

Abstract
The de novo fatty acid synthesis catalyzed by key lipogenic enzymes, including fatty acid synthase (FASN) has emerged as one of the novel targets of anti-cancer approaches. The present study explored the possible inhibitory efficacy of [6]-gingerol on de novo fatty acid synthesis associated with mitochondrial-dependent apoptotic induction in HepG2 cells. We observed a dissipation of mitochondrial membrane potential accompanied by a reduction of fatty acid levels. [6]-gingerol administration manifested inhibition of FASN expression, indicating FASN is a major target of [6]-gingerol inducing apoptosis in HepG2 cells. Indeed, we found that increased ROS generation could likely be a mediator of the anti-cancer effect of [6]-gingerol. A reduction of fatty acid levels and induction of apoptosis were restored by inhibition of acetyl-CoA carboxylase (ACC) activity, suggesting an accumulation of malonyl-CoA level could be the major cause of apoptotic induction of [6]-gingerol in HepG2 cells. The present study also showed that depletion of fatty acid following [6]-gingerol treatment caused an inhibitory effect on carnitine palmitoyltransferase-1 activity (CPT-1), whereas C75 augmented CPT-1 activity, indicating that [6]-gingerol exhibits the therapeutic benefit on suppression of fatty acid β-oxidation.
Keywords: Carnitine palmitoyltransferase-1 (CPT-1)/de novo fatty acid synthesis/fatty acid synthase (FASN)/[6]-gingerol/malonyl-CoA
Introduction
Diets rich in vegetables and fruits supplemented with spices offer protection against malignancies [1,2]. Polyphenols extracted from such sources inhibit tumor cell proliferation [3-7] although their mechanisms of action are less well delineated.
Obesity is associated with metabolic syndrome and deregulation of de novo synthesis of lipids leading to numerous consequences, including tumorigenesis and tumor progression [8]. Many research studies have proposed the beneficial actions of polyphenols extracted for reductions of hepatic fat accumulation, excess weight, and obesity levels by inhibiting the de novo lipid synthesis that leads to reducing the risk of carcinogenesis without disturbing food appetite [9-11], suggesting the therapeutic action of these compounds targeting the de novo synthesis of lipid pathway. However, this pathway is usually over-expressed in cancers to provide precursors for their metabolism and membrane synthesis to support their proliferative phenotype [8]. Inhibiting the endogenous fatty acid biosynthesis pathway in cancer cells promotes cancer cell death via induction of the apoptosis pathway [12-17]. However, the mechanisms of the action of polyphenols targeting the endogenous fatty acid biosynthesis pathway in cancers are less well characterized. Thus, the inhibition of lipogenesis will provide therapeutic efficiency for prevention of obesity-induced carcinogenesis and an alternative strategy for anti-cancer therapy.
There is reprogramming of energy pathways in cancers favoring glycolytic ATP production (60-90% of ATP needs: aerobic glycolysis or Warburg effect) to ensure a high tumor progression rate with the remainder coming from oxidative phosphorylation even though oxygen supply may be adequate [18,19] and elevated mitochondria competency [20]. This metabolic alteration results from aerobic and glycemic conditions through the induction of the oncogenes (c-m yc, H-Ras, v-src) and transcription factors, hypoxia-inducible factor (HIF-1α) [21,22]. To confer rapid proliferation and survival, cancer also redirects acetyl-CoA from the oxidative phosphorylation (OXPHOS) to develop over-expression of the de novo fatty acid synthesis pathway. Besides the production of ATP, enhanced glycolysis in cancer cells is necessary for providing substrates, including acetyl-CoA and malonyl-CoA for this lipogenesis pathway [22]. The enzymes participating in de novo fatty acid synthesis are up-regulated or constitutively expressed in most types of cancer cells [23-25]. De novo fatty acid synthesis uses cytosolic citrate exported from mitochondria into the cytoplasm, which is then converted to acetyl-CoA by ATP-citrate lyase (ACLY) followed by carboxylation to form malonyl-CoA by acetyl-CoA carboxylase (ACC). Fatty acid synthase (FASN) uses acetyl-CoA, malonyl-CoA, and NADPH to elaborate long chain saturated fatty acids (LCFAs), especially 16-C palmitate, which is desaturated to monounsaturated fatty acids (MUFAs) by stearoyl-CoA desaturase (SCD-1). MUFAs are the most important constituent of membrane phospholipids [26]. De novo LCFAs play important roles in serving as precursors for macromolecule synthesis for highly proliferative mammalian cancer cells, more than in most normal cells for which their lipids come from the abundant levels in the circulation [27]. Enrichment of the cell membrane with these fatty acid forms makes the plasma membrane, creating more resistance to peroxidation and to chemo-therapy [28]. Thus, the over-expression of de novo fatty acid synthesis becomes an important requirement and is essential for carcinogenesis and the progression of cancer. Anticancer therapy targeting the de novo LCFA synthesis enzymes has been extensively studied to become one of the most efficient cancer therapies [13,29] by promoting cancer cell apoptosis without affecting non-transformed cells [30,31].
Synthetic FASN inhibitors, such as orlistat, cerulenin and its analogue C75 are potential cancer treatments [29]. However, C75 stimulates carnitine palmitoyltransferase-1 (CPT-1) activity which contra-indicates its clinical application [31]. This effect leads to activation of β-oxidation of fatty acid, consequently enhancing anorexia and loss of body weight. Naturally extracted substances inhibiting CPT-1 activity [6] will open a new research perspective in the attempt to study their mechanisms targeting fatty acid synthesis to be alternative and effective anticancer drugs. [6]-gingerol (1-[4’-hydroxy-3’-methoxyphenyl]-5-hydroxy-3-decanone) is a major phenolic in ginger rhizomes (Zingiber officinale Roscoe, Zingiberaceae) acting against several cancer types [32-34]. Thus, [6]-gingerol implicating inhibition of lipid synthesis will result in cancer cytotoxicity and lead to its being challenged as an optional therapy for cancers or as an adjunct treatment to modulate the potential antitumor therapy of other chemotherapeutic drugs.
The present study examined the antiproliferative effect of [6]-gingerol on de novo fatty acid synthesis in HepG2 cells. [6]-gingerol decreased the de novo fatty acid synthesis correlating with an enhanced mitochondrial-dependent apoptotic pathway. An accumulation of malonyl-CoA following depletion of fatty acid levels by [6]-gingerol was found to inhibit CPT-1 activity, leading to suppression of β-oxidation in HepG2 cells. The first discovery of [6]-gingerol as a new FASN inhibitor will provide one of the potential perspective anticancer treatments and lipogenesis inhibitors to protect obesity-induced carcinogenesis.
Materials and methods
Equipment used
All flow cytometry used a FACScalibur flow cytometry (Becton Dickinson (BD), Franklin Lakes, New Jersey, USA) and the data was analyzed using CellQuestPro software (BD). Confocal microscopy used a Fluoview FV10i-DOC confocal laser scanning microscopy system (Olympus), using universal plan apochomat 60x phase contrast oil-immersion objective and equipped with Fluoview software version 3.0. For Western blots, the fluorescence intensity of the protein band was determined using an ImageQuant LAS 4000 CCD camera (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Cell culture and [6]-gingerol treatment
Human hepatocellular carcinoma, HepG2 cell line (HB-8065) was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). HepG2 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) (ATCC (Manassas, VA, USA and Corning, Tewksbury MA, USA) supplemented with 10% fetal bovine serum and 1% of penicillin/streptomycin solution (100 IU/mL of penicillin and 100 µg/ml of streptomycin) (Gibco BRL, Grand Island, NY, USA) under a humidified 5% CO2 at 37°C. For each experiment, HepG2 cells were treated with different concentrations of [6]-gingerol (Sigma Chemical Co., St. Louise, MO, USA), which was diluted from the stock solution dissolved in DMSO to the culture medium at the maximum final concentration of DMSO at 0.1%.
MTT analysis of cell viability
The cytotoxic effect of [6]-gingerol on the growth and proliferation of HepG2 cells was determined by MTT assay. Briefly, at the end of the treatment period of HepG2 cells with [6]-gingerol, cells were incubated with MTT solution (5 mg/mL) for 2 h at 37°C. The formazan dyes were dissolved in DMSO and measured the absorbance at 595 nm under the microplate reader.
Flow cytometric measurement of apoptosis
After treatment of HepG2 cells cultured in 60 mm3 petri dishes with different concentrations of [6]-gingerol for 24 and 48 h, both the adherent and floating cells were harvested, centrifuged and then double stained with Alexa Fluor488 annexin V and PI (Life Technologies, Invitrogen, Grand Island, NY, USA). The stained cells were analyzed by flow cytometry.
Flow cytometric analysis of cell cycle distribution
After treatment of HepG2 cells cultured in 60 mm3 petri dishes with different concentrations of [6]-gingerol for 24 and 48 h, both the adherent and floating cells were harvested, fixed and incubated with ice-cold 70% ethanol at 4°C for 24 h. The fixed cells were centrifuged at 200xg for 5 min. Cell pellets were incubated with a solution containing 100 µg/mL of RNase A in PBS incubated at 37°C for 30 min. Then, cells were stained with 20 µg/mL of PI, then incubated for 2.5 h in the dark at room temperature. The stained cells were subsequently analyzed by flow cytometry.
Western blotting and immunocytochemistry
After treatment of HepG2 cells cultured in 60 mm3 petri dishes with different concentrations of [6]-gingerol for 24 and 48 h, both the adherent and floating cells were harvested followed by lysed and quantified protein concentration by the BCA (bicinchoninic acid) protein assay reagent (Thermo Scientific, Rockford, IL, USA). FASN (Abcam, Biomed Diagnostics Co., Ltd, Thailand), ACC (Merck Millipore, Darmstadt, Germany) and ACLY (Cell Signaling Technology Inc., Boston, MA, USA) expressions were determined by SDS-PAGE system and immunoblotting assay. Extracted proteins were subjected to 8% SDS-polyacrylamide gel. Following electrophoresis, protein blots were transferred to polyvinyldienedifluoride membranes, (PVDF) membrane. To block non-specific binding, the membrane was subsequently incubated with “Rapidblock” solution (AMRESCO, Solon, OH, USA) for 30 minutes at room temperature, followed by an overnight incubation at 4°C with primary antibodies against FASN, ACC and ACLY (Dilution 1:1000). Membranes were then incubated with horseradish peroxidase-conjugated goat anti-Rabbit IgG secondary antibody (Life Technologies) diluted 1:5000 in PBS solution containing 5% non-fat dry milk for 1 h at room temperature. The blots were developed using the Novex ECL Chemiluminescent Substrate Reagent Kit (Life Technologies). The fluorescence intensity of each band was measured using a ImageQuant CCD camera.
For FASN protein immunofluorescence analysis, both the adherent and floating cells were incubated with anti-FASN antibody at 4°C overnight followed by Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L) Antibody (Molecular Probe) at room temperature for 1 h in the dark. Cells were imaged by a confocal laser scanning microscope using 405 and 473 nm laser lines and emission bandpass filters of 461 nm (for DAPI) and 520 nm (for Alexa Fluor488).
Mitochondrial membrane potential (DYm) analysis by flow cytometry and confocal microscopy
After treatment of HepG2 cells cultured in 60 mm3 petri dishes with different concentrations of [6]-gingerol for 24 and 48 h, both the adherent and floating cells were harvested and incubated with “JC-1 Dye Mitochondrial Membrane Potential Probe” (5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanineiodide) (Life Technologies) at 37°C and 5% CO2 for 30 minutes. The JC-1 dye exhibits an aggregated form which accumulates in the mitochondria in a response to ΔΨm. A healthy ΔΨm exhibits a high aggregated JC-1 form accumulated in mitochondria and emits a strong red fluorescence intensity (~590 nm) while a low monomeric JC-1 form existing in cytoplasm emits a low green fluorescence intensity (~529 nm). The red/green fluorescence intensity ratio indicates the potential of the mitochondrial membrane. The disruption of ΔΨm shifts a fluorescence emission from red to green. The level of ΔΨm in HepG2 cells was measured by flow cytometry and a confocal laser scanning microscope.
Measurement of free fatty acid level
This depends on estimating free fatty acid produced by de novo fatty acid synthesis using a free fatty acid quantification kit (Abcam AB65341) for which the manufacturer’s protocol was followed. Briefly, after HepG2 cells were incubated with different concentrations of [6]-gingerol, both the adherent and floating cells were harvested, homogenized with chloroform-Triton-X 100 solution (1% Triton-X 100 in pure chloroform) and then centrifuged at the top speed to collect the organic phase. The supernatant was vacuum dried to remove the chloroform and then residues dissolved in fatty acid assay buffer. The acyl-CoA synthase reagent and the palmitic acid (as standard) were added to the samples. This converted the long chain fatty acids to CoA derivatives which were then oxidized. To this reaction mix in assay buffer, the kit “enzyme mix”, the “enhancer”, and the fluorescence probe were added. FFAs were measured fluorometrically at Ex/Em 535/590 nm using a microplate reader. Results were expressed in percentage of intracellular long chain fatty acid.
Estimation of intracellular reactive oxygen species (ROS)
Assay of intracellular ROS relied on the membrane-permeable fluorescent dye 5-(and-6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Molecular Probes). [6]-gingerol treated cells were collected, incubated in PBS containing CM-H2DCFDA (10 μM) for 30 min at 37°C and then ROS was estimated by the fluorescence emission (excitation 485 nm, emission 525 nm) by flow cytometry and confocal laser microscopy.
Carnitine palmitoyl transferase-1 (CPT-1) activity assay
CPT-1 activity was determined by a spectrophotometric method as previously described [35]. Briefly, [6]-gingerol treated cells were harvested and lyzed in Tris-Hcl buffer (pH 7.4) containing 1 mM EDTA and 0.25 M sucrose. The lysate was subsequently centrifuged at (500 g, 4°C, 10 min). To collect mitochondria, the supernatant was re-centrifuged at 14,000 g (4°C, 15 min). The mitochondria were then resuspended in lysis buffer. Equal mitochondrial protein was mixed with Tris buffer (100 mM, pH 8.0, 0.1% Triton X-100, 1 mM EDTA) and 0.5 mM DTNB and 0.01 mM palmitoyl CoA were added. L-Carnitine at 1.25 mM was then added and measured O.D. at 412 nm.
Statistical analysis
The data were expressed as mean ± SD of at least three independent experiments. One way analysis of variance (ANOVA) with Turkey’s post-hoc analysis was used to determine the statistically significant differences for all treated and vehicle control samples using Graph Prism Software, version 5. P<0.05 was considered significant.
Results
[6]-gingerol reduced HepG2 cell viability
HepG2 cells cultured with vehicle alone remained viable throughout the incubation period (24 and 48 h), but in the presence of 0.1 to 0.5 mM [6]-gingerol dose- and time-dependently reduced cell viability (Figure 1) (IC50 0.56 mM and 0.42 mM after 24 h and 48 h exposure, respectively).
Figure 1.
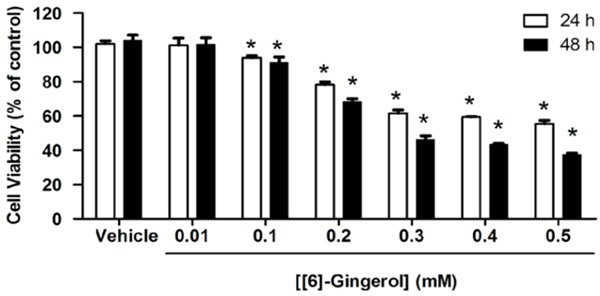
[6]-gingerol reduced HepG2 cell viability.HepG2 cells were treated with different concentrations of [6]-Gingerol for 24 h and 48 h and then MTT assay was performed. Data are expressed as percentage of cell viability compared with 100% of the control. Cells treated with a medium or 0.1% DMSO vehicle without [6]-gingerol were considered as the control. Three independent experiments of triplicate samples were performed for statistical analysis and expressed as mean ± SD. *denotes statistically significant difference from the control at P<0.05.
[6]-gingerol promoted apoptosis and cell cycle arrest in HepG2 cells
Investigations were performed to determine the mechanisms of [6]-gingerol to suppress cell viability which occurred by apoptosis induction. Apoptosis was determined by Alexa Fluor488 Annexin V binding to externalized plasma membrane phosphatidylserine (PS), an early apoptotic event, while both Annexin V and PI staining which identified cells late apoptosis. HepG2 cells treated with 0.1 and 0.5 mM [6]-gingerol for 24 h contained apoptotic cells (Figure 2AI, 2AII). Increasing the exposure period of [6]-gingerol to 48 h, apoptosis was more pronounced (increased to 33 and 54%) (Figure 2BI, 2BII). In contrast, very few vehicle treated cells were stained (5%) cells. It was noticed that increased concentration and incubation period of [6]-gingerol showed an increase of the apoptosis and a reduction of the cell viability population.
Figure 2.
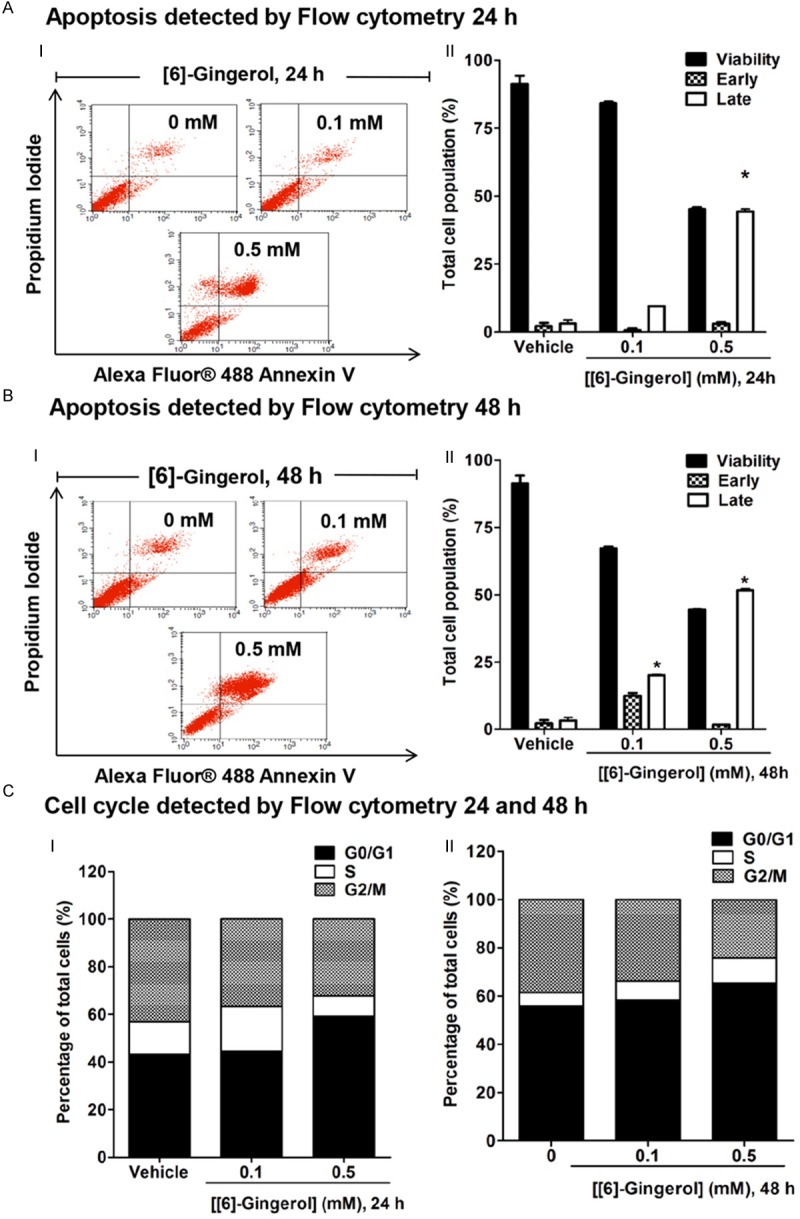
[6]-gingerol promoted apoptosis and cell cycle arrest in HepG2 cells. Cells were treated with 0.1 and 0.5 mM [6]-gingerol for 24 h and 48 h. Cell cycle and apoptosis analysis were determined by flow cytometry. The control was defined as cells treated with a medium or 0.1% DMSO vehicle without [6]-gingerol. AI. Cell death and apoptosis following [6]-gingerol treatment for 24 h identified as the events that were single stained for Alexa Fluor488-Annexin V (lower right quadrant or early apoptisis) or double stained for both Alexa Fluor® 488-Annexin V and PI (upper right quadrant or late apoptosis). II. The distribution of viable, early, and late apoptotic cells were evaluated relative to the whole cell populations (set as 100%). BI. Cell death and apoptosis following [6]-gingerol treatment for 48 h. II. The distribution of viable, early, and late apoptotic cells were evaluated relative to the whole cell populations (set as 100%). C. PI staining was used to determine the percentages of cells in each phase of the cell cycle relative to the whole cell populations (set as 100%) and expressed in histogram profile of cell cycle distribution in Go/G1, S, and G2/M phases after 24 h I and 48 h II of [6]-gingerol treatment. Three independent experiments were performed for statistical analysis and expressed as mean ± SD. *denotes statistically significant difference from the control at P<0.05.
Flow cytometry of PI stained permeabilized cells was used to determine cell cycle progression. [6]-gingerol dose and time dependently increased the proportion of cells in the G0/G1 (quiescent) phase, suggesting that cell division was slowed (Figure 2C) by 55% and 74%, respectively as compared with the control group which showed approximately 52% of cells in the G0/G1 phase. Notably, [6]-gingerol caused a decrease in the percentage of cells in S and G2/M phases observed at 24 h of treatment. Increase in cell cycle arrest in the G0/G1 phase and decrease in cells in S and G2/M phases were monitored as incubation time increased to 48 h of [6]-gingerol treatment.
[6]-gingerol reduced FASN protein expression and fatty acid synthesis
We then explored whether the apoptosis could result from [6]-gingerol interfering with DNF by down-regulating FASN protein. HepG2 cells incubated in [6]-gingerol for 24 (Figure 3AI) and 48 h (Figure 3AII) showed that the expression level of FASN was slightly decreased after treatment with [6]-gingerol at 0.1, 0.25 for 24 h. The expression was significantly decreased after 24 and 48 h of treatment at 0.5 mM [6]-gingerol as compared with the vehicle only. In contrast, other lipogenic enzymes (ACC and ACLY) remained unchanged following 24 and 48 h of [6]-gingerol treatment. Immunofluorescence in Figure 3BI showed a similar FASN time dependent decrease with 0.5 mM [6]-gingerol. The expression level of protein in Figure 3BI was calculated and expressed as the relative expression level of FASN compared with β-actin in Figure 3BII. [6]-Gingerol at 0.5 mM for 24 and 48 h incubation showed reduction of FASN protein expression by 60% and 80%, respectively.
Figure 3.
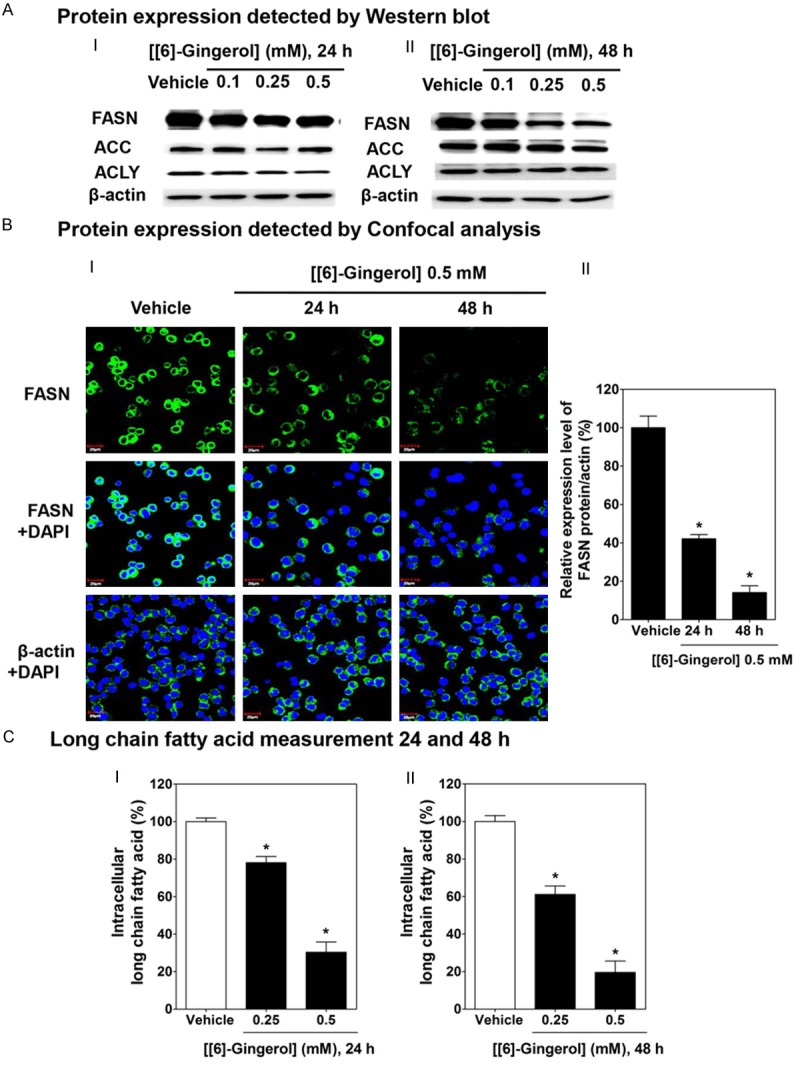
[6]-gingerol decreased FASN protein expression and de-novo fatty synthesis in HepG2 cells. Cells were treated with [6]-gingerol for 24 h and 48 h or 0.1% DMSO vehicle. A. Western blots of [6]-gingerol treatment for 24 I or 48 h II for FASN, ACC, ACLY antibodies and β-actin. BI. Confocal images of HepG2 cell cultures treated with vehicle or with 0.5 mM [6]-gingerol for 24 or 48 h. Cells were fixed and labelled with either anti-FASN or anti-β-actin antibodies and Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L) antibody (green) and DAPI (blue). BII Quantitation of FASN fluorescence relative to β-actin. C. Long chain fatty acid contents of HepG2 cells after treatment with [6]-gingerol (0.25 or 0.5 mM) for 24 I or 48 h II expressed relative to that in cells treated with vehicle only. Each data point is the mean ± SD of 3 independent experiments. *P<0.05 comparing vehicle only.
Free fatty acid level was also depressed with 0.5 mM [6]-gingerol by 69 % at 24 h (Figure 3CI) and 81% after 48 h (Figure 3CII) as compared with vehicle alone. This data accords with the loss of FASN protein expression and shows reduced de novo fatty acid synthesis.
[6]-gingerol induces mitochondrial-dependent apoptosis and ROS production
Dissipation of ΔΨm is a prelude to the mitochondrial dependent apoptosis [15]. This was tested using JC-1 dye whose distribution across the mitochondrial membrane depends on ΔΨm, and then was detected the depolarization of ΔΨm by immunofluorescence staining with laser confocal microscopy and also quantified by the emission wavelength shift using flow cytometry [36]. CCCP was used as a positive control to increase green fluorescence intensity, indicating a depolarized ΔΨm [37]. The immunofluorescence results in Figure 4A showed that cells with a normal ΔΨm containing healthy mitochondria exhibited many JC-1 aggregated in their mitochondria and gave high red fluorescence. After 24 and 48 h treatment with 0.5 mM of [6]-gingerol, JC-1 dye formed a monomer JC-1, exhibiting an increase of green fluorescence in the cytoplasm and a decrease of red JC-1 fluorescence in the mitochondria, indicating a disruption of ΔΨm. Flow cytometry in Figure 4B showed that the healthy mitochondria had red/green fluorescence ratio of ~90/10 or 10% of disruption. HepG2 cells exposed to [6]-gingerol at 0.1, 0.25, and 0.5 mM for 24 h disrupted ΔΨm by 16%, 18%, and 52%, respectively. Increasing the treatment time period to 48 h showed an augmentation of the disruption of ΔΨm. Thus, apoptosis induction by [6]-gingerol in HepG2 cells was shown to be dependent on disruption of ΔΨm.
Figure 4.
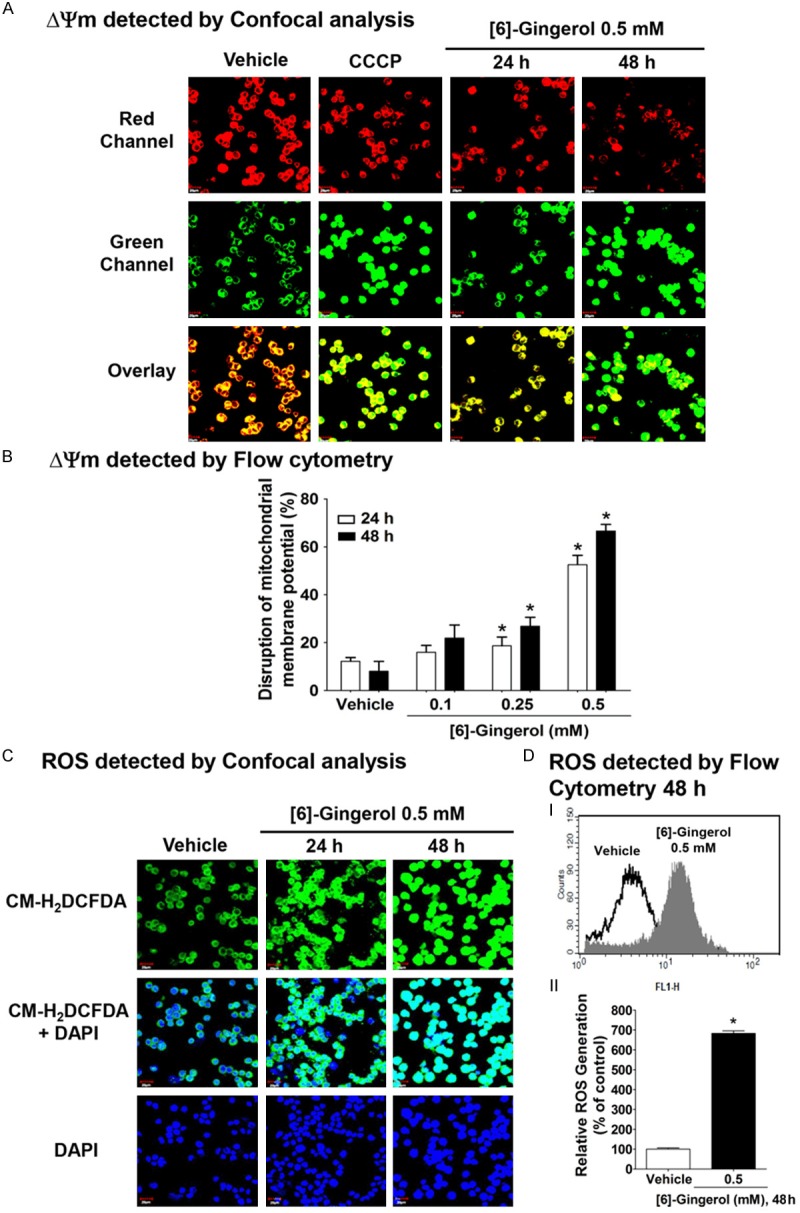
[6]-Gingerol disrupted the mitochondrial membrane potential (ΔΨm) and promoted ROS production in HepG2 cells. A. Confocal imaging of ΔΨm of HepG2 cells treated with vehicle (0.1% DMSO), 0.5 mM [6]-gingerol for 24 h and 48 h and then labeled with JC-1 fluorochrome and examined by confocal microscopy. Cells incubated for 10 min with carbonyl cyanide m-chlorophenylhydrazone (CCCP) is a proton ionophore that dissipates the mitochondrial membrane potential (predominantly green) was used as a positive control. B. Flow cytometery of ΔΨm in HepG2 cells treated with vehicle, 0.1, 0.25, or 0.5 mM [6]-gingerol for 24 or 48 h and then labeled with JC-1. C. Confocal imaging of ROS production after treatment with vehicle or 0.5 mM [6]-gingerol for 24 and 48 h. and then visualised using the ROS sensitive dye CM-H2DCFDA (green) and counterstained with DAPI (blue). DI. Flow cytometery showing ROS generation after treatment with vehicle or 0.5 mM [6]-gingerol for 48 h. II. Quantitation of ROS generation from flow cytometry of CM-H2DCFDA stained cells relative to vehicle alone (100%). Each data point is the mean ± SD of 3 independent experiments. *P <0.05 comparing vehicle only.
We further verified our understanding about the mechanism involved in mitochondrial-dependent [6]-gingerol-induced apoptosis in HepG2 cells; we investigated ROS production by staining cells with CM-H2DCFDA fluorescence and analyzed them by confocal laser microscopy and flow cytometry. Confocal laser microscopy analysis (Figure 4C) showed an increase of intracellular ROS generation as observed with an increase of green fluorescence of CM-H2DCFDA after exposure to 0.5 mM [6]-gingerol for 24 and 48 h in HepG2 cells. Figure 4DI showed the histogram of an overlay appearance of an increase of CM-H2DCFDA fluorescence intensity after HepG2 cells were treated with 0.5 mM of [6]-gingerol for 48 h, indicating an increase of intracellular ROS generation. Likewise, Figure 4DII additionally confirmed an increase of intracellular ROS after exposure to 0.5 mM [6]-gingerol for 48 h up to 684% compared with 100% of the control group.
The present study showed consistent results with that obtained from C75 (4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid) (Sigma Chemical Co., St. Louis, MO, USA), which is identified as the FASN inhibitor [6,38,39]. C75 induced a reduction of red/green fluorescence intensity ratio by 40%, indicating the damage of ΔΨm compared with the control group, without any alteration in the level of FASN protein expression (Figure 5). C75 caused approximately 70% decrease of fatty acid synthesis.
Figure 5.
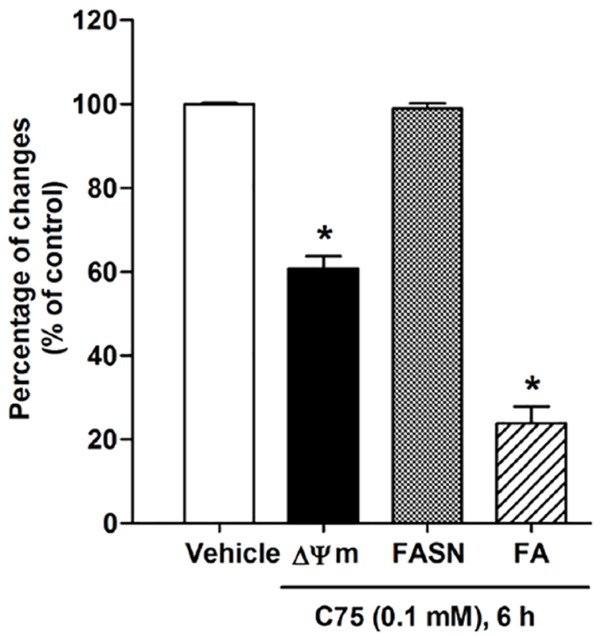
C75 induced apoptosis and inhibited fatty acid synthesis in HepG2 cells. The effect was not mediated by FASN downregulation. Cells were treated with 0.1 mM C75 for 6 h. ΔΨm was detected by flow cytometry and quantified by calculating the relative level of the red to green fluorescence emission of JC-1 fluorochrome. The disruption of ΔΨm decreased the red/green fluorescence intensity ratio of JC-1 fluorochrome. FASN protein expression was quantified as a ratio of the individual band protein intensity relative to the β-actin band intensity in the same blot (e.g. FASN/β-actin). Intracellular long chain fatty acid was determined by using the free fatty acid quantification kits. Three independent experiments were performed for statistical analysis and expressed as mean ± SD. *denotes statistically significant difference from the control at P<0.05.
[6]-gingerol promoting mitochondrial-dependent apoptosis is mediated by ROS production in HepG2 cells
To obtain a better understanding of a possible mediator participating in [6]-gingerol-induced apoptosis in HepG2 cells, the present study investigated if increased ROS generation resulted in the onset of mitochondrial disruption and FASN inhibition. Cells treated with 0.5 mM [6]-gingerol for 3 h showed an increase of the percentage of intracellular ROS generation from 100% of the control to approximately 128% without any alteration of ΔΨm (Figure 6A, 6B). A 0.5 mM [6]-gingerol treatment for 3 h did not alter intracellular fatty acid synthesis (Figure 6C), whereas an increase of ΔΨm disruption and a decrease of fatty acid synthesis were initially detected at 6 h of [6]-gingerol treatment (Figure 6B, 6C). A disruption of ΔΨm was measured by an increase of JC-1 green fluorescence intensity which was initially changed from 10% of the control up to 37% after 6 h of [6]-gingerol treatment (Figure 6B). Likewise, a long chain fatty acid level began to be decreased by 30% (Figure 6C) after 6 h of [6]-gingerol treatment. [6]-gingerol did not alter the expression of FASN protein within 6 h of incubation (Figure 6D). The percentage of ROS generation, ΔΨm disruption, and intracellular fatty acid depletion were observed in HepG2 cells treated with 0.1 mM C75 for 6 h (Figure 6A-C).
Figure 6.
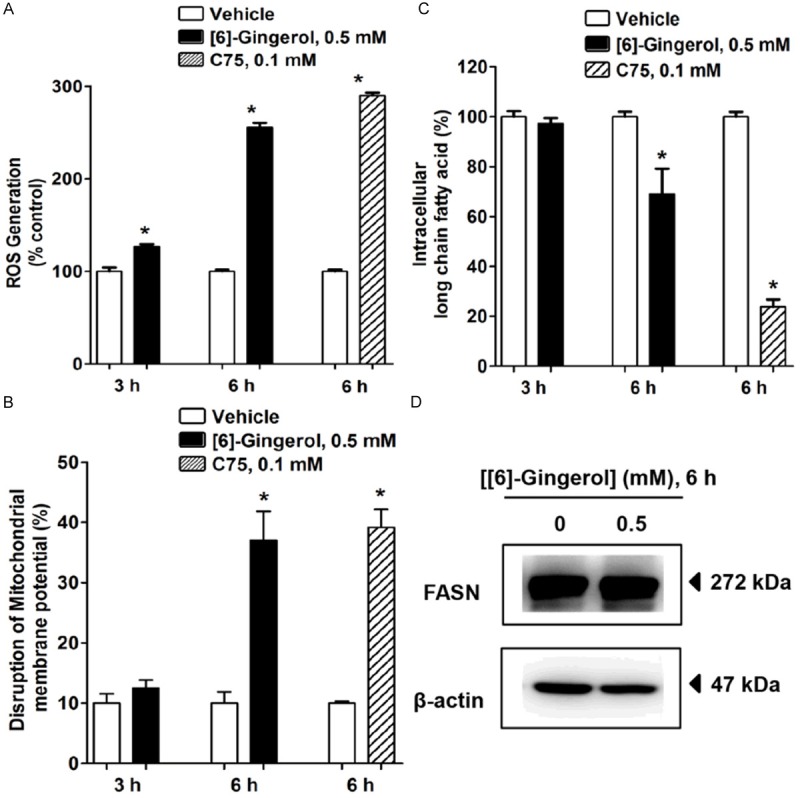
[6]-gingerol inhibited FASN activity mediated by an increase of ROS generation in HepG2 cells. Cells were treated with vehicle, 0.5 mM [6]-gingerol or C75 for 3 h and 6 h. Data was normalised to vehicle only (100%). A. ROS production was stained with CM-H2DCFDA dye and detected by flow cytometry. ROS generation in the control group was quantified and expressed as 100%. B. The disruption of ΔΨm was measured by labeling the cells with JC-1 dye and detected by flow cytometry. Histograms show the percentages of disruption of ΔΨm relative to the whole cell populations. C. Intracellular long chain fatty acid levels were determined in cell lysates by a free fatty acid quantification kit. D. FASN protein expression was detected by immunoblotting with anti-FASN antibody after treatment with 0.5 mM [6]-gingerol for 6 h. The band results are representative of those obtained from at least three independent experiments. β-actin was used as an internal standard to confirm the integrity and equal protein loading and transferring. Each data point is the mean ± SD of 3 independent experiments. *P<0.05 comparing vehicle only.
Taken together, these findings suggest that [6]-gingerol enhances ROS generation, which in turn triggers the inhibition of the de novo fatty acid synthesis pathway, which is accompanied by an induction of mitochondrial-dependent apoptosis in HepG2 cells.
An accumulation of malonyl-CoA leading to a decrease of CPT-1 activity causes apoptosis in HepG2 cells following de novo fatty acid inhibition by [6]-gingerol treatment
To evaluate whether an accumulation of malonyl-CoA, as a result of FASN inhibition, is one of the major causes of apoptosis, in addition to a depletion of end product fatty acids [40,41] after [6]-gingerol treatment. Treated cells with a competitive inhibitor of ACC, 5-(tetradecyloxy)-2-furoic acid (TOFA) at 10 µg/ml for 6 h caused an inhibitory effect on intracellular fatty acid synthesis without an induction of damage of ΔΨm, as shown in Figure 7A and 7B. TOFA treatment decreased the intracellular fatty acid level more than 60% as compared with the control. The degree of the decrease of intracellular fatty acid after cells treated with [6]-gingerol was found to be augmented by TOFA. We suggest that a reduction of fatty acid per se seemed not to be the major cause of apoptosis following inhibition of the fatty acid synthesis pathway. To evaluate the possibility that an accumulation of malonyl-CoA resulted in disruption of ΔΨm after inhibition of fatty acid synthesis, this study exposed cells to TOFA for 1 h prior to 0.5 mM [6]-gingerol and 0.1 mM C75 treatment for 6 h. TOFA reduced the cytotoxic effect of [6]-gingerol on damaging of ΔΨm from approximately 40% to 10% (Figure 7B). This result demonstrates that inhibiting an accumulation of malonyl-CoA by TOFA inhibits fatty acid synthesis and rescues the damage of ΔΨm from FASN inhibition by [6]-gingerol. An accumulation of malonyl-CoA following FASN inhibition appears to play a prominent role instead of a depletion of end product fatty acids on induction of damage of ΔΨm and apoptosis in HepG2 cells. We observed a similar cytotoxic effect of an accumulation of malonyl-CoA following treatment with 0.1 mM C75 for 6 h. This pharmacological FASN inhibitor reduced the intracellular fatty acid level to approximately 20% from the control (Figure 7A) and caused damage of ΔΨm to approximately 40% compared with 10% of the control (Figure 7B). Exposure of HepG2 cells with TOFA before C75 treatment completely rescued cells from C75-induced damage of ΔΨm (Figure 7B).
Figure 7.
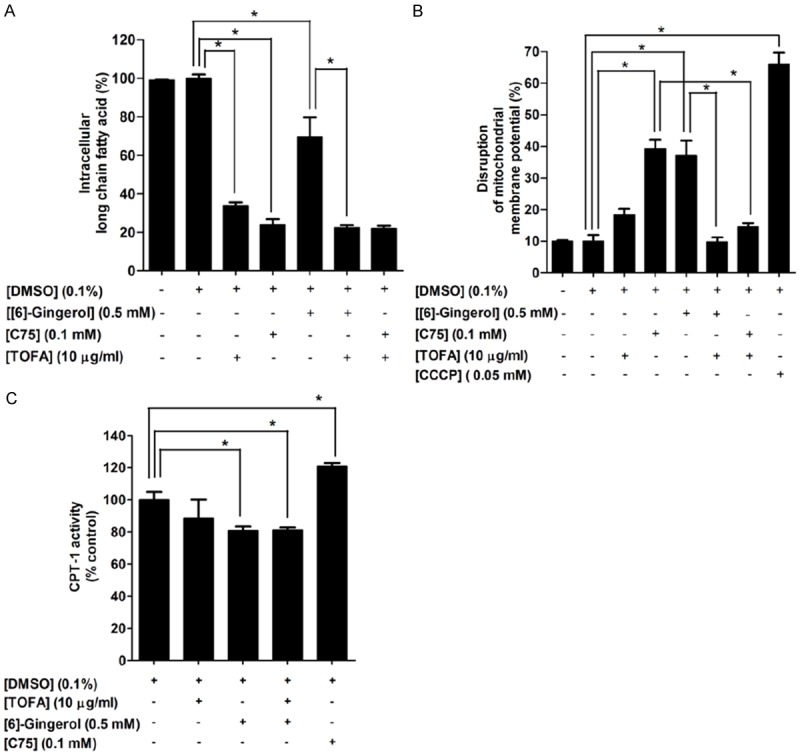
An accumulation of malonyl-CoA and inhibition of CPT-1 activity participated in [6]-gingerol-induced apoptosis and inhibition of FASN in HepG2 cells. Cells were treated with 0.5 mM [6]-gingerol or 0.1 mM C75 or 10 µg/ml TOFA for 6 h. 10 µg/ml TOFA was pre-incubated for 1 h prior to [6]-gingerol or C75 treatment for 6 h. The control was defined as cells treated with a medium or 0.1%DMSO vehicle alone. A. Percentages of intracellular fatty acid levels were determined using the free fatty acid quantification kit. B. ΔΨm was measured by JC-1 dye fluorochrome and detected by flow cytometry. The disruption of ΔΨm was quantified by calculating an increase of a monomer JC-1 green fluorescence in the cytoplasm and a decrease of an aggregated JC-1 red fluorescence in the mitochondria. CCCP was used as a positive control to induce depolarization of ΔΨm. C. Cells were treated with 0.5 mM [6]-gingerol, 0.1 mM C75, and 10 µg/ml TOFA for 24 h. CPT-1 activity was assayed and presented as percentage of CPT-1 (nmol/min/mg protein). Three independent experiments were performed for statistical analysis and expressed as mean ± SD. *denotes statistically significant difference from the control at P<0.05.
To investigate if an accumulation of malonyl-CoA following [6]-gingerol-induced fatty acid deprivation caused apoptosis via modulation of CPT-1 activity, we determined the CPT-1 activity after treatment of HepG2 cells with 0.5 mM of [6]-gingerol for 24 h. As shown in Figure 7C, [6]-gingerol decreased CPT-1 activity by approximately 20% as compared with the control. In contrast, 24 h of 0.1 mM C75 increased CPT-1 activity by 20%, which is consistent with the previous report demonstrating that C75 stimulates CPT and fatty acid β-oxidation activity in many cancer cells, such as lung cancer [6,42]. TOFA treatment showed no alteration of CPT-1 activity. Thus, our result indicates that increased malonyl-CoA as a result of inhibition of fatty acid synthesis inhibits CPT-1 activity.
Discussion
[6]-gingerol has been known to possess a potential anti-cancer agent via induction of apoptosis in various types of cancer cells [43-45]. However, the contribution of de novo fatty acid synthesis to this apoptosis is unclear. The present study has demonstrated that [6]-gingerol reduces de novo fatty acid synthesis by downregulating FASN resulting in mitochondrial dysfunction and culminating in HepG2 cell death. An early consequence of this is the raised ROS generation whose effects were manifest as [6]-gingerol-induced cytotoxicity. This accords with the notion that many other polyphenols also induce tumor cell apoptosis through excessive ROS generation [46-51]. This study proposed that [6]-gingerol provides a promising anticancer agent, whose apoptotic action results from CPT-1 inhibition by accumulation of pathologically high malonyl-CoA concentrations. Down regulation of FASN also promotes ceramide formation [29] mediation lipotoxicity.
The mechanism by which [6]-gingerol enhances ROS production has not yet been studied. The associations with reduced ΔΨm, fatty acid synthesis, and malonyl-CoA accumulation coupled to CPT-1 inhibition support a mitochondrial mediated production of ROS. Mitochondrial dependent apoptotic induction was found following ROS generation, which implied that mitochondria is defined as target of ROS-triggered apoptosis in HepG2 cells. ROS is primarily created as superoxide and then dismutated by mitochondrial SOD (Mn) to the less reactive H2O2, However, there is the increasing realization that H2O2 is a ubiquitous signaling molecule regulating many redox and tissue functions including mitochondrial function [52]. Thus, the high and sustained H2O2 also disrupts mitochondrial energy and redox homeostasis. However, fluorometric detection of ROS in the present study did not distinguish various reactive species thus limiting the interpretation about these processes. Mitochondria is also defined as a potential vulnerable target of ROS-induced oxidative stress to trigger apoptosis via up-regulation of Bcl-2/Bax ratio-mediated pore forming in the mitochondrial membrane, which consequently promotes mitochondrial-dependent apoptosis in carcinoma cells [44]. Our present study suggests that ROS level significantly reaches severe oxidative damage to many cellular biomolecule functions that work in concerted action to participate in growth arrest and cell death [53,54], leading to apoptotic induction following [6]-gingerol exposure. The disturbance of redox homeostasis by an imbalance between an elevation of ROS formation and a decrease of ROS scavenging ability makes cancer cells more vulnerable to damage by further increase of ROS generation than in normal cells [55]. Additionally, ROS-induced oxidative stress results in the releasing of cathepsin D from the reduced lysosomal membrane stability into the cytosol to trigger apoptosis by increasing permeabilization and the releasing of cytochrome c after treatment with [6]-gingerol [47]. The generation of ROS can also trigger apoptosis via promoting cells exposed to pro-apoptotic activity of mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinases (ERK) and c-Jun N-terminal kinases (JNK) pathway [56,57].
Besides the dissipation of ΔΨm, the present study also showed that [6]-gingerol treatment caused excessive ROS production that resulted in a decrease of protein expression, negatively regulating the fatty acid synthesis pathway. ROS caused a decrease of FASN protein expression is still not fully understood and needed to be further evaluated. An increase of ROS production is proposed to modulate intracellular signaling pathways that regulate transcriptional and translational processes of FASN expression, since FASN is highly regulated by the interplay of intracellular multiple signaling pathways [58-62].
FASN expression is remarkably overexpressed in most cancer cells to support a high requirement of fatty acid product for membrane phospholipid and signaling molecule biosynthesis in regulating cancer cell metabolism and proliferation [15,17]. Its expression is generally undetectable in normal tissues with the notable exception of the lipogenic liver [63,64]. Inhibition of FASN expression and activity results in apoptosis in several cancer cells without generating a cytotoxic effect on normal cells [15,64,65], supporting the notion that FASN is therefore an effective target for anticancer therapeutic drugs. Our findings showed that [6]-gingerol had no effect on the ACC and ACLY protein expression levels, suggesting that ACC and ACLY might not be the promising anti-cancer therapeutic targets of [6]-gingerol in HepG2 cells. The present study showed a correlation between mitochondrial-dependent apoptosis induction and depletion of cellular protein levels of FASN and fatty acid synthesis in HepG2 cells caused by [6]-gingerol treatment. There might be multiple possible mechanisms to explain the apoptosis induction by a decrease of fatty acid synthesis. It has been reported that during FASN inhibition, a depletion of saturated and monounsaturated fatty acid levels promotes cancer cells to oxidative stress-induced lipid peroxidation, leading to increased apoptotic cell death [29]. In addition, depletion of fatty acid level by itself abolishes cancer cells to synthesize new cell membranes, membrane-anchored lipid rafts that facilitate growth factor signal transmission, and downstream signaling molecules for undergoing proliferative pathways, such as the PI3K signaling pathway [29,40]. Inhibition of FASN by C75 inhibits the PI3K/AKT/mTOR pathway, which subsequently activates the eukaryotic translation initiation complex 4F (eIF4F) and p70S6K-phosphorylated ribosomal protein S6. These events finally inhibit transcription of oncogenic genes that regulate cell growth and apoptosis in ovarian carcinoma cells [39].
Additionally, the cytotoxic ability of fatty acid depletion may be secondary to an accumulation of toxic intermediates of this pathway [29,40]. We demonstrate that an inhibition of FASN and fatty acid synthesis results in decreasing malonyl-CoA utilization, indicating an upstream accumulation of substrate for FASN enzyme activity. The present study found that TOFA counteracted the apoptotic induction effect of C75 and [6]-gingerol treatment in HepG2 cells. However, an inhibition of fatty acid synthesis by TOFA failed to induce apoptosis. Thus, a depletion of fatty acid product per se following FASN inhibition may not fulfill a major cause of apoptosis. An excessive malonyl-CoA accumulation as a result of FASN inhibition is an additional mechanism to potentially play an important role as mediator in apoptosis in [6]-gingerol-treated HepG2 cells. An overproduction of malonyl-CoA is characterized to function as a pro-apoptotic factor to promote cancer cell death following FASN inhibition [65,66].
Malonyl-CoA consequently participates in apoptosis induction by competitively inhibiting CPT-1 activity, which serves as the outer mitochondrial membrane enzyme for stimulating mitochondria fatty acid β-oxidation [40,64]. We found that inhibition of FASN by [6]-gingerol inhibited CPT-1 activity following increase of malonyl-CoA level. An induction of apoptosis by an inhibition of CPT-1 activity following an excess malonyl-CoA level is at least in part mediated by an accumulation of a sphingolipid ceramide level [40]. This cytotoxicity induced by overproduction of ceramide following FASN inhibition has been reported to be the result of up-regulated expression of pro-apoptotic factors, including BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and death-associated protein kinase 2 (DAPK2) for the initiation of an apoptotic pathway in cancer cells [13,29]. Our result showed that an inhibition of malonyl-CoA synthesis by TOFA suppressed fatty acid synthesis but did not disrupt ΔΨm and an inhibition on CPT-1 activity in HepG2 cells. Thus, taken together, we suggest an inhibitory role of malonyl-CoA accumulation on CPT-1 activity-induced apoptosis by [6]-gingerol treatment. Our results demonstrate that inhibition of fatty acid synthesis together with accumulation of malonyl-CoA by [6]-gingerol has a promising apoptotic effect on inhibition of CPT-1 activity in HepG2 cells. In contrast, C75 caused stimulation of CPT-1 activity with inhibition of fatty acid synthesis and increased malonyl-CoA level. This direct stimulation on CPT-1 is related to consequent side effects on induction of severe weight loss and decrease of food intake of C75 as a result of acceleration of β-oxidation and inhibition of the hypothalamic neuropeptide-Y production, respectively [6,42]. Experimental studies suggest that the direct CPT-1 stimulation by C75 abolishes cytotoxicity of accumulated malonyl-CoA on inhibition of CPT-1 activity [67]. Apoptosis is mediated by inhibition of FASN-mediated downregulation of the PI3K/AKT/mTOR cascade [39] and inhibition of the human epidermal growth factor receptor 2 (HER2)-meditated FASN synthesis via the PI3K pathway [68].
In conclusion, the present study suggests that a reduction of de novo synthesis of fatty acid following [6]-gingerol treatment promotes apoptosis of HepG2 cancer cells. These studies show an excess level of malonyl-CoA, an important proapoptotic stimulant to activate apoptosis, instead of a depletion of LCFAs level after exposure to the inhibitory ability of [6]-gingerol on FASN activity. An inhibition on CPT-1 activity triggered by an accumulation of toxic malonyl-CoA level reinforces [6]-gingerol as the new potential anti-cancer therapy to overcome the drawback effects on CPT-1 and weight loss activation. Such side effects hinder the further clinical application of some compounds including C75. [6]-Gingerol will be a promising anti-cancer molecule for further studies either alone or in combination with other anti-cancer drugs.
Acknowledgements
Authors are thankful to Assoc. Prof. Mary Sarawit, PhD., Distinguished Specialist for Language and International Affairs, Office of the President, Naresuan University for helpful reviewing and editing the manuscript. This work has been accomplished with supporting funds from Naresuan University, Thailand.
Disclosure of conflict of interest
None.
Abbreviations
- ACC
acetyl-CoA carboxylase
- ACLY
ATP-citrate lyase
- CPT-1
carnitine palmitoyltransferase-1
- EGCG
(-)-epigallocatechin-3-gallate
- FASN
fatty acid synthase
- C75
4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid
- DYm
mitochondrial membrane potential
- MUFAs
monounsaturated fatty acids
- ROS
reactive oxygen species
- TOFA
5-(tetradecyloxy)-2-furoic acid
- LCFAs
saturated long chain fatty acids
- SCD-1
stearoyl-CoA desaturase
References
- 1.Kaur M, Agarwal C, Agarwal R. Anticancer and cancer chemopreventive potential of grape seed extract and other grape-based products. J Nutr. 2009;139:1806S–1812S. doi: 10.3945/jn.109.106864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr. 2001;21:381–406. doi: 10.1146/annurev.nutr.21.1.381. [DOI] [PubMed] [Google Scholar]
- 3.Diaz-Laviada I, Rodriguez-Henche N. The potential antitumor effects of capsaicin. Prog Drug Res. 2014;68:181–208. doi: 10.1007/978-3-0348-0828-6_8. [DOI] [PubMed] [Google Scholar]
- 4.Rahman AA, Makpol S, Jamal R, Harun R, Mokhtar N, Ngah WZ. Tocotrienol-rich fraction, [6] -gingerol and epigallocatechin gallate inhibit proliferation and induce apoptosis of glioma cancer cells. Molecules. 2014;19:14528–14541. doi: 10.3390/molecules190914528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daker M, Bhuvanendran S, Ahmad M, Takada K, Khoo AS. Deregulation of lipid metabolism pathway genes in nasopharyngeal carcinoma cells. Mol Med Rep. 2013;7:731–741. doi: 10.3892/mmr.2012.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Relat J, Blancafort A, Oliveras G, Cufi S, Haro D, Marrero PF, Puig T. Different fatty acid metabolism effects of (-)-epigallocatechin-3-gallate and C75 in adenocarcinoma lung cancer. BMC Cancer. 2012;12:280. doi: 10.1186/1471-2407-12-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YK, Hwang JT, Lee MS, Kim YM, Park OJ. Kidney bean husk extracts exert antitumor effect by inducing apoptosis involving AMP-activated protein kinase signaling pathway. Ann N Y Acad Sci. 2009;1171:484–488. doi: 10.1111/j.1749-6632.2009.04697.x. [DOI] [PubMed] [Google Scholar]
- 8.Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism. 2014;63:895–902. doi: 10.1016/j.metabol.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Wang Y, Xie Z, Zhou Y, Zhang Y, Wan X. The anti-obesity effects of green tea in human intervention and basic molecular studies. Eur J Clin Nutr. 2014;68:1075–87. doi: 10.1038/ejcn.2014.143. [DOI] [PubMed] [Google Scholar]
- 10.Kang OH, Kim SB, Seo YS, Joung DK, Mun SH, Choi JG, Lee YM, Kang DG, Lee HS, Kwon DY. Curcumin decreases oleic acid-induced lipid accumulation via AMPK phosphorylation in hepatocarcinoma cells. Eur Rev Med Pharmacol Sci. 2013;17:2578–2586. [PubMed] [Google Scholar]
- 11.Figarola JL, Singhal P, Rahbar S, Gugiu BG, Awasthi S, Singhal SS. COH-SR4 reduces body weight, improves glycemic control and prevents hepatic steatosis in high fat diet-induced obese mice. PLoS One. 2013;8:e83801. doi: 10.1371/journal.pone.0083801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agostini M, Almeida LY, Bastos DC, Ortega RM, Moreira FS, Seguin F, Zecchin KG, Raposo HF, Oliveira HC, Amoedo ND, Salo T, Coletta RD, Graner E. The fatty acid synthase inhibitor orlistat reduces the growth and metastasis of orthotopic tongue oral squamous cell carcinomas. Mol Cancer Ther. 2014;13:585–595. doi: 10.1158/1535-7163.MCT-12-1136. [DOI] [PubMed] [Google Scholar]
- 13.Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Furuta E, Iiizumi M, Mohinta S, Watabe M, Chalfant C, Watabe K. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res. 2006;66:5934–5940. doi: 10.1158/0008-5472.CAN-05-3197. [DOI] [PubMed] [Google Scholar]
- 14.Cheng X, Li L, Uttamchandani M, Yao SQ. In situ proteome profiling of C75, a covalent bioactive compound with potential anticancer activities. Org Lett. 2014;16:1414–1417. doi: 10.1021/ol500206w. [DOI] [PubMed] [Google Scholar]
- 15.Rossato FA, Zecchin KG, La Guardia PG, Ortega RM, Alberici LC, Costa RA, Catharino RR, Graner E, Castilho RF, Vercesi AE. Fatty acid synthase inhibitors induce apoptosis in non-tumorigenic melan-a cells associated with inhibition of mitochondrial respiration. PLoS One. 2014;9:e101060. doi: 10.1371/journal.pone.0101060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan C, Wei H, Minjuan Z, Yan X, Jingyue Y, Wenchao L, Sheng H. The mTOR inhibitor rapamycin synergizes with a fatty acid synthase inhibitor to induce cytotoxicity in ER/HER2-positive breast cancer cells. PLoS One. 2014;9:e97697. doi: 10.1371/journal.pone.0097697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao P, Mao JM, Zhang SY, Zhou ZQ, Tan Y, Zhang Y. Quercetin induces HepG2 cell apoptosis by inhibiting fatty acid biosynthesis. Oncol Lett. 2014;8:765–769. doi: 10.3892/ol.2014.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006;66:5977–5980. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 19.Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ, Rodriguez-Enriquez S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol. 2014;50:10–23. doi: 10.1016/j.biocel.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 20.Djafarzadeh S, Vuda M, Takala J, Jakob SM. Effect of remifentanil on mitochondrial oxygen consumption of cultured human hepatocytes. PLoS One. 2012;7:e45195. doi: 10.1371/journal.pone.0045195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marin-Hernandez A, Lopez-Ramirez SY, Del Mazo-Monsalvo I, Gallardo-Perez JC, Rodriguez-Enriquez S, Moreno-Sanchez R, Saavedra E. Modeling cancer glycolysis under hypoglycemia, and the role played by the differential expression of glycolytic isoforms. FEBS J. 2014;281:3325–3345. doi: 10.1111/febs.12864. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Enriquez S, Marin-Hernandez A, Gallardo-Perez JC, Moreno-Sanchez R. Kinetics of transport and phosphorylation of glucose in cancer cells. J Cell Physiol. 2009;221:552–559. doi: 10.1002/jcp.21885. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira LM. Cancer metabolism: the Warburg effect today. Exp Mol Pathol. 2010;89:372–380. doi: 10.1016/j.yexmp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Hopperton KE, Duncan RE, Bazinet RP, Archer MC. Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Exp Cell Res. 2014;320:302–310. doi: 10.1016/j.yexcr.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52:585–589. doi: 10.1016/j.plipres.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biswas S, Lunec J, Bartlett K. Non-glucose metabolism in cancer cells--is it all in the fat? Cancer Metastasis Rev. 2012;31:689–698. doi: 10.1007/s10555-012-9384-6. [DOI] [PubMed] [Google Scholar]
- 27.Mounier C, Bouraoui L, Rassart E. Lipogenesis in cancer progression (review) Int J Oncol. 2014;45:485–492. doi: 10.3892/ijo.2014.2441. [DOI] [PubMed] [Google Scholar]
- 28.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 29.Vandhana S, Coral K, Jayanthi U, Deepa PR, Krishnakumar S. Biochemical changes accompanying apoptotic cell death in retinoblastoma cancer cells treated with lipogenic enzyme inhibitors. Biochim Biophys Acta. 2013;1831:1458–1466. doi: 10.1016/j.bbalip.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007;67:8180–8187. doi: 10.1158/0008-5472.CAN-07-0389. [DOI] [PubMed] [Google Scholar]
- 31.Zhou W, Han WF, Landree LE, Thupari JN, Pinn ML, Bililign T, Kim EK, Vadlamudi A, Medghalchi SM, El Meskini R, Ronnett GV, Townsend CA, Kuhajda FP. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res. 2007;67:2964–2971. doi: 10.1158/0008-5472.CAN-06-3439. [DOI] [PubMed] [Google Scholar]
- 32.Lee HS, Seo EY, Kang NE, Kim WK. [6] -Gingerol inhibits metastasis of MDA-MB-231 human breast cancer cells. J Nutr Biochem. 2008;19:313–319. doi: 10.1016/j.jnutbio.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 33.Poltronieri J, Becceneri AB, Fuzer AM, Filho JC, Martin AC, Vieira PC, Pouliot N, Cominetti MR. [6] -gingerol as a cancer chemopreventive agent: a review of its activity on different steps of the metastatic process. Mini Rev Med Chem. 2014;14:313–321. doi: 10.2174/1389557514666140219095510. [DOI] [PubMed] [Google Scholar]
- 34.Saha A, Blando J, Silver E, Beltran L, Sessler J, DiGiovanni J. 6-Shogaol from dried ginger inhibits growth of prostate cancer cells both in vitro and in vivo through inhibition of STAT3 and NF-kappaB signaling. Cancer Prev Res (Phila) 2014;7:627–638. doi: 10.1158/1940-6207.CAPR-13-0420. [DOI] [PubMed] [Google Scholar]
- 35.Kant S, Kumar A, Singh SM. Fatty acid synthase inhibitor orlistat induces apoptosis in T cell lymphoma: role of cell survival regulatory molecules. Biochim Biophys Acta. 2012;1820:1764–1773. doi: 10.1016/j.bbagen.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Li Z, Liu C, Yin L. The apoptotic effect of apigenin on human gastric carcinoma cells through mitochondrial signal pathway. Tumour Biol. 2014;35:7719–7726. doi: 10.1007/s13277-014-2014-x. [DOI] [PubMed] [Google Scholar]
- 37.Zhang K, Li H, Song Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014;15:576–585. doi: 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grube S, Dunisch P, Freitag D, Klausnitzer M, Sakr Y, Walter J, Kalff R, Ewald C. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J Neurooncol. 2014;118:277–287. doi: 10.1007/s11060-014-1452-z. [DOI] [PubMed] [Google Scholar]
- 39.Tomek K, Wagner R, Varga F, Singer CF, Karlic H, Grunt TW. Blockade of fatty acid synthase induces ubiquitination and degradation of phosphoinositide-3-kinase signaling proteins in ovarian cancer. Mol Cancer Res. 2011;9:1767–1779. doi: 10.1158/1541-7786.MCR-10-0467. [DOI] [PubMed] [Google Scholar]
- 40.Fritz V, Benfodda Z, Henriquet C, Hure S, Cristol JP, Michel F, Carbonneau MA, Casas F, Fajas L. Metabolic intervention on lipid synthesis converging pathways abrogates prostate cancer growth. Oncogene. 2013;32:5101–5110. doi: 10.1038/onc.2012.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason P, Liang B, Li L, Fremgen T, Murphy E, Quinn A, Madden SL, Biemann HP, Wang B, Cohen A, Komarnitsky S, Jancsics K, Hirth B, Cooper CG, Lee E, Wilson S, Krumbholz R, Schmid S, Xiang Y, Booker M, Lillie J, Carter K. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS One. 2012;7:e33823. doi: 10.1371/journal.pone.0033823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turrado C, Puig T, Garcia-Carceles J, Artola M, Benhamu B, Ortega-Gutierrez S, Relat J, Oliveras G, Blancafort A, Haro D, Marrero PF, Colomer R, Lopez-Rodriguez ML. New synthetic inhibitors of fatty acid synthase with anticancer activity. J Med Chem. 2012;55:5013–5023. doi: 10.1021/jm2016045. [DOI] [PubMed] [Google Scholar]
- 43.Lin CB, Lin CC, Tsay GJ. 6-Gingerol Inhibits Growth of Colon Cancer Cell LoVo via Induction of G2/M Arrest. Evid Based Complement Alternat Med. 2012;2012:326096. doi: 10.1155/2012/326096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nigam N, Bhui K, Prasad S, George J, Shukla Y. [6] -Gingerol induces reactive oxygen species regulated mitochondrial cell death pathway in human epidermoid carcinoma A431 cells. Chem Biol Interact. 2009;181:77–84. doi: 10.1016/j.cbi.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 45.Nigam N, George J, Srivastava S, Roy P, Bhui K, Singh M, Shukla Y. Induction of apoptosis by [6] -gingerol associated with the modulation of p53 and involvement of mitochondrial signaling pathway in B[a] P-induced mouse skin tumorigenesis. Cancer Chemother Pharmacol. 2010;65:687–696. doi: 10.1007/s00280-009-1074-x. [DOI] [PubMed] [Google Scholar]
- 46.Chen C, Han X, Zou X, Li Y, Yang L, Cao K, Xu J, Long J, Liu J, Feng Z. 4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75), an inhibitor of fatty-acid synthase, suppresses the mitochondrial fatty acid synthesis pathway and impairs mitochondrial function. J Biol Chem. 2014;289:17184–17194. doi: 10.1074/jbc.M114.550806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang G, Wang S, Zhong L, Dong X, Zhang W, Jiang L, Geng C, Sun X, Liu X, Chen M, Ma Y. 6-Gingerol induces apoptosis through lysosomal-mitochondrial axis in human hepatoma G2 cells. Phytother Res. 2012;26:1667–1673. doi: 10.1002/ptr.4632. [DOI] [PubMed] [Google Scholar]
- 48.Rastogi N, Gara RK, Trivedi R, Singh A, Dixit P, Maurya R, Duggal S, Bhatt ML, Singh S, Mishra DP. (6)-Gingerolinduced myeloid leukemia cell death is initiated by reactive oxygen species and activation of miR-27b expression. Free Radic Biol Med. 2014;68:288–301. doi: 10.1016/j.freeradbiomed.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 49.Chen G, Chen Z, Hu Y, Huang P. Inhibition of mitochondrial respiration and rapid depletion of mitochondrial glutathione by beta-phenethyl isothiocyanate: mechanisms for antileukemia activity. Antioxid Redox Signal. 2011;15:2911–2921. doi: 10.1089/ars.2011.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deeb D, Gao X, Liu YB, Gautam SC. Inhibition of cell proliferation and induction of apoptosis by CDDO-Me in pancreatic cancer cells is ROS-dependent. J Exp Ther Oncol. 2012;10:51–64. [PMC free article] [PubMed] [Google Scholar]
- 51.Valenti D, de Bari L, Manente GA, Rossi L, Mutti L, Moro L, Vacca RA. Negative modulation of mitochondrial oxidative phosphorylation by epigallocatechin-3 gallate leads to growth arrest and apoptosis in human malignant pleural mesothelioma cells. Biochim Biophys Acta. 2013;1832:2085–2096. doi: 10.1016/j.bbadis.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 52.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. 2014;289:8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palanivel K, Kanimozhi V, Kadalmani B, Akbarsha MA. Verrucarin A Induces Apoptosis Through ROS-Mediated EGFR/MAPK/Akt Signaling Pathways in MDA-MB-231 Breast Cancer Cells. J Cell Biochem. 2014;115:2022–2032. doi: 10.1002/jcb.24874. [DOI] [PubMed] [Google Scholar]
- 54.Pan MH, Hsieh MC, Kuo JM, Lai CS, Wu H, Sang S, Ho CT. 6-Shogaol induces apoptosis in human colorectal carcinoma cells via ROS production, caspase activation, and GADD 153 expression. Mol Nutr Food Res. 2008;52:527–537. doi: 10.1002/mnfr.200700157. [DOI] [PubMed] [Google Scholar]
- 55.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 56.Biswas N, Mahato SK, Chowdhury AA, Chaudhuri J, Manna A, Vinayagam J, Chatterjee S, Jaisankar P, Chaudhuri U, Bandyopadhyay S. ICB3E induces iNOS expression by ROS-dependent JNK and ERK activation for apoptosis of leukemic cells. Apoptosis. 2012;17:612–626. doi: 10.1007/s10495-011-0695-9. [DOI] [PubMed] [Google Scholar]
- 57.Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 58.Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, Hirota S, Hosobe S, Tsukada T, Miura K, Kamada S, Saito K, Iiizumi M, Liu W, Ericsson J, Watabe K. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68:1003–1011. doi: 10.1158/0008-5472.CAN-07-2489. [DOI] [PubMed] [Google Scholar]
- 59.Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. 2012;10:133–142. doi: 10.1158/1541-7786.MCR-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qiu C, Dongol S, Lv QT, Gao X, Jiang J. Sterol regulatory element-binding protein-1/fatty acid synthase involvement in proliferation inhibition and apoptosis promotion induced by progesterone in endometrial cancer. Int J Gynecol Cancer. 2013;23:1629–1634. doi: 10.1097/IGC.0000000000000004. [DOI] [PubMed] [Google Scholar]
- 61.Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, Wakelam M, Harris AL, Schulze A. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1:3. doi: 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kant S, Kumar A, Singh SM. Tumor growth retardation and chemosensitizing action of fatty acid synthase inhibitor orlistat on T cell lymphoma: implication of reconstituted tumor microenvironment and multidrug resistance phenotype. Biochim Biophys Acta. 2014;1840:294–302. doi: 10.1016/j.bbagen.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 63.Impheng H, Pongcharoen S, Richert L, Pekthong D, Srisawang P. The selective target of capsaicin on FASN expression and de novo fatty acid synthesis mediated through ROS generation triggers apoptosis in HepG2 cells. PLoS One. 2014;9:e107842. doi: 10.1371/journal.pone.0107842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puig T, Turrado C, Benhamu B, Aguilar H, Relat J, Ortega-Gutierrez S, Casals G, Marrero PF, Urruticoechea A, Haro D, Lopez-Rodriguez ML, Colomer R. Novel Inhibitors of Fatty Acid Synthase with Anticancer Activity. Clin Cancer Res. 2009;15:7608–7615. doi: 10.1158/1078-0432.CCR-09-0856. [DOI] [PubMed] [Google Scholar]
- 65.Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A. 2000;97:3450–3454. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000;60:213–218. [PubMed] [Google Scholar]
- 67.Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, Pinn ML, Ronnett GV, Kuhajda FP. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 2003;63:7330–7337. [PubMed] [Google Scholar]
- 68.Jin Q, Yuan LX, Boulbes D, Baek JM, Wang YN, Gomez-Cabello D, Hawke DH, Yeung SC, Lee MH, Hortobagyi GN, Hung MC, Esteva FJ. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010;12:R96. doi: 10.1186/bcr2777. [DOI] [PMC free article] [PubMed] [Google Scholar]