting practical routes to the discovery of less toxic, resistance-evasive therapies.
Less selective pharmacological action is generally associated with decreased vulnerability to resistance, but also with increased toxicity1,2. The classic example is amphotericin B (AmB), an exceptionally resistance-evasive but also highly toxic antifungal agent that has remained the last line of defense in treating invasive fungal infections for over half a century3. An excess of 1.5 million people die from such infections each year, in large part because the extreme toxicity of AmB is dose-limiting4. Extensive efforts to develop a clinically viable less toxic amphotericin have been made, but without success5. Moreover, it has remained unclear whether such a decrease in toxicity would come at the cost of an increase in vulnerability to pathogen resistance.
For decades the pursuit of a less toxic amphotericin was guided by the widely accepted model in which AmB (Fig. 1a) kills cells via ion channel-mediated membrane permeabilization5,6. This model suggests that improving the therapeutic index of this drug requires the selective self-assembly of oligomeric ion channels in yeast vs. human cells, a problem that has been very challenging to approach rationally. Contrary to this model, it was recently shown that AmB primarily exists as a large extramembranous aggregate which kills yeast by simply binding7 and extracting8 ergosterol, and may kill human cells by similarly binding cholesterol9. Ergosterol is critical for many different aspects of yeast physiology10–14, and mutations that alter sterol biosynthesis in a manner that confers resistance abrogate fungal virulence15, explaining the failure of fungi to evolve AmB resistance in the clinic. This new sterol sponge model enabled efforts to improve the therapeutic index of AmB to focus on the simpler problem of selectively binding sterols, and this yielded the recent discovery of a new derivative, C2′deoxyAmB (C2′deOAmB) that binds ergosterol but not cholesterol and is toxic to yeast but not human cells9. Limited synthetic access to this derivative, however, has hindered its further development and the determination of whether this improvement in therapeutic index is coupled to a decreased capacity to evade resistance.
Figure 1. Synthesis of AmB urea derivatives.
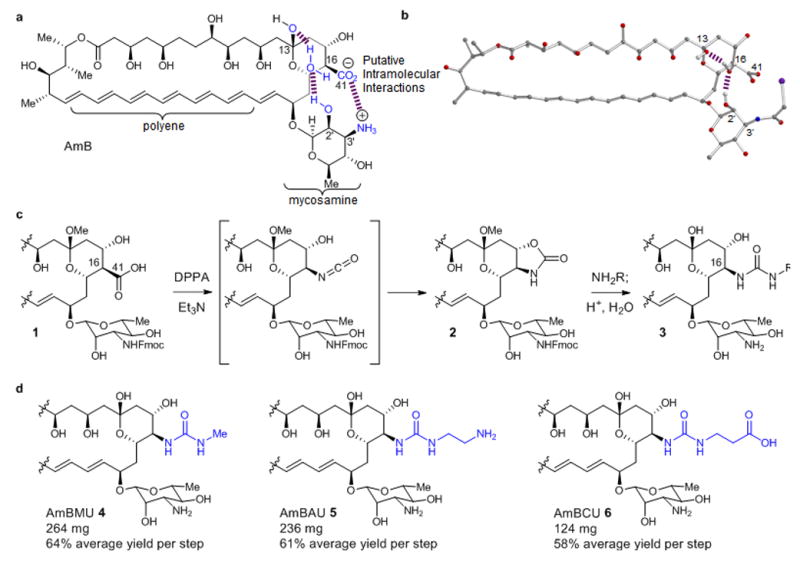
a. Chemical structures of AmB and C2′deOAmB. b. X-ray crystal structure of N-iodoacylAmB showing an intramolecular water bridged hydrogen bond between the C2′ and C-13 hydroxyl groups. c. General scheme for synthesis of AmB ureas. d. Synthesis of AmBMU, AmBAU and AmBCU from AmB in only 3 or 4 steps.
To rationalize the greater ergosterol-selective binding observed with C2′deOAmB we took into consideration several new structural insights regarding these prototypical small molecule-small molecule interactions. First, the mycosamine appendage is critical for binding both ergosterol and cholesterol16. Recent solid-state NMR evidence also confirms direct contact between the A and B rings of ergosterol and the AmB polyene motif in the sterol sponge complex8. Previous work suggested a relationship between activities of AmB and rotational conformers of the mycosamine sugar17,18, and a recent crystal structure of an AmB derivative19 (Fig. 1b) reveals a water-bridged hydrogen bond between the C2′ and the C13 hydroxyl groups and also suggests an intramolecular salt bridge between what would correspond to C41 carboxylate and C3′ ammonium ions in AmB (Fig. 1a). We propose that this pair of intramolecular polar interactions collectively stabilize the relative positions of the mycosamine appendage and the polyene motif in a ground state conformation of AmB that binds both ergosterol and cholesterol, and that deletion of the C2′ hydroxyl group disrupts this stabilization and thus favors a shift to an alternate conformer that selectively binds ergosterol. Alternatively stated, this model predicts that a ligand-selective allosteric effect underlies these small molecule-small molecule interactions, similar to that which has been observed in a number of proteins20,21. Guided by this model, and further encouraged by previous reports of modest but promising improvements in therapeutic index5, we pursued more synthetically accessible disruptions of the putative intramolecular salt bridge between the C41 carboxylate and C3′ ammonium ions.
RESULTS
Three-step synthesis of AmB ureas
For more than 50 years a less toxic AmB derivative has been sought through semisynthesis and more recently via genetic manipulations of the producing organism5,22–25. Ease of manipulation of the C16 carboxylate has made this position an especially attractive target for derivatization. However, all previously reported derivatives have maintained a C16–C41 carbon–carbon bond. Enabling us to explore a new chemotype, we discovered that treatment of a minimally protected variant of AmB (1) with diphenyl phosphoryl azide (DPPA) cleanly promotes a stereospecific Curtius rearrangement in which the C16–C41 bond is cleaved and the resulting isocyanate is intramolecularly trapped by the neighboring C15 alcohol26 to form an oxazolidinone 2 (Fig. 1c). This particular oxazolidinone, in turn, is surprisingly reactive to ring-opening with primary amines under mild conditions to yield a new class of urea containing amphotericins (3) having a C16-nitrogen bond. Interestingly, the parent heterocycle, 2-oxazolidinone, is unreactive under the same conditions.
We further found that 1 can be directly converted to 3 in a scalable one-pot operation involving serial addition of DPPA, an amine, and aqueous acid. Starting with 1 g of fermented AmB and using methyl amine as the nucleophile, this overall three-step sequence yields 264 mg of AmB methyl urea (AmBMU, 4) (64% average yield per step, 25% overall yield) (Fig. 1d). Employing ethylene diamine produces 236 mg of AmB amino urea (AmBAU, 5) (61% average yield per step, 22% overall yield), and in a four step-variant, reaction with β-alanine allylester followed by deallylation yields 124 mg AmB carboxylatoethyl urea (AmBCU, 6) (58% average yield per step, 12% overall yield). This chemistry thus provides rapid, efficient, and scalable access to these new derivatives starting with the natural product that is already fermented on the metric ton scale.
AmB ureas are selectively toxic to yeast
With this new series of AmB derivatives in hand, we determined their sterol binding properties. We next asked if, like AmB8, the urea derivatives act as a sterol sponge that extracts ergosterol from the membranes of yeast cells. Using an ultracentrifugation-based membrane isolation assay8, we quantified the amount of ergosterol remaining in the membranes of S. cerivisiae after treatment with AmB or the urea derivatives. As seen previously with AmB8, the majority of ergosterol was removed from yeast membranes upon treatment with each of the urea derivatives (Fig. 2a). We next probed sterol binding selectivity via isothermal titration calorimetry (ITC) (Fig. 2b)9. AmB binds both ergosterol and cholesterol, but the aglycone, amphoteronolide B (AmdeB, 7), binds neither sterol16. Like C2′deOAmB9, all of the new C16 urea-containing derivatives retain the capacity to bind ergosterol but, within the limits of detection of this experiment, show no binding to cholesterol.
Figure 2. Sterol extraction and binding capacities of AmB ureas.

a. Percent ergosterol remaining in S. cerevisiae membranes after extraction by AmB or its derivatives. The percent ergosterol remaining was normalized to DMSO-only treated controls. Values represent the mean of at least three experiments ± SD. *, p≤0.05, **, p≤0.001; NS, not significant. b. Total net isotherms from ITC showing AmB or derivatives binding to ergosterol (top) or cholesterol (bottom).
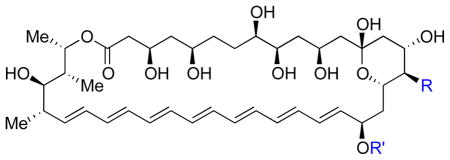
This sterol binding selectivity translated into a major improvement in therapeutic index in vitro (Table 1). Specifically, we determined the minimum inhibitory concentration (MIC) against Saccharomyces cerevisiae and the minimum hemolytic concentration (MHC) against human red blood cells for AmB, a series of previously reported C41 and/or C3′-modified derivatives5, and the new AmB ureas. AmB is a potent antifungal (MIC 0.5 μM), but is also highly toxic to human red blood cells (MHC 8.6 μM). AmdeB is non-toxic to both9,27. Previously reported modifications including esterification to a methyl ester (AmBME, 8)28,29, reduction of the carboxylic acid (C41MeAmB, 9)27, conversion to a methyl amide (AmBMA, 10)30, and double modification to form a triazoloethyl amide bis-aminopropyl derivative (AmBTABA, 11)31 produce modest improvements in therapeutic index, with the best results obtained with AmBTABA (MIC 0.25 μM, MHC 49 μM). In contrast, all of the AmB urea derivatives retained antifungal activity but show dramatically reduced toxicity to human red blood cells. The MHC of AmBMU and AmBAU exceeds the limits of solubility in this assay (500 μM). Remarkably, the only structural difference between AmBMA and AmBMU is the insertion of a protonated nitrogen atom between the C16 and C41 carbons (Fig. 1a,b).
Table 1. Antifungal and human cell toxicity.
Minimum inhibitory concentration (MIC) against S. cerevisiae and minimum hemolytic concentration (MHC) causing 90% hemolysis against human red blood cells. Previously reported compounds were independently synthesized and HPLC purified and characterization data matched literature values.
| ||||
|---|---|---|---|---|
| Compound | R | R′ | MIC (μM) S. cerevisiae | MHC (μM) red blood cells |
| AmB |
|
mycosamine | 0.5 | 8.55 ± 2.20 |
| AmdeB (7) |
|
|
>500 | >500 |
| AmBME (8) |
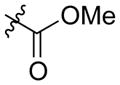
|
mycosamine | 0.25 | 30.67 ± 5.38 |
| C41MeAmB (9) |

|
mycosamine | 0.5 | 22.03 ± 6.26 |
| AmBMA (10) |
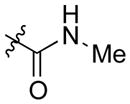
|
mycosamine | 0.25 | 15.32 ± 3.39 |
| AmBTABA (11) |
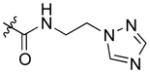
|
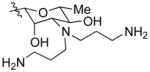
|
0.25 | 48.5 ± 8.7 |
| AmBMU |
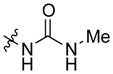
|
mycosamine | 0.5 | >500 |
| AmBAU |
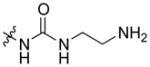
|
mycosamine | 0.25 | >500 |
| AmBCU |
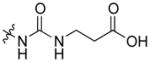
|
mycosamine | 3 | 323.8 ± 30.2 |
The activities of the AmB urea derivatives were further tested against a series of pathogenic yeast strains (Table 2)32. Both AmBMU and AmBAU demonstrate potent antifungal activity against all strains tested including invasive Candida, Cryptococcus, and Aspergillus. Notably, Cryptococcus neoformans strains 89–610 and T1 are fluconazole resistant33 while Aspergillus fumigatus strains 11628 and 14532 possess CYP51 mutations and are thus voriconazole resistant34. AmBCU also retained activity, but was in general somewhat less potent. The compounds were also tested for toxicity against human renal proximal tubule epithelial cells (RPTEC), the critical site of toxicity for AmB in patients. They all showed little or no toxicity to hTERT1 RPTEC35, and substantially reduced toxicity to the more sensitive primary RPTEC36.
Table 2. Antifungal and human cell toxicity against clinically relevant cell lines.
MIC against yeast pathogens (C. albicans, C. glabrata, C. tropicalis, C. parapsilosis, C. neoformans, and A. fumigatus) and minimum toxic concentration (MTC) causing 90% toxicity against human hTERT1 renal proximal tubule epithelial cells (RPTEC) and human primary RPTECs
| AmB | AmBMU | AmBAU | AmBCU | ||
|---|---|---|---|---|---|
| Candida albicans K1 | 0.25 | 0.5 | 0.5 | 0.5 | MIC (μg/mL) |
| Candida glabrata 760 | 0.06 | 0.25 | 0.25 | 1 | |
| Candida tropicalis 5810 | 0.06 | 1 | 1 | 2 | |
| Candida parapsilosis 22019 | 0.06 | 0.25 | 0.25 | 1 | |
| Cryptococcus neoformans H99 | 0.06 | 1 | 1 | 1 | |
| Cryptococcus neoformans 89–610 | 0.125 | 1 | 1 | 1 | |
| Cryptococcus neoformans T1 | 0.125 | 1 | 1 | 1 | |
| Aspergillus fumigatus 41 | 1 | 2 | 4 | 4 | |
| Aspergillus fumigatus 293 | 1 | 2 | 2 | 4 | |
| Aspergillus fumigatus 11628 | 0.5 | 0.5 | 1 | 2 | |
| Aspergillus fumigatus 14532 | 0.5 | 1 | 2 | 2 | |
|
| |||||
| hTERT1 RPTEC | 6.4 ± 1.3 | >80 | 37.6 ± 4.8 | >80 | MTC (μM) |
| Primary RPTEC | 2.4 ± 0.3 | 44.4 ± 2.1 | 11.3 ± 0.4 | >80 | |
AmB ureas are more efficacious and less toxic in mice
Based on all of these results, AmBMU and AmBAU were judged to be especially promising and thus further evaluated for efficacy and toxicity in vivo (Fig. 3a–d)37. In a mouse model of disseminated candidiasis both AmBMU and AmBAU were substantially more effective than AmB at reducing fungal burden in the kidneys at all three tested doses (1, 4 and 16 mg/kg, intraperitoneal injection). The differences in efficacy were most pronounced at the 16 mg/kg dose at 24 hours post inoculation. Relative to AmB, AmBMU reduced the fungal burden by 1.2 log units (p ≤ 0.0001), and AmBAU reduced the fungal burden by nearly 3 log units (p ≤ 0.0001). We speculate that an improved pharmacological profile, potentially due to a >20 fold increase in water solubility relative to AmB, may contribute to this unexpected dramatic improvement in antifungal activity for the new compounds in vivo.
Figure 3. Efficacy and toxicity of AmB ureas in mice.

Quantification of the fungal burden in the kidneys of C. albicans infected neutropenic mice 2, 6, 12 and 24 hours after a single IP injection of AmB, AmBMU or AmBAU at dosages of a. 1 mg/kg b. 4 mg/kg or c. 16 mg/kg. P-values are relative to AmB at each indicated time point, *, p≤0.05, **, p≤0.001, ***, p≤0.0001, d. Dose response toxicity assessed via determination of lethality upon single IV injection of AmB, AmBMU and AmBAU to healthy mice at doses of ranging from 0.5 to 64 mg/kg (5 mice per dosage). Mice were monitored for survival up to one day.
Acute mouse toxicity was determined by single intravenous administration of AmB or its derivatives to healthy, uninfected mice and monitoring for lethality (Fig. 3d). All mice in the 4 mg/kg AmB dosage group died within seconds. AmBAU was drastically less toxic with >50% lethality only observed at the 64 mg/kg dosage group. Strikingly, all mice dosed with even 64 mg/kg AmBMU survived with no observable toxicity.
AmB ureas still evade resistance
We next investigated the impacts of increased ergosterol selectivity on the development of resistance to these analogs. Due to its unique mode of action7, AmB is not susceptible to most of the common mechanisms of resistance to other antimicrobials. Its lipid target is not as readily mutable as proteins or RNA; it is unaffected by efflux pumps; and its polyene macrolide structure is not a substrate for secretion via drug-detoxifying enzymes38. Moreover, ergosterol plays a central role in many aspects of yeast physiology10–14. Mutations in genes involved in ergosterol biosynthesis can change sterol structures in ways that confer AmB resistance in vitro39, however, these mutations have enormous fitness costs in vivo, crippling fungal virulence15. Consequently, resistance rarely, if ever appears in the clinic40. We asked if the improved sterol selectivity of AmBMU and AmBAU rendered them more vulnerable to the evolution of resistance.
We first determined the MIC of AmB, AmBMU and AmBAU against a panel of lab-generated strains carrying mutations in all seven of the non-essential C. albicans late-stage ergosterol biosynthesis genes (Fig. 4a)15. Surprisingly, AmBMU and AmBAU displayed an in vitro resistance profile that was very similar to AmB (Fig. 4a). As for AmB, only erg2, erg6 or erg3/erg11 mutants showed substantial resistance to AmBMU and AmBAU, and all of these mutants are known to be avirulent15. Thus, known ergosterol biosynthesis mutations do not appear to be a threat to the efficacy of AmBAU and AmBMU.
Figure 4. Characterization of mechanisms and costs of resistance to AmB ureas.
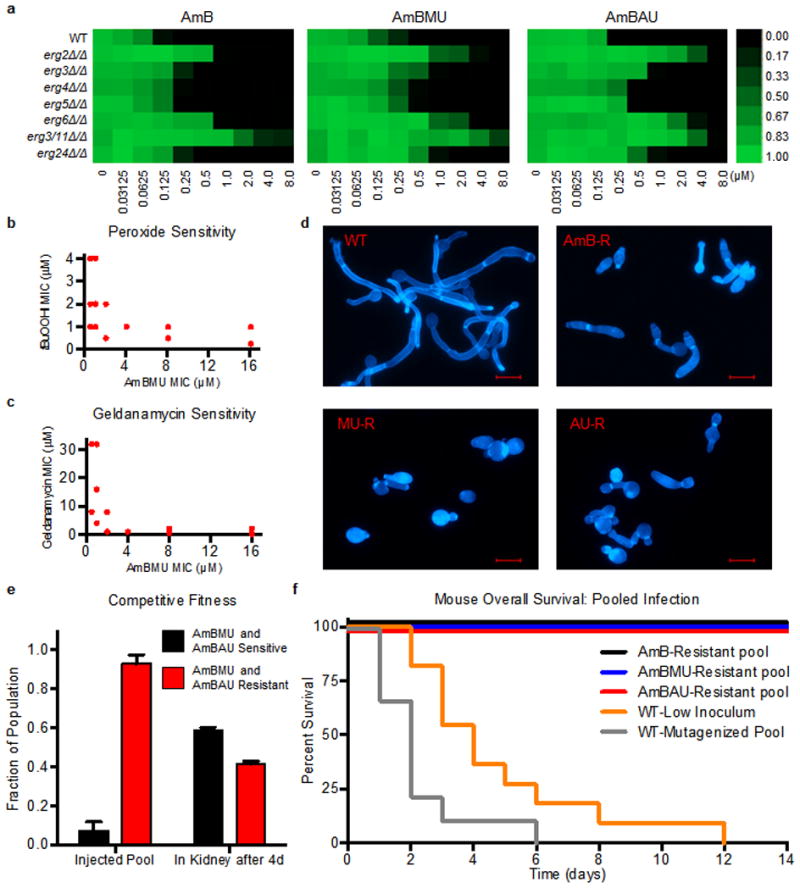
a. Activity of AmB, AmBMU and AmBAU against C. albicans ergosterol biosynthesis mutants. Growth (as judged by OD600 at 24 h) is shown relative to WT with no compound added. Scale bar indicates relative growth ranging from bright green (equal to WT growth, 1.00) to black (zero growth, 0.00). b,c. MIC of tert-butyl peroxide and geldanamycin compared to AmBMU for all resistant isolates selected; each point indicates one or more isolates. d. Filamentation in response to serum at 37°C. Representative images of wild-type and mutants selected in AmB, AmBMU or AmBAU, (all cross-resistant), stained with Calcofluor White. Scale bar = 10 μm e. Competitive infection of mice with 16 different strains (1 WT and 15 that are resistanct to AmBMU and AmBAU). 3×104 cells of each strain injected per mouse (4.8×105 total inoculum). Fraction of pool sensitive or resistant to AmBMU and AmBAU determined before tail-vein injection and 4d later after isolation from kidneys. f. Mouse overall survival after tail-vein injection. Mice were injected with a pool of 5 passaged and mutagenized WT strains each at 1.6×105 (8×105 total inoculum), 1.6×105 cells of parental WT (WT-Low), or pools of 5 resistant mutants with 1.6×105 cells of each mutant.
We next used exhaustive unbiased selections for survival in the presence of the compounds to ask if any other mutations could confer resistance to AmBMU or AmBAU. One-step selections on plates containing 8X the MIC of AmB, AmBMU or AmBAU yielded no colonies with stable resistance to any of the drugs, even after ethyl methanesulfonate mutagenesis. We then utilized a gradual resistance-selection protocol in liquid culture, with serial two-fold increases in drug concentration. Most serial selections ended in extinction of the lineage. However, we did recover 5–8 mutants for each drug that exhibited a ≥4-fold increase in MIC. Importantly, all of these substantially resistant mutants were cross-resistant to all three compounds, suggesting no new mechanisms of resistance unique to AmBMU or AmBAU (Supplementary Results, Supplementary table 1).
To identify the mutations responsible for resistance in these selections, we used whole genome sequencing of the WT strain as well as all 11 of the in vitro-evolved resistant mutants, including both strongly and weakly-resistant isolates (Supplementary table 1). Most mutants with strong resistance to AmB or the derivatives contained mutations in the ERG2 or ERG6 locus that subsequently underwent a loss of heterozygosity (Supplementary Fig. 1)15. Unexpectedly, we also identified 3 independent mutants with low-level (~2-fold) AmB resistance mediated through a recurrent substitution in ORF19.7285 (D216Y), an uncharacterized WD40 repeat protein conserved across fungi. However, these mutants were no more resistant to AmBMU and AmBAU than to AmB (Supplementary table 1).
We then asked if any of the mutants with substantial resistance to AmBMU or AmBAU (>4-fold MIC increase) could elude the dramatic fitness defects previously demonstrated for AmB-resistance. As previously reported15, all AmB-resistant mutants are extremely sensitive to oxidative stressors, which are continually encountered during the course of infection. In addition, they become highly dependent on the molecular chaperone Hsp90, which supports diverse fungal stress responses15. All of the mutants resistant to AmBMU or AmBAU were likewise severely sensitized to the oxidative stressor tert-butyl hydrogen peroxide and the Hsp90 inhibitor geldanamycin (Fig. 4b,c). Previous work has also demonstrated that AmB-resistance mutations disable filamentation, a key driver of virulence in Candida15. Again, in response to stimulation with fetal bovine serum at 37°C, all mutants with strong resistance to AmBMU or AmBAU were unable to form the filaments observed in the wild-type (Fig. 4d).
We next asked if resistance to AmBMU or AmBAU reduces competitive fitness in vivo. To do this, we infected mice with a pool of strains, consisting of the wild-type parent (AmBMU and AmBAU-sensitive) and 15 AmBMU or AmBAU-resistant mutants (with each strain comprising 1/16th of the total population). After allowing the infection to proceed for four days (in the absence of drug treatment), we euthanized the mice and tested the drug sensitivity of fungal colonies isolated from in the kidneys to determine the AmBMU and AmBAU-resistant fraction of the final population. Indeed, even over this short period of infection, the percentage of the surviving population resistant to AmBMU or AmBAU dropped dramatically and the drug-sensitive parent rapidly overtook the population (Fig. 4e).
Finally, we tested if any of our AmBMU or AmBAU-resistant mutants retained the capacity to cause lethal infection. To do so, we inoculated mice with pools of resistant mutants and compared their survival to mice infected with wild-type strains. At a low inoculum (to match that of an individual resistant strain) the wild-type strain killed all infected mice (Fig. 4f). Wild-type strains subjected to the same mutagenesis and in vitro passaging used to generate the resistant strains also killed all of the mice. In stark contrast, all mice infected with pools of mutants selected for resistance to AmB, AmBMU or AmBAU survived the infection (Fig. 4f). Thus, AmBMU and AmBAU are no more vulnerable to resistance than AmB, which has evaded resistance despite widespread clinical utilization for over half a century.
DISCUSSION
Our findings collectively reveal that selective antimicrobial action and evasion of resistance are not mutually exclusive. Here, compounds that bind with high selectivity to a pathogen-specific lipid evade the emergence of virulent resistant strains, suggesting major costs in fitness are caused by even small changes in the structure of this lipid. This is likely because ergosterol is a central molecular node41 in yeast physiology, being critical for the function of membrane proteins10, endocytosis11, vacuole fusion12, membrane compartmentalization13, and pheromone signaling14.
Our results further suggest that AmBMU and AmBAU are exceptionally promising candidates for replacing AmB as a less toxic treatment for invasive fungal infections. These new derivatives bind ergosterol but not cholesterol, maintain potent activity against a broad range of pathogenic fungi, are substantially more effective and less toxic than AmB in vivo, yet still evade resistance. Moreover, these compounds are accessed in just three steps from AmB, which is already fermented and commercially available on the metric ton scale. All of the reagents used in their synthesis are already employed on the process scale to prepare other pharmaceuticals, including diphenyl phosphoryl azide42.
Our results also support a novel ligand-selective allosteric-effect model for guiding the rational development of other non-toxic amphotericins. This model predicts that disruption of intramolecular polar interactions between functional groups on the macrolide core and the mycosamine appendage cause a conformational shift in the molecule, from one that binds both ergosterol and cholesterol to one that selectively binds ergosterol. Strikingly, the portion of AmB that contains all of these functional groups, i.e., the module comprised of C13–C23, is 100% conserved in every member of the mycosamine-bearing polyene macrolide family of natural products43. This includes a collection of “aromatic polyenes” that are reported to be orders of magnitude more potent than AmB44 and effective against even very challenging to treat Aspergillus infections45. Thus, synthesizing the analogous urea derivatives of other polyene macrolide natural products could lead to ultrapotent and/or broader spectrum yet still minimally toxic new antifungal agents.
More broadly, our results show that targeting pathogen specific and polyfunctional lipids represents a promising blueprint for achieving the highly sought combination of low toxicity and evasion of resistance. Intriguingly, nisin, which binds a bacterial polyfunctional lipid, lipid II, has similarly evaded resistance despite half a century of use as a food preservative46, and the recently discovered antibiotic teixobactin also binds lipid II and thus far has evaded resistance in vitro47. It was recently discovered that binding of the same lipid underlies the action of defensin peptides48, critical components of innate immunity that have retained antibiotic activity over more than two billion years of evolution. Other recent studies increasingly show that specific lipid-transmembrane protein interactions are critical for diverse cellular functions49. Thus, as new microbe-specific and polyfunctional lipids are discovered50, they present outstanding targets for the rational development of other non-toxic and resistance-evasive antimicrobials.
Online Methods
General Reaction Conditions
Due to the light and air sensitivity of amphotericin B (AmB), all reactions were performed in oven or flame dried glassware under an atmosphere of argon under low light conditions. Compounds were stored under an anaerobic atmosphere. All solvents were dispensed from a solvent purification as described by Pangborn and coworkers51 (THF, Et2O : dry neutral alumina; DMSO, DMF, CH3OH : activated molecular sieves). Triethylamine and pyridine were freshly distilled under nitrogen from CaH2. Camphorsulfonic acid was recrystallized from ethanol. Water was obtained from a Millipore (Billerica, MA) MilliQ water purification system. Reactions were monitored by RP-HPLC using an Agilent 1200 Series HPLC system equipped with an Agilent Zorbax Eclipse C18 3.5-μm, 4.6 × 75 mm column with UV detection at 383 nm at 1.2 mL/min, or an Agilent 6230 ESI TOF LC/MS system equipped with an Agilent Zorbax Eclipse C18 1.8-μm, 2.1 × 50 mm column with UV detection at 383 nm at 0.4 mL/min. Full experimental details and characterization for new compounds appear in the Supplementary Note.
Extinction Coefficient Determination
Extinction coefficients (L mol−1 cm−1) were determined as previously reported9 and were as follows: AmB (ε406 = 164,000), AmdeB (ε406 = 102,000), AmBME (ε406 = 117,000), C41MeAmB (ε406 = 102,000), AmBMA (ε406 = 114,000), AmBTABA (ε406 = 121,000), AmBMU (ε406 = 87,000), AmBAU (ε406 = 87,000), AmBCU (ε406 = 58,000).
In Vivo Sterol Extraction Studies and Membrane Isolation
This assay was performed similar to that previously described8. Specifically, 75 mL overnight cultures of Saccharomyces cerevisiae were grown to stationary phase (OD ~1.7) in YPD media at 30°C, shaking. 49.5 mL of this culture was transferred to a 50 mL Falcon centrifuge tubes.
Cells were treated with 500 μL of DMSO, 500 μM AmB, 500 μM AmBAU, 500 μM AmBMU, or 500 μM AmBCU (final compound concentration of 5 μM). Falcon tubes were incubated in the shaking incubator at 30°C for 2 hours. Tubes were inverted at the 1 hour timepoint to resuspend.
Yeast membranes were isolated using a modified version of Haas’ spheroplasting and isosmotic cell lysis protocol and differential ultracentrifugation. After treatment time, tubes were centrifuged for 5 minutes at 3000 g at 23°C. The supernatant was decanted and 5 mL of wash buffer (milliQ H2O (89%), 1M aq. DTT (1%), and 1M aq. Tris buffer pH 9.4 (10%)) was added. Tubes were vortexed to resuspend and incubated in a 30°C water bath for 10 minutes. Tubes were then centrifuged for 5 minutes at 3000 g at 23°C and the supernatant decanted.
1 mL of spheroplasting buffer (1M aq. potassium phosphate buffer pH 7.5 (5%), 4M aq. sorbitol (15%), and YPD media (80%)) and 100 μL of a 5 mg/mL aq. solution of lyticase from Arthrobacter luteus (L2524 Sigma-Aldrich) was added to each tube, vortexed to resuspend. Tubes were incubated in a 30°C shaking incubator for 30 minutes. After incubation, tubes were centrifuged for 10 minutes at 1080 g at 4°C and the supernatant decanted.
1 mL of PBS buffer and 20 μL of a 0.4 mg/ml dextran in 8% Ficoll solution was added to each tube, mixed very gently to resuspend. This suspension was placed in an ice bath for 4 minutes and then transferred to a 30°C water bath for 3 minutes.
The suspensions were transferred to 2 mL Eppendorf tubes, vortexed to ensure complete lysis, and centrifuged at 15,000 g at 4°C to remove un-lysed cells and cell debris. The resulting supernatants were transferred to thick-wall polycarbonate ultracentrifuge tubes (3.5 mL, 13 × 51 mm, 349622 Beckman Coulter). PBS buffer was added to the tubes to bring the volume up to ~3 mL. The tubes were centrifuged for 1 hour at 100,000 g at 4°C in a Beckman Coulter TLA-100.3 fixed-angle rotor in a tabletop ultracentrifuge. The supernatant was poured off. The remaining membrane pellet was resuspended in 1 mL PBS buffer. 750 μL of the suspension was transferred to a 7 mL vial and stored at −80°C until further analysis.
Gas chromatography quantification of sterols
The suspension was allowed to warm to room temperature and 20 μL of internal standard (4 mg/mL cholesterol in chloroform) was added. They were dissolved in 3 mL 2.5% ethanolic KOH, which was vortexed gently, capped, and heated in a heat block on a hot plate at 90°C for 1 hour. The vials were allowed to cool to room temperature. 1 mL of brine was added to the contents of each vial. Extraction was performed three times, each with 2 mL of hexane. Organic layers were combined, dried over MgSO4, filtered through Celite® 545, and transferred to another 7 mL vial. The contents of the vial were concentrated in vacuo. The lipid films were dried on high vac with P2O5 for 30 minutes to remove residual water.
To the resulting lipid films, 100 μL pyridine and 100 μL N,O-Bis(trimethylsilyl)trifluoroacetamide with 1% trimethylchlorosilane (T6381-10AMP Sigma-Aldrich) was added and vortexed gently. This solution was heated at 60°C for 1 hour to produce TMS ethers. The vials were placed in an ice bath and the solvent was evaporated off by nitrogen stream. Vials were kept at low temperature to prevent evaporation of the sterol ethers along with the solvent. The resulting films were resuspended in 100 μL of decane, filtered using a Supelco ISO-Disc PTFE Filter (4 mm × 0.2 μm) and transferred to a GC vial insert for analysis.
Gas chromatography analysis was carried out on an Agilent 7890A gas chromatograph equipped with FID and Agilent GC 7693 Autosampler. Samples were separated on a 30 m, 0.320 mm ID, 0.25 um film HP-5 capillary column (19091J-413 Agilent). Hydrogen was employed as a carrier gas with a flow rate of 4 mL/min. Nitrogen make-up gas, hydrogen gas, and compressed air were used for the FID. A split/splitless injector was used in a 20:1 split. The injector volume was 2 μL. The column temperature was initially held at 250°C for 0.5 min, then ramped to 265°C at a rate of 10°C/min with a final hold time of 12.5 min. The injector and detector temperature were maintained at 270°C and 290°C, respectively.
Isothermal Titration Calorimetry
ITC was performed as previously reported9.
Growth Conditions for S. cerevisiae
S. cerevisiae were grown following known procedures9.
Growth Conditions and MIC Assay for C. albicans, C. tropicalis, C. parapsilosis, and C. glabarata
The organisms were maintained, grown, subcultured, and quantified on Sabouraud dextrose agar (SDA; Difco Laboratories, Detroit, MI). 24 hours prior to the study, the organisms were subcultured at 35°C. MIC determinations were performed in duplicate on at least two occasions using the Clinical and Laboratory Standards Institute M27-A3 microbroth methodology52.
Growth Conditions and MIC Assay for C. neoformans
C. neoformans MIC was determined as previously reported after 48 hours33.
Growth Conditions and MIC Assay for C. fumigatus
The organisms were maintained, grown, subcultured, and quantified on potato dextrose agar (PDA; Difco Laboratories, Detroit, MI). MIC determinations were performed in duplicate on at least two occasions using the Clinical and Laboratory Standards Institute M28-A2 microbroth methodology53 at 48 hours.
Hemolysis Assays
Hemolysis experiments were performed following known procedures9.
WST-8 Cell Proliferation Assays
Primary Renal Proximal Tubule Epithelial Cells Preparation
Primary human renal proximal tubule epithelial cells (RPTECs) were prepared following known procedures9.
TERT1 Renal Proximal Tubule Epithelial Cells Preparation
TERT1 human renal proximal tubule epithelial cells (RPTECs) were purchased from ATCC (CRL-4031, Manassas, VA) and immediately cultured upon receipt. Complete growth media was prepared using DMEM:F12 media (ATCC, 30-2006), triiodo-L-thyronine (Sigma, T6397), recombinant human EGF (Life Technologies, PHG0311), ascorbic acid (Sigma, A4403), human transferrin (Sigma, T8158), insulin (Sigma I9278), prostaglandin E1 (Sigma, P7527), hydrocortisone (Sigma, H0888), sodium selenite (Sigma, S5261), and G418 (Sigma, A1720). Complete media was stored at 4°C and used within 28 days. TERT1 RPTECs were grown in CO2 incubator at 37°C with an atmosphere of 95% air/5% CO2.
WST-8 Reagent Preparation
WST-8 reagent was prepared and stored following known procedures9.
WST-8 Assay
A suspension of primary or TERT1 RPTECs in complete growth media was brought to a concentration of 1 × 105 cells/mL. A 96-well plate was seeded with 99 μL of the cell suspension and incubated at 37°C with an atmosphere of 95% air/5% CO2 for 3 hours. Positive and negative controls were prepared by seeding with 100 μL of the cell suspension or 100 μL of the complete media. Compounds were prepared as 5 mM (AmB) and 8 mM (AmBAU, AmBMU, AmBCU, and AmdeB) stock solutions in DMSO and serially diluted to the following concentrations with DMSO: 8000, 6000, 4000, 3000, 2000, 1500, 1000, 800, 600, 400, 300, 200, 100, 50, 25, 10, 5, 2.5, 1, 0.5, 0.25, and 0.1 μM. 1 μL aliquots of each solution were added to the 96-well plate in triplicate, with each column representing a different concentration of the test compound. The 96-well plate was incubated at 37°C with an atmosphere of 95% air/5% CO2 for 24 hours. After incubation, the media was aspirated and 100 μL of serum-free media was added and 10 μL of the WST-8 reagent solution was added to each well. The 96-well plate was mixed in a shaking incubator at 200 rpm for 1 minute and incubated at 37°C with an atmosphere of 95% air/5% CO2 for 2 hours. Following incubation, the 96-well plate was mixed in a shaking incubator at 200 rpm for 1 minute and absorbances were read at 450 nm using a Biotek H1 Synergy Hybrid Reader (Wanooski, VT). Experiments were performed in triplicate and the reported cytotoxicity represents an average of three experiments.
Data Analysis
Percent hemolysis was determined according to the following equation:
Concentration vs. percent hemolysis was plotted and fitted to 4-parameter logistic (4PL)54 dose response fit using OriginPro 8.6. The MTC was defined as the concentration to cause 90% loss of cell viability.
Ethics Statement
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin according to the guidelines of the Animal Welfare Act, The Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals, and Public Health Service Policy.
In Vivo Murine Efficacy Study
All studies were approved by the Animal Research Committee of the William S. Middleton Memorial VA Hospital (Madison, WI). Efficacy was assessed by CFU count in the kidneys of neutropenic mice with a disseminated fungal infection as described previously by Andes et al40,55,56. A clinical isolate of Candida albicans (K-1) was grown and quantified on SDA. For 24 hours prior to infection, the organism was subcultured at 35°C on SDA slants. A 106 CFU/mL inoculum (CFU, colony forming units) was prepared by placing six fungal colonies into 5 mL of sterile, depyrogenated normal (0.9%) saline warmed to 35°C. Six-week-old ICR/Swiss specific-pathogen-free female mice were obtained from Harlan Sprague Dawley (Madison, WI). The mice were weighed (23–27 g) and given intraperitoneal injections of cyclophosphamide to render neutropenia (defined as <100 polymorphonuclear leukocytes/mm3). Each mouse was dosed with 150 mg/kg of cyclophosphamide 4 days prior to infection and 100 mg/kg 1 day before infection. Disseminated candidiasis was induced via tail vein injection of 100 μL of inoculum. AmB, AmBAU, or AmBMU were reconstituted with 1.0 mL of 5% dextrose. Each animal in the treatment group was given a single 200 μL intraperitoneal (ip) injection of reconstituted AmB, AmBAU, or AmBMU 2 hours post-infection. Doses were calculated in terms of mg of compound/kg of body weight. At each time point (6, 12, and 24 hours post-infection), three animals per experimental condition were sacrificed by CO2 asphyxiation. The kidneys from each animal were removed and homogenized. The homogenate was diluted serially 10-fold with 9% saline and plated on SDA. The plates were incubated for 24 hours at 35°C and inspected for CFU viable counts. The lower limit of detection for this technique is 100 CFU/mL. All results are expressed as the mean log10 CFU per kidney for three animals.
In Vivo Murine Toxicity Study
All studies were approved by the Animal Research Committee of the William S. Middleton Memorial VA Hospital (Madison, WI). Uninfected Swiss ICR mice were used for assessment of infusion toxicity. Groups of five mice were treated with single intravenous doses of AmB, AmBAU, AmBMU (reconstituted with 1.0 mL of 5% dextrose), or sterile pyrogen-free 0.85% NaCl administered via the lateral tail vein over 30 seconds. Dose levels studies included 0.5, 1, 2, 4, 8, 16, 32, and 64 mg/kg. Following administration mice were observed continuously for one hour and then every 6 hours up to 24 hours for signs of distress or death.
Resistance Studies
Minimum Inhibitory Concentration and Growth Assays
Susceptibility of wild-type and resistant strains to AmB, AmBAU, AmBMU, tert-butyl peroxide (Sigma-Aldrich), geldanamycin and radicicol (A.G. Scientific) was determined in flat bottom, 96-well microtiter plates (Costar) using a broth microdilution protocol adapted from CLSI M27-A3. Overnight cultures (14–20hr) were grown at 30°C in YPD, and approximately 5×103 cells were seeded per well. For AmB, AmBAU, and AmBMU, MIC assays were performed at 37°C in RPMI buffered with MOPS (0.165M) with 10% fetal bovine serum (Sigma-Aldrich) added; for tert-butyl peroxide, geldanamycin, and radicicol, MIC’s were determined in YPD at 30°C. MIC’s were determined after 24h incubation as the concentration of compound resulting in no visible growth in wells. For quantitative display of growth at drug dilutions, OD600 was measured in a spectrophotometer (Tecan) and displayed as heat maps using Java TreeView 1.1.3 (http://jtreeview.sourceforge.net).
Media and Growth Conditions
C. albicans was generally grown and maintained as described previously15. Stocks were stored in 15% glycerol at −80°C; strains were generally grown in YPD media at 30°C. Drugs were added directly to media from DMSO stocks.
In Vitro Gradual Selection of AmB, AmBAU, or AmBMU Resistance
Selection of resistance to AmB, AmBAU, and AmBMU was performed as follows. 1 mL overnight (14–20hr) cultures of SC5314 (WT) were washed in PBS, then treated with 3% ethyl methanesulfonate (EMS) for 45 min. Cells were then washed 4x in YPD and resuspended in YPD and allowed to recover for 3h. Cells were then inoculated to an OD600 of approximately 0.025–0.05 in 100mL YPD containing 0.25μM AmB or AmB-AU, or 0.375μM AmB-MU. After 24–72 hours, a 1mL aliquot was removed from any culture that had grown to saturation and subjected to another round of mutagenesis in the same manner as described above. After recovery, cells were then inoculated into a new YPD flask containing 2x higher concentration of the same drug. Cultures that grew were subjected to one more round of EMS mutagenesis before inoculating into a 2-fold higher drug concentration (total of 3 rounds of EMS mutagenesis) and then passaged at 2-fold higher increments of drug concentration until reaching 2μM AmB or AmB-AU, or 3μM AmB-MU. Cultures were passaged once more at 2μM AmB or AmBAU or 3μM AmBMU, then plated onto YPD media and frozen in glycerol stocks before further evaluation.
Filamentation Assay
Hyphal induction was performed by growing C. albicans overnight at 30°C in YPD, washing in PBS, and diluting 1:100 into RPMI+10% fetal bovine serum (Sigma-Aldrich) at 37°C in a culture tube on a rotating wheel. After 3h, cultures were washed in PBS and resuspended in 250μg/mL Calcofluor white in a microcentrifuge tube, and shaken at 30°C for 10 min. Cells were then washed twice in PBS, concentrated 10-fold, briefly sonicated in a water bath, and mounted on slides for visualization under a DAPI filter set at 60X magnification.
Murine Model of Systemic Infection
All animal protocols were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animals were maintained according to the guidelines of the MIT Committee on Animal Care (CAC). These studies were approved by the MIT CAC (protocol #0312-024-15). We used 7–12-week-old female Balb/c mice ordered from Taconic farms for all mouse virulence studies. All strains were prepared for inoculation by diluting overnight cultures (14–20h) 1:100 into YPD and growing into log phase for 4–5 hours, then washing 3x in PBS before. Strains were injected into the lateral tail vein in a volume of 100 μl. For mouse survival experiments, strains were grouped as follows: The wild-type Mutagenized pool consisted of 5 SC5314 colonies subjected in parallel to mutagenesis and passaging (as described above) without drug exposure, injected as of 1.6×105 cfu per strain (8×105 cfu total inoculum per mouse); the Wild-type low inoculum was the SC5314 parental strain injected at 1.6×105 cfu. AmB-Resistant, AmBAU-Resistant, and AmBMU-resistant pools were comprised of strains isolated from each selection in the presence of the indicated drug, using strains that exhibited >4-fold MIC increase for the drug used. Individual resistant strains were present in the pools at 1.6×105 cfu per mouse (8×105 total inoculum per mouse when pooled). Each strain or pool of strains was tested in at least two independent experiments, and data were pooled. Mice were weighed daily and monitored for signs of morbidity and sacrificed when body weight decreased by 20%, or when signs of extreme distress were apparent. For the competitive infection with quantification of kidney burden, a pool comprised of 16 strains at equal fraction of the population, one SC5314 wild-type and 5 strains each from selections for resistance to AmB, AmBAU, and AmBMU was used, with 3×104 cells of each strain inoculated per mouse (4.8×105 total inoculum). Three mice were used per experiment, in a total of two experiments. 4d after infection, mice were sacrificed and kidneys were removed aseptically, homogenized, and plated onto YPD plates. Pools of the inoculum immediately before injection were also plated. 184 colonies were randomly selected from the pre-infection and 184 from the post-infection plates and tested for growth in 96-well plates in the presence of 1 μM AmBAU or 1.5 μM AmBMU, and the fraction of wells from the pre and post-infection pools exhibiting growth in either drug was determined.
Whole Genome Sequencing, Alignment, Mapping, and Variant Calling
Whole genome sequencing and analysis was performed as previously described15.
Supplementary Material
Acknowledgments
Portions of this work were supported by the National Institutes of Health (R01GM080436, R01GM080436-S), Howard Hughes Medical Institute (HHMI), and The Mathers Foundation. M.D.B is an HHMI Early Career Scientist and S.L. is an HHMI Investigator.
Footnotes
Author Contributions S.A.D. and M.D.B. conceived the study and oversaw design of synthesis, biophysical, and several biological experiments, B.M.V., L.W., and S.L. designed resistance studies, and D.R.A. designed mouse toxicity and efficacy studies. S.A.D. synthesized all compounds, B.M.V. executed all resistance studies, M.M.E. performed sterol binding and designed and performed cell toxicity assays, and K.M. performed efficacy and toxicity studies in mice. S.A.D., B.M.V., S.L., and M.D.B. wrote the manuscript.
Competing Financial Interest The University of Illinois has filed patents on compounds and chemistry reported herein. These have been licensed to REVOLUTION Medicines, a company for which M.D.B. is a founder.
Supplementary Information is available in the online version of the paper
References
- 1.Li J, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 2.Cortes JE, et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome–Positive Leukemias. New Engl J Med. 2013;369:1783–1796. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis D. Amphotericin B: spectrum and resistance. J Antimicrob Chemoth. 2002;49:7–10. doi: 10.1093/jac/49.suppl_1.7. [DOI] [PubMed] [Google Scholar]
- 4.Brown GD, et al. Hidden Killers: Human Fungal Infections. Sci Transl Med. 2012;4:165rv113. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 5.Volmer AA, Szpilman AM, Carreira EM. Synthesis and biological evaluation of amphotericin B derivatives. Nat Prod Rep. 2010;27:1329–1349. doi: 10.1039/b820743g. [DOI] [PubMed] [Google Scholar]
- 6.Ermishkin LN, Kasumov KM, Potzeluyev VM. Single ionic channels induced in lipid bilayers by polyene antibiotics amphotericin B and nystatine. Nature. 1976;262:698–699. doi: 10.1038/262698a0. [DOI] [PubMed] [Google Scholar]
- 7.Gray KC, et al. Amphotericin primarily kills yeast by simply binding ergosterol. Proc Natl Acad Sci USA. 2012;109:2234–2239. doi: 10.1073/pnas.1117280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson TM, et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol. 2014;10:400–406. doi: 10.1038/nchembio.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilcock BC, Endo MM, Uno BE, Burke MD. C2′-OH of Amphotericin B Plays an Important Role in Binding the Primary Sterol of Human Cells but Not Yeast Cells. J Am Chem Soc. 2013;135:8488–8491. doi: 10.1021/ja403255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YQ, et al. Requirement for Ergosterol in V-ATPase Function Underlies Antifungal Activity of Azole Drugs. PLoS Pathog. 2010;6:e1000939. doi: 10.1371/journal.ppat.1000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heese-Peck A, et al. Multiple Functions of Sterols in Yeast Endocytosis. Mol Biol Cell. 2002;13:2664–2680. doi: 10.1091/mbc.E02-04-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato M, Wickner W. Ergosterol is required for the Sec18/ATP-dependent priming step of homotypic vacuole fusion. The EMBO Journal. 2001;20:4035–4040. doi: 10.1093/emboj/20.15.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klose C, et al. Yeast Lipids Can Phase-separate into Micrometer-scale Membrane Domains. J Biol Chem. 2010;285:30224–30232. doi: 10.1074/jbc.M110.123554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin H, McCaffery JM, Grote E. Ergosterol promotes pheromone signaling and plasma membrane fusion in mating yeast. J Cell Biol. 2008;180:813–826. doi: 10.1083/jcb.200705076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincent BM, Lancaster AK, Scherz-Shouval R, Whitesell L, Lindquist S. Fitness Trade-offs Restrict the Evolution of Resistance to Amphotericin B. PLoS Biol. 2013;11:e1001692. doi: 10.1371/journal.pbio.1001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palacios DS, Dailey I, Siebert DM, Wilcock BC, Burke MD. Synthesis-enabled functional group deletions reveal key underpinnings of amphotericin B ion channel and antifungal activities. Proc Natl Acad Sci USA. 2011;108:6733–6738. doi: 10.1073/pnas.1015023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann A, Baginski M, Czub J. How Do Sterols Determine the Antifungal Activity of Amphotericin B? Free Energy of Binding between the Drug and Its Membrane Targets. J Am Chem Soc. 2010;132:18266–18272. doi: 10.1021/ja1074344. [DOI] [PubMed] [Google Scholar]
- 18.Matsumori N, Sawada Y, Murata M. Mycosamine orientation of amphotericin B controlling interaction with ergosterol: Sterol-dependent activity of conformation-restricted derivatives with an amino-carbonyl bridge. J Am Chem Soc. 2005;127:10667–10675. doi: 10.1021/ja051597r. [DOI] [PubMed] [Google Scholar]
- 19.Jarzembska KN, et al. Controlled Crystallization, Structure, and Molecular Properties of Iodoacetylamphotericin B. Cryst Growth Des. 2012;12:2336–2345. [Google Scholar]
- 20.Neant-Fery M, et al. Molecular basis for the thiol sensitivity of insulin-degrading enzyme. Proc Natl Acad Sci USA. 2008;105:9582–9587. doi: 10.1073/pnas.0801261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duggan KC, et al. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chem Biol. 2011;7:803–809. doi: 10.1038/nchembio.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacPherson DT, et al. Recent Advances in the Chemistry of Antiinfective Agents. Vol. 119. Royal Society of Chemistry; 1993. pp. 205–222. [Google Scholar]
- 23.Power P, et al. Engineered Synthesis of 7-Oxo- and 15-Deoxy-15-Oxo-Amphotericins: Insights into Structure-Activity Relationships in Polyene Antibiotics. Chem Biol. 2008;15:78–86. doi: 10.1016/j.chembiol.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Carmody M, et al. Biosynthesis of Amphotericin Derivatives Lacking Exocyclic Carboxyl Groups. J Biol Chem. 2005;280:34420–34426. doi: 10.1074/jbc.M506689200. [DOI] [PubMed] [Google Scholar]
- 25.Byrne B, Carmody M, Gibson E, Rawlings B, Caffrey P. Biosynthesis of Deoxyamphotericins and Deoxyamphoteronolides by Engineered Strains of Streptomyces nodosus. Chem Biol. 2003;10:1215–1224. doi: 10.1016/j.chembiol.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Maeda H, Suzuki M, Sugano H, Matsumoto K. A Facile Synthesis of (S)-Isoserine from (S)-Malic Acid. Synthesis. 1988;5:401–402. [Google Scholar]
- 27.Palacios DS, Anderson TM, Burke MD. A Post-PKS Oxidation of the Amphotericin B Skeleton Predicted to be Critical for Channel Formation Is Not Required for Potent Antifungal Activity. J Am Chem Soc. 2007;129:13804–13805. doi: 10.1021/ja075739o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonner DP, Mechlinski W, Schaffner CP. Polyene Macrolide Derivatives. 3 Biological Properties of Polyene Macrolide Ester Salts. J Antibiot. 1972;25:261–262. [PubMed] [Google Scholar]
- 29.Keim GR, et al. Comparative Toxicological Studies of Amphotericin B Methyl Ester and Amphotericin B in Mice, Rats, and Dogs. Antimicrob Agents Chemother. 1976;10:687–690. doi: 10.1128/aac.10.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tevyashova AN, et al. Structure-Antifungal Activity Relationships of Polyene Antibiotics of the Amphotericin B Group. Antimicrob Agents Chemother. 2013;57:3815–3822. doi: 10.1128/AAC.00270-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paquet V, Volmer AA, Carreira EM. Synthesis and in vitro biological properties of novel cationic derivatives of amphotericin B. Chem Eur J. 2008;14:2465–2481. doi: 10.1002/chem.200701237. [DOI] [PubMed] [Google Scholar]
- 32.Pfaller M, et al. Epidemiology and outcomes of candidemia in 3648 patients: data from the Prospective Antifungal Therapy (PATH Alliance®) registry, 2004–2008. Diagn Micr Infec Dis. 2012;74:323–331. doi: 10.1016/j.diagmicrobio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Cruz MC, et al. Immunosuppressive and Nonimmunosuppressive Cyclosporine Analogs Are Toxic to the Opportunistic Fungal Pathogen Cryptococcus neoformans via Cyclophilin-Dependent Inhibition of Calcineurin. Antimicrob Agents Chemother. 2000;44:143–149. doi: 10.1128/aac.44.1.143-149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lepak AJ, Marchillo K, VanHecker J, Andes DR. Posaconazole Pharmacodynamic Target Determination against Wild-Type and Cyp51 Mutant Isolates of Aspergillus fumigatus in an In Vivo Model of Invasive Pulmonary Aspergillosis. Antimicrob Agents Chemother. 2013;57:579–585. doi: 10.1128/AAC.01279-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis JK, et al. Metabolic response to low-level toxicant exposure in a novel renal tubule epithelial cell system. Mol Biosyst. 2011;7:247–257. doi: 10.1039/c0mb00146e. [DOI] [PubMed] [Google Scholar]
- 36.Zager RA. Polyene antibiotics: Relative degrees of in vitro cytotoxicity and potential effects on tubule phospholipid and ceramide content. Am J Kidney Dis. 2000;36:238–249. doi: 10.1053/ajkd.2000.8967. [DOI] [PubMed] [Google Scholar]
- 37.Andes D, Stamsted T, Conklin R. Pharmacodynamics of Amphotericin B in a Neutropenic-Mouse Disseminated-Candidiasis Model. Antimicrob Agents Chemother. 2001;45:922–926. doi: 10.1128/AAC.45.3.922-926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfaller MA. Antifungal Drug Resistance: Mechanisms, Epidemiology, and Consequences for Treatment. Am J Med. 2012;125:S3–S13. doi: 10.1016/j.amjmed.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Sanglard D, Ischer F, Parkinson T, Falconer D, Bille J. Candida albicans Mutations in the Ergosterol Biosynthetic Pathway and Resistance to Several Antifungal Agents. Antimicrob Agents Chemother. 2003;47:2404–2412. doi: 10.1128/AAC.47.8.2404-2412.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfaller MA, et al. Wild-Type MIC Distributions and Epidemiological Cutoff Values for Amphotericin B, Flucytosine, and Itraconazole and Candida spp. as Determined by CLSI Broth Microdilution. J Clin Microbiol. 2012;50:2040–2046. doi: 10.1128/JCM.00248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinto JP, Machado R, Xavier JG, Futschik ME. Targeting molecular networks for drug research. Front Genet. 2014;5 doi: 10.3389/fgene.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grongsaard P, et al. Convergent, Kilogram Scale Synthesis of an Akt Kinase Inhibitor. Organic Process Research & Development. 2012;16:1069–1081. [Google Scholar]
- 43.Dailey I. PhD thesis. University of Illinois; Urbana-Champaign: 2012. Synthesis and Function of the Conserved Motif of Mycosamine Containing Polyene Macrolides. [Google Scholar]
- 44.Kotler-Brajtburg J, et al. Classification of Polyene Antibiotics According to Chemical Structure and Biological Effects. Antimicrob Agents Chemother. 1979;15:716–722. doi: 10.1128/aac.15.5.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moonis M, Ahmad I, Bachhawat BK. Liposomal Hamycin in the control of experimental Aspergillosis in mice - relative toxicity, therapeutic efficacy and tissue distribution of free and liposomal Hamycin. Indian Journal of Biochemistry & Biophysics. 1992;29:339–345. [PubMed] [Google Scholar]
- 46.Hasper HE, et al. An Alternative Bactericidal Mechanism of Action for Lantibiotic Peptides That Target Lipid II. Science. 2006;313:1636–1637. doi: 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- 47.Ling LL, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider T, et al. Plectasin, a Fungal Defensin, Targets the Bacterial Cell Wall Precursor Lipid II. Science. 2010;328:1168–1172. doi: 10.1126/science.1185723. [DOI] [PubMed] [Google Scholar]
- 49.Laganowsky A, et al. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han X, Yang K, Gross RW. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev. 2012;31:134–178. doi: 10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 52.Clinical Laboratory Standards Institute (CLSI) Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard. 3. Clinical and Laboratory Standards Institute; Wayne, PA: 2008. CLSI document M27–A3. [Google Scholar]
- 53.Clinical Laboratory Standards Institute (CLSI) Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard. 2. Clinical Laboratory Standards Institute; Wayne, PA: 2007. CLSI document M38-A2. [Google Scholar]
- 54.Sebaugh JL. Guidelines for accurate EC50/IC50 estimation. Pharm Stat. 2011;10:128–134. doi: 10.1002/pst.426. [DOI] [PubMed] [Google Scholar]
- 55.Andes D, van Ogtrop M. Characterization and Quantitation of the Pharmacodynamics of Fluconazole in a Neutropenic Murine Disseminated Candidiasis Infection Model. Antimicrob Agents Chemother. 1999;43:2116–2120. doi: 10.1128/aac.43.9.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andes D, van Ogtrop M. In Vivo Characterization of the Pharmacodynamics of Flucytosine in a Neutropenic Murine Disseminated Candidiasis Model. Antimicrob Agents Chemother. 2000;44:938–942. doi: 10.1128/aac.44.4.938-942.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.