

Abstract
Standard combined modality therapies for aerodigestive tract malignancies have suboptimal outcomes, and targeting cancer-specific molecular pathways in combination with radiation could improve the therapeutic ratio. Dysregulation of epigenetic modulators such as histone deacetylases (HDACs), and developmental morphogens such as the hedgehog (HH) pathway have been implicated in aerodigestive tumor progression and metastasis. We hypothesized that simultaneous targeting of HDACs and the HH-pathway mediator Smoothened (Smo) represents an opportunity to overcome therapeutic resistance in these cancers. We evaluated the effects of the HDAC inhibitor SAHA and Smo inhibitor GDC-0449 with radiation in multiple aerodigestive cancer cell lines. Isobologram analyses showed that SAHA and GDC-0449 synergistically suppressed cancer cell proliferation in vitro. SAHA and GDC-0449 cooperatively enhanced G0/G1 cell cycle arrest which was associated with up-regulation of p21waf. GDC-0449 prevented SAHA-induced up-regulation of Gli-1 and Gli-2. Both Smo and Ptc-1 expression was cooperatively suppressed by SAHA and GDC-0449. The combination of SAHA and GDC-0449 induced radiation sensitization with 2 Gy as determined by colony formation assays and cytogenetic analyses, which correlated with higher residual γ-H2AX and 53BP1 foci. In mouse tumor xenografts of the SqCC/Y1 cell line, SAHA and GDC-0449 delayed tumor growth longer and prolonged survival more than either agent alone. In summary, we have identified synergistic effect of HDAC and HH signaling for radiosensitization to improve therapeutic outcomes for aerodigestive malignancies.
Keywords: Histone deacetylases, hedgehog, radiation sensitization, lung cancer, head and neck cancer, lysine deacetylases, smoothened, homologous recombination
Introduction
Aerodigestive tract tumors are the leading cause of cancer morbidity and mortality worldwide, with over 240,000 respiratory, 289,000 digestive, and 42,000 oropharynx cancers expected to be diagnosed in the United States in 2014 [1]. For the majority of aerodigestive tumors in the locally-advanced, recurrent, or metastatic setting, oncologic outcomes remain poor due to resistance to conventional cytotoxic chemotherapeutics and radiation therapy. Despite the identification and production of an array of clinically available small molecular targeted agents, few biologic agents have been identified that improve survival or confer radiation sensitization in combined modality therapy. Thus, there is a need to apply targeted biologic agents in a rational way to improve the efficacy of combined modality therapy in order to improve both local control and overall survival.
Epigenetic dysregulation of gene expression by histone deacetylases (HDACs) is an established phenomenon in a multiple cancers. HDACs remove acetyl groups from lysine amino acid residues of histone and non-histone proteins, thereby affecting signal transduction pathways and the expression of tumor suppressor genes such as p21waf. Inhibition of HDACs with clinically available therapeutics such as valproic acid, suberoylanilide hydroxamic acid (SAHA), and LBH-589 have been shown to have anti-neoplastic effects in a variety of tumor histologies [2]. In normal cellular physiology, HDACs and histone acetyl transferases (HATs) dynamically regulate the post-translational acetylation status of multiple proteins, and HDAC inhibitors allow unopposed HAT-induced hyperacetylation. This in turn, leads to multiple pleiotropic cellular effects including cell cycle arrest, apoptosis, differentiation, and autophagy in cancer cells [2]. There is also substantial evidence that HDAC inhibitors such as SAHA induce radiation sensitization through regulation of free radical reactive oxygen species (ROS), suppression of homologous recombination, and chromatin remodeling [3-5]. These lines of pre-clinical evidence have lead to multiple clinical trials evaluating the effect of HDAC inhibition in combination with conventional chemotherapeutics and radiation therapy in clinical trials [6-10].
Pathways of embryologic development such as the hedgehog (HH) pathway have also been shown to be active in aerodigestive tract tumors, contributing to the initiation, progression, and metastasis of tumors [11,12]. The HH pathway is involved in the patterning of the aerodigestive tract in development [13], and is recapitulated in the cancer state where it promotes tumor growth, invasion, and metastasis. This signaling pathway involves the binding of the HH ligand to the Patched (Ptc) transmembrane protein, thereby relieving constitutive inhibition of Smoothened (Smo) signal transduction. In response to HH binding to Ptc, Smo activates the Gli family of transcription factors which up-regulate multiple genes including Ptc-1, Gli-1, cyclin D1, IGF-2, and c-myc [11]. Aberrant HH signaling is well established as an oncogenic driver in multiple cancers such as basal cell carcinoma, medulloblastoma, and aerodigestive tract tumors [14-17]. Clinical inhibitors of Smo such as GDC-0449 (Vismodegib), produce dramatic regression of HH addicted malignancies such as basal cell carcinomas [18]. However, HH pathway inhibition by GDC-0449 does not produce durable tumor control [19], suggesting other mechanisms of tumor resistance. Furthermore, results of clinical trials using Smo inhibitors in combination with cytotoxic chemotherapeutic agents have been disappointing, underlining the need to develop rational target based combined modality strategies for HH pathway suppression.
An interaction between HDACs and the HH pathway has recently been identified that cooperatively regulates signal transduction and survival in multiple tumor types [20,21]. Such interaction could represent an opportunity to overcome resistance to either HDAC or Smo inhibition in isolation. In pancreatic cancer cell lines, SAHA and the Smo antagonist SANT-1 have been shown to cooperatively up-regulate p21waf and down-regulate the HH pathway, which in turn promotes cell cycle arrest and subsequent death [21,22]. This interaction has also been evaluated in medulloblastoma, where removal of acetyl groups from the transcription factor Gli by HDAC1 is a rate limiting step in HH pathway signal activation [20]. Moreover, as targeting of these pathways cooperatively induces cell cycle arrest [21], there is also a potential mechanism for radiation sensitization since homologous recombination (HR)-mediated repair of DNA damage occurs primarily in the S and G2/M phases of the cell cycle. Therefore, combined targeting of HDACs and HH signaling may offer an opportunity to improve therapeutic responses when used with radiotherapy for aerodigestive cancers.
Based on the aforesaid lines of evidence, we hypothesized that there is a cooperative interaction between HDACs and the HH pathway in aerodigestive tract tumors contributing to therapeutic resistance that can be exploited for combined modality therapy with ionizing radiation (IR). To test this hypothesis, we evaluated the effect of the HDAC inhibitor SAHA and Smo inhibitor GDC-0449 on tumor growth and radiation responses in multiple aerodigestive tract cancer cell lines.
Materials and methods
Cell culture
The head and neck cancer (HNC) cell lines SqCC/Y1, HN30, and HN31 were obtained from Dr. Jeffrey Myers at the MD Anderson Comprehensive Cancer Center (Houston, TX). The cell lines FaDu and H1299 were obtained from the American Type Culture Collection (ATCC). All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco), supplemented with 5% fetal bovine serum (FBS, Sigma-Aldrich), 1% non-essential amino acid (NEAA), 1% sodium pyruvate (Sigma-Aldrich), 1% modified Eagle’s medium (MEM) vitamin (Gibco), and 1% L-glutamine (Gibco), unless otherwise specified. Cell culture and experiments were carried out in the absence of antibiotics in standard cell culture conditions of 37°C with 5% CO2.
Small molecules
SAHA and GDC-0449 were obtained from Selleck Chemicals. These agents were dissolved in dimethysulfoxide (DMSO) stock solution obtained from Sigma-Aldrich, and stored at -20°C. The DMSO vehicle was used alone as control to account for its potential effect on any experimental variable.
Cellular proliferation
The effect of SAHA and GDC-0449 on cellular proliferation was quantified using the IN Cell Analyzer 2000 (General Electric). In 48 well plates (Costar), cells were seeded at a concentration of 500-1000 cells per well. Cells were exposed to the indicated treatment for 72 hours (h), and then fixed and permeabilized by freezing the cells in de-ionized water at -80°C for at least one hour. After thawing the cells at room temperature (RTP), the cells were stained with Hoechst 33342 dye (Sigma-Aldrich) for 1 hour at RTP. The number of Hoechst 33342 positive nuclei were counted in representative images from each well using the IN Cell Analyzer 2000, and compared to appropriate controls. The concentration of SAHA and GDC-0449 to produce 50% effect of the maximum effect on cellular proliferation (IC50) was calculated based on a dose response curve, and an isobologram analysis was done using the Chou-Talalay Method with the Calcusyn (Biosoft) software program as previously described [23]. All experiments were performed at least 3 times in quadruplicate.
Colony formation assay
Cells were seeded in 6-well tissue culture plates (Costar) at a concentration of 100 cells per well. After being allowed to adhere to the wells, cells were treated with the indicated treatment for 24 h. The medium with the indicated treatment was removed and replaced with fresh medium, and cells were immediately irradiated with the indicated radiation dose. Cells were then allowed to grow for 10 days. To visualize colonies, cells were stained with Crystal Violet (Sigma-Aldrich), and colonies were counted if they consisted of at least 50 cells using an inverted light microscope. The number of colonies per well was compared to appropriate controls. Experiments were repeated 3 times in triplicate.
Flow cytometric analysis of cell cycle distribution
SqCC/Y1 and H1299 were seeded in 6-well tissue culture plates at a concentration of 250,000 cells per well and allowed to attach for at least 12 hours. Next, they were serum starved for 24 h in DMEM without FBS. After 24 h of serum starvation, DMEM supplemented with 5% FBS and treated with the indicated small molecule was added to the cells for 24 h. Cells were then trypsinized from the plate using 0.05% trypsin solution (Sigma-Aldrich), and fixed in 100% ice cold ethanol for 1 h. Following fixation, cells were stained with a 50 µg/mL solution of propidium iodide (Roche) and 1% RNase A (Thermo Scientific). Prior to flow cytometric analysis, cells were disaggregated by aspirating through a 0.4 x 13 mm hypodermic needle (Beckton Dickinson) with a syringe. Cellular DNA content was determined using a Beckman Coulter Flow Cytometer, and 10,000 events were captured per histogram. Experiments were repeated at least 3 times with similar results each time.
Immunoblot analysis
Standard immunoblot analysis was done on whole cell extracts obtained from cells in 10 cm tissue culture dishes at 70-80% confluence. Briefly, radioimmunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors (Roche) was used to lyse cells and extract protein. Protein concentration was determined using the BCA protein Assay kit (BioRad) based upon a bovine serum albumin (BSA) standard curve. In each well, 20 µg of protein was added and run on a 10% Tris sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. Next, protein was transferred to a polyvinylidene difluoride membrane (Thermo Scientific). The membrane was then blocked with 4% skim milk for 1 h. Membranes were incubated with primary antibody in 4% BSA at 4°C overnight at a concentration of 1:1000-1:10,000. The primary antibodies rabbit anti-acetyl H3, anti-total H3, anti-p21waf, and anti-γ-H2AX were obtained from Cell Signaling Technology, and the primary antibodies rabbit anti-ptc-1 and anti-Rad51, and mouse anti-GAPDH were obtained from Santa Cruz Biotechnology. Membranes were then incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies against mouse or rabbit (Santa Cruz Biotechnology) for 1 h at RTP at a concentration of 1:1000-1:10,000 in 4% skim milk. HRP signal to detect protein antigens was activated using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific), and visualized with UltraCruz autoradiography film (Santa Cruz Biotechnology). The molecular weight of the detected protein was determined by comparing the band to an adjacent well loaded with the Precision Plus Protein Kaleidoscope (BioRad) as a size marker.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted using TRIzol reagent (Life technologies) according to the manufacturer’s protocol. First-strand cDNA was synthesized with SuperScripIII cDNA Synthesis kit (Invitrogen) using about 2 μg total RNA as template. For real-time PCR, we obtained TaqMan FAM-MGB probes for Gli-1, Gli-2, Smo, and GAPDH (Life technologies). PCR was performed in a total volume of 20 μl containing TaqMan Gene Expression Master Mix (Life technologies), each probes and cDNA using the CFX96 real-time PCR detection system (BioRad). Gene expression levels were expressed relative to appropriate controls.
Immunofluorescence
The cell line H1299 was grown on sterile glass cover slips and treated as indicated. Cells were fixed with 4% paraformaldehyde (USB Corporation) and permeabilized with 0.5% Triton-X (Sigma-Aldrich). Next, cells were blocked with a solution of 4% FBS. Cover slips were then incubated at 4°C overnight in primary antibody solution of rabbit anti-γ-H2AX or anti-53BP1 (Santa Cruz Biotechnology) at a concentration of 1:1000. After rinsing the cells with phosphate buffered saline (PBS), cells were incubated with an anti-rabbit secondary antibody conjugated to Alexa Fluor 594 (red) or Alexa Fluor 488 (green) obtained from Invitrogen at a concentration of 1:1000 at RTP for 1 h. Cells were then counterstained with 4’, 6 diamino-2-phenylindose (DAPI), and the coverslips were mounted on glass slides and sealed with nail polish. Nuclear foci were visualized using an AX10 (Zeiss) fluorescent microscope and images taken with AxioCam (Zeiss).
Homologous recombination reporter system
HR in the H1299 cell line was assessed using the green fluorescent protein (GFP) linked HR reporter system previously developed and described [24]. The 18-bp-I-Sce I site was inserted within one copy of the GFP gene, inactivating GFP expression [25]. DNA double strand breaks (DSBs) induced by the I-Sce I endonuclease that occur during the S/G2 phase of the cell cycle are repaired by HR [24,26], thereby allowing a donor GFP gene fragment to restore GFP expression and fluorescence, which can be directly monitored as a measure of HR activity. Relative levels of HR in the presence of SAHA and GDC-0449 for 24 h was assessed by flow cytometric quantification of cells expressing GFP using restored GFP expression as a marker of HR activity after DSB induction by the I-Sce I endonuclease. Experiments were repeated at least 3 times, capturing 50,000 events per histogram.
Metaphase chromosomal aberrations analysis
Chromosome aberration analyses were performed by the previously described procedure [27,28]. In brief, cells treated with drug and IR were analyzed for G2-type chromosome aberrations. Cells were irradiated with 1 Gy and those in metaphase were collected 1.5 hour post-treatment [29]. Chromosome spreads were prepared after hypotonic treatment of cells, fixed in acetic acid methanol, and stained with Giemsa [30]. G2-phase chromosomal aberrations were measured by counting chromatid breaks and gaps per metaphase as previously described [29,31]. Fifty metaphase sets were scored for each post irradiation time point and the results show are the mean of three experiments.
Orthotopic xenograft model
An orthotopic HNC xenograft model was used to evaluate the efficacy of combined treatments of SAHA, GDC-0449, and IR, as well as each agent alone. Head and neck tumors were generated by injecting 1 x 106 SqCC/Y1 cells into the left neck area of female severe combined immunodeficiency-non-obese diabetic (SCID-NOD) mice weighing 20-25 grams (g) obtained from the UT Southwestern institutional breeding core. Tumor masses were measured twice per week and mice were stratified so that tumor volumes in each group (5 mice per group) were not statistically different prior to the experiment. SAHA and GDC-0449 were administered daily concurrently with radiation therapy at a dose of 25 mg/kg of SAHA, and 12.5 mg/kg GDC-0449, dissolved in 50 µL of DMSO via intraperitoneal injection with a hypodermic needle daily for 10 days. Mice were sacrificed when tumor burdens reached approximately 500 mm3. Survival and tumor volume data were graphed from two separate studies, with 10 mice per group. Tumor tissues were removed for histological examination. All animal studies were carried out under a University of Texas at Southwestern Medical Center Institutional Animal Care and Use Committee approved protocol and in accordance with the guidelines for ethical conduct in the care and use of animals in research. Experiments were repeated three times.
Irradiation technique
Cells in vitro were irradiated using the Mark I Irradiator (Shephard and Associates). Dose was calculated and delivered in units of Gray (Gy, joules per kilogram).
Statistical analysis
Groups of data were tested for statistical significance using a two-sided student’s t-test. Log-rank tests were applied to Kaplan-Meier survival analyses censored for death for in vivo experiments. All in vivo statistical analyses were performed using Graph Pad Prism software. Results were considered to be statistically significant for a P-value < 0.05.
Results
Suppression of cellular proliferation by HDAC and Smo blockade
The potential anti-proliferative effects of HDAC and HH pathway inhibition in aerodigestive tract cancers were evaluated by determining the effect of increasing doses of SAHA and GDC-0449 on cellular proliferation in the SqCC/Y1, FADU, HN30, HN31, and H1299 cell lines for 72 h. The absolute number of Hoechst 33342 stained nuclei was counted using the IN Cell Analyzer 2000 at 72 h, and IC50 values were calculated with respect to cellular proliferation (Figure 1A). Varying concentrations of SAHA and GDC-0449 were then evaluated in combination with respect to cellular proliferation at 72 h, and isobologram analysis was performed using the Chou-Talalay method as previously described [23]. A syngergistic anti-proliferative effect was observed in all cell lines, defined as a combination index (CI) less than 1. Moreover, at low concentrations of SAHA and GDC-0449, there was an enhanced anti-proliferative effect seen in all cell lines (Figure 1B). In SqCC/Y1, the effect of 0.5 µM of SAHA in combination with 1 µM of GDC-0449 significantly enhanced suppression of cellular proliferation (P < 0.01). For the cell lines FADU, HN30, HN31, and H1299, 1 µM of SAHA in combination with 1 µM of GDC-0449 similarly enhanced suppression of cellular proliferation. Thus, in all cell lines, there was a statistically significant decrease in cellular proliferation when SAHA and GDC-0449 were combined.
Figure 1.

Isobologram analysis showing synergistic suppression on cellular proliferation by combined targeting of HDACs and HH signaling. A. Characteristics of aerodigestive tumor cell lines and dose responses to SAHA and GDC-0449. B. Proliferation with SAHA 1 µM and GDC-0449 1 µM. *P < 0.05.
To determine the basis for the suppression of cellular proliferation, cell cycle analysis was performed on the SqCC/Y1 and H1299 cell lines by staining the DNA content of cells with propidium iodide. Flow cytometric evaluation of DNA content revealed that the combination of SAHA and GDC-0449 enhanced G0/G1 cell cycle arrest after 24 h of exposure (Figure 2A). In the SqCC/Y1 cell line, neither 0.5 µM SAHA nor 5 µM GDC-0449 caused significant changes in cell cycle distribution, but in combination, SAHA and GDC-0449 induced a significant 10% increase in G0/G1 cell cycle accumulation compared to control (P = 0.0029), with a corresponding reduction of cells in the S phase (P = 0.0067). As a single agent, 1 µM SAHA caused a significant 13% increase of cells in the G0/G1 phase compared to control (P = 0.03) in the H1299 cell line at 24 h, whereas 5 µM GDC-0449 had no significant effect. However, in H1299, when SAHA and GDC-0449 were combined, there was a 32% increase in G0/G1 cell cycle distribution (P = 0.0015), which was statistically significant compared to SAHA alone (Figure 2A, P < 0.05). Representative histograms are provided showing the effect of SAHA and GDC-0449 on cell cycle distribution in the H1299 cell line (Figure 2B). Thus, consistent with their effects on cellular proliferation, SAHA and GDC-0449 similarly enhanced cell cycle arrest in the G0/G1 phase of the cell cycle in SqCC/Y1 and H1299.
Figure 2.

Enhanced G0/G1 cell cycle arrest with SAHA and GDC-0449. A. Effect of SAHA and GDC-0449 on cell cycle distribution in SqCC/Y1 and H1299. B. Representative flow cytometry histograms of DNA content for H1299. C. Immunoblot analysis of histone H3 acetylation status and p21 expression.
To gain insight into the mechanistic basis for suppressed cellular proliferation and impaired cell cycle progression, immunoblot analysis was done on relevant signaling pathways. Exposure to 0.5 µM SAHA caused hyperacetylation of histone H3 alone or in combination with 5 µM GDC-0449 (Figure 2C). Expression of the cyclin-dependent kinase inhibitor p21waf , a known regulator of the G0/G1 checkpoint, was also evaluated in SqCC/Y1. Consistent with enhanced G0/G1 cell cycle arrest [21], p21waf was enhanced in response to the combination of 0.5 µM SAHA and 5 µM GDC-0449 (Figure 2C). HH pathway activity was also analyzed in response to SAHA and GDC-0449 (Figure 3). Expression of the known product of the HH pathway marker Ptc-1 was determined to evaluate HH pathway activity [11,32]. In SqCC/Y1 and H1299 (Figure 3A), SAHA and GDC-0449 cooperatively suppressed Ptc-1 expression. Using RT-PCR, expression of the HH pathway genes Gli-1, Gli-2, and Smo were analyzed (Figure 3B). Both Gli-1 and Gli-2 were found to be induced by SAHA, which was abrogated by GDC-0449. Consistent with their effects on Ptc-1 protein expression, SAHA and GDC-0449 also cooperatively down-regulated Smo expression.
Figure 3.
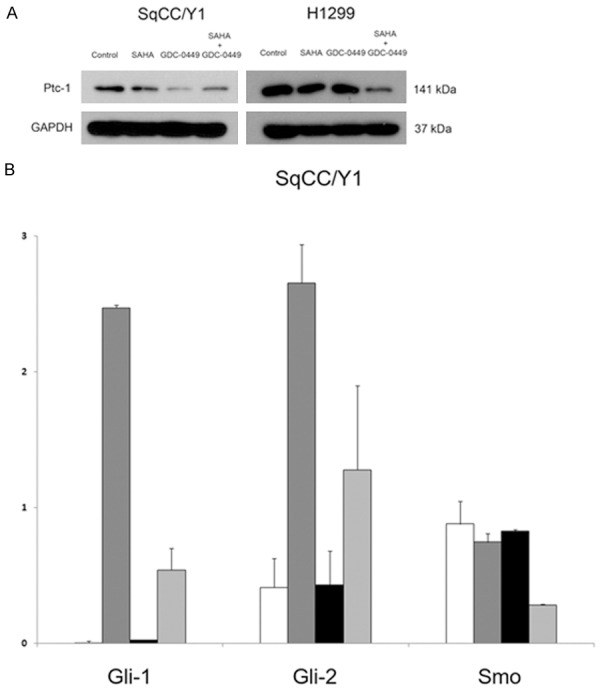
Hedgehog pathway modulation in response to HDAC and Smo inhibition. A. Immunoblot analysis of HH pathway gene Ptc-1 expression. B. Expression of HH pathway genes Gli-1, Gli-2, and Smo by RT-PCR. White bar, DMSO; dark grey bar, SAHA; black bar, GDC-0449; light grey bar, SAHA + GDC-0449.
Taken together, the cellular proliferation and cell cycle analyses demonstrated a synergistic suppression of cellular proliferation by combined HDAC and HH pathway inhibition. This effect was associated with cooperative up-regulation of the cyclin-dependent kinase inhibitor p21waf by SAHA and GDC-0449. Because of this cooperative induction of cell cycle arrest, we hypothesized that the combination of SAHA and GDC-0449 could confer a radiation sensitization effect through the suppression of HR-directed DNA repair, which occurs primarily in the S and G2/M phase of the cell cycle when a homologous chromosome is available as a DNA template [26,33].
Radiation sensitization by combined targeting of HDACs and HH signaling
To assess the potential radiation sensitization effect of targeted inhibition of HDAC and Smo, the effects of 0.1 µM SAHA, 1 µM GDC-0449, and 2 Gy of IR were evaluated alone and in combination on SqCC/Y1 and H1299 by colony formation assay (Figure 4A). In SqCC/Y1, 0.1 µM of SAHA enhanced the effect of 2 Gy, while 1 µM of GDC-0449 did not enhance the effect of 2 Gy as a single agent. However, the most significant suppression of colony formation in SqCC/Y1 occurred in the combination of SAHA, GDC-0449, and 2 Gy (P < 0.05), resulting in nearly a 10-fold reduction in colony formation with 13% of colonies compared to the control. In H1299, both SAHA and GDC-0449 enhanced radiation-induced suppression of colony formation as single agents. However, as seen for SqCC/Y1, the combination of SAHA and GDC-0449 caused significantly more radiation sensitization with 2 Gy (P < 0.05), compared to either SAHA or GDC-0449 alone with only 29% of colonies compared to controls.
Figure 4.
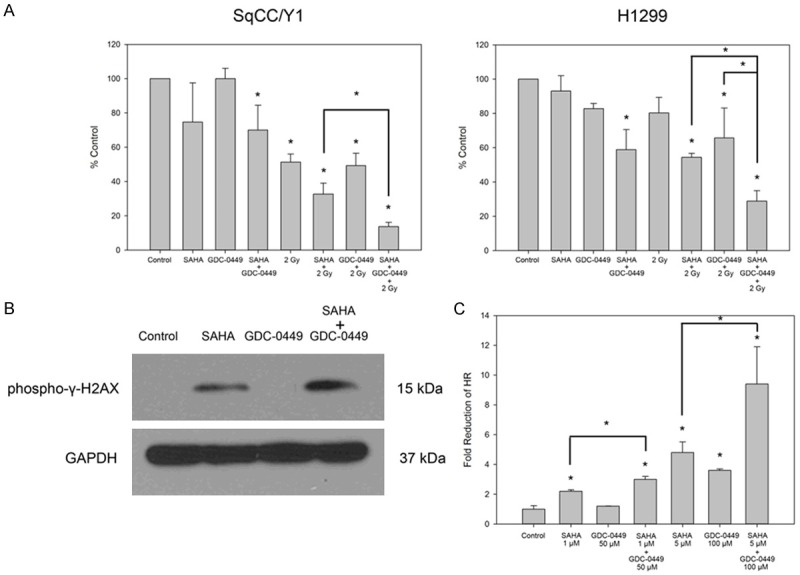
Radiation sensitization by SAHA and GDC-0449. A. SAHA and GDC-0449 had a cooperative anti-proliferative effect and enhanced radiation responses in colony formation assays. B. Expression of the DNA damage marker phospho-γ-H2AX in response to 2 Gy with control, SAHA, GDC-0449, and SAHA + GDC-0449. C. SAHA and GDC-0449 cooperatively suppressed homologous recombination (HR) in combination in an HR reporter assay. *P < 0.05.
Because of the radiation sensitization effect seen in colony formation assay, we evaluated SqCC/Y1 for evidence of inability to repair DNA double strand breaks (DSBs). Evidence for DSBs was determined by assessing γ-H2AX expression in SqCC/Y1 (Figure 4B). SqCC/Y1 was treated for 24 h with control (DMSO), 0.5 µM SAHA, 5 µM GDC-0449, and SAHA + GDC-0449, and then exposed to 2 Gy of IR. At 4 h after exposure to 2 Gy, protein was extracted from SqCC/Y1, and expression of γ-H2AX was visualized by immunoblot. Consistent with colony formation data, SAHA was associated with up-regulation of γ-H2AX foci at 4 h post-IR, which was further enhanced by combining SAHA with GDC-0449, suggesting enhanced formation and prolongation of potently lethal DNA DSBs by through the combination of HDAC and HH pathway inhibition concurrently with clinical doses of IR.
Since the number of DNA DSBs is increased with combination therapy, we next evaluated the effect of SAHA and GDC-0449 on HR, which is a critical pathway of DNA DSB repair in mammalian cells [34]. A GFP-linked HR reporter system in H1299 was used to assess HR activity after 24 h exposure to SAHA and GDC-0449 (Figure 4C). Doses of 1 µM SAHA and 50 µM GDC-0449 caused suppression of HR as single agents, but this effect was significantly more pronounced when SAHA and GDC-0449 were combined (P < 0.05). HR was reduced by 2.2 fold by 1 µM SAHA (P < 0.05), 1.2 fold by 50 µM GDC-0449, and 3 fold by 1 µM SAHA + 50 µM GDC-0449, which was significant compared to SAHA alone (P < 0.05). A similar effect was seen when high doses of 5 µM SAHA and 100 µM GDC-0449 were combined. As a single agent, 5 µM SAHA reduced HR by 4.8 fold (P < 0.05), 100 µM GDC-0449 reduced HR by 3.6 fold (P < 0.05), and the combination of 5 µM SAHA and 100 µM GDC-0449 reduced HR by 9.4 fold (P < 0.05). The effect of 5 µM SAHA combined with 100 µM GDC-0449 was also significant (P < 0.05) compared to either SAHA or GDC-0449 alone.
In order to further quantify the burden of DNA DSBs associated with the combination of SAHA, GDC-0449, and 2 Gy, immunofluorescence was done to detect evidence of γ-H2AX and 53BP1 foci formation in H1299 (Figure 5A). H1299 was treated with DMSO (control), 1 µM SAHA, 50 µM GDC-0449, and 1 µM SAHA and 50 µM GDC-0449 for 24 h. Cells were then irradiated with 2 Gy IR as indicated, and 4 h post-IR, immunofluorescence was done for γ-H2AX and 53BP1. The combination of SAHA and GDC-0449 without IR did not significantly induce large foci formation of phospho-γ-H2AX (green fluorescence) or 53BP1 (red fluorescence). As a single modality, 2 Gy increased the formation of large γ-H2AX and 53BP1 foci (P < 0.05). However, when 1 µM SAHA, 50 µM GDC-0449, and 2 Gy were combined, there was a pronounced significant increase in both γ-H2AX and 53BP1 foci compared to 2 Gy alone (P < 0.05). Representative DAPI stained nuclei showing large γ-H2AX and 53BP1 foci formation is shown in Figure 5B.
Figure 5.
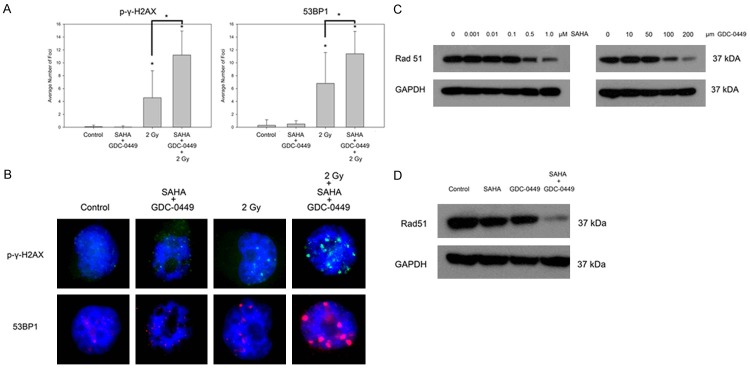
Mechanisms of radiation sensitization by SAHA and GDC-0449. A. SAHA and GDC-0449 enhance formation of phospho-γ-H2AX and 53BP1 with 2 Gy of radiation. B. Immunofluorescence of representative cells with DNA damage foci phospho-γ-H2AX and 53BP1. C. Expression of the HR recombinase Rad51 in response to a dose titration of SAHA or GDC-0449. D, the combination of SAHA and GDC-0449 cooperatively down-regulates Rad51 expression. *P < 0.05.
To better understand the mechanism of HR suppression, we next evaluated expression levels of the Rad51 protein which is recruited to DNA DSBs and is required for HR-directed repair [34]. A dose titration was done exposing H1299 to increasing concentrations of either SAHA or GDC-0449 for 24 h with respect to Rad51 protein expression (Figure 5C). Consistent with previous reports showing SAHA-induced down-regulation of Rad51 [35], both SAHA and GDC-0449 caused a dose dependent down-regulation of Rad51 in H1299. At doses of 0 to 0.1 µM SAHA, there was no perceptible change in Rad51 expression, but doses of 0.5 to 1 µM caused dose dependent down-regulation of Rad51. GDC-0449 caused a dose dependent down-regulation of Rad51 from 50-200 µM. Importantly, although 0.5 µM SAHA or 50 µM GDC-0449 caused a slight down-regulation of Rad51, the combination caused a prominent and cooperative down-regulation of Rad51 (Figure 5D).
Further evidence for enhanced DNA damage was then determined from metaphase chromosomal aberration analyses (Figure 6). Representative chromosomal aberrations are shown with H1299 cells treated with 1 Gy in combination with SAHA, GDC-0449, and SAHA + GDC-0449 (Figure 6A). Consistent with the results of colony formation, DNA damage foci responses, and homologous reporter assays, the combination of SAHA + GDC-0449 with 1 Gy IR caused more significantly more chromosomal aberrations than any other treatment group (Figure 6B).
Figure 6.
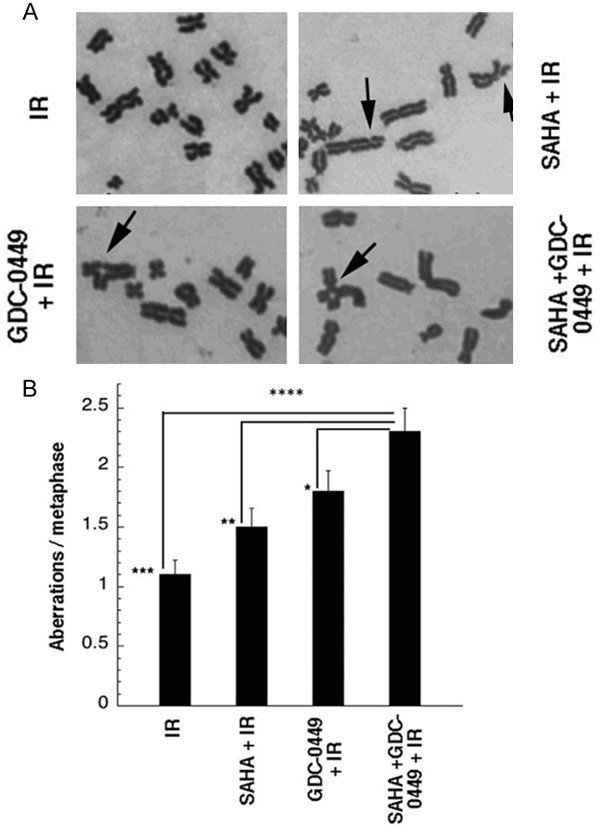
Enhanced formation of radiation induced chromosomal aberrations by SAHA and GDC-0449 in H1299 cell line. A. Representative chromosome aberrations induced by combining SAHA, GDC-0449, and SAHA + GDC-0449 with 1 Gy IR. B. The combination of SAHA + GDC-0449 significantly increased formation of chromosomal aberrations in comparison to all other treatment groups. Arrows, chromosomal aberrations. *P < 0.05.
These radiation and HR assays taken together demonstrate that combined targeting of HDACs and HH signaling enhance radiation sensitization in vitro. This was associated with cooperative repression of HR and down-regulation of Rad51, which is essential for HR-mediated DNA repair. In addition, there was a corresponding increase of large γ-H2AX and 53BP1 foci, which are markers of increased DNA DSBs, when SAHA and GDC-0449 were combined with 2 Gy. This data provided us with an impetus to evaluate the SAHA and GDC-0449 in vivo with respect to tumor growth delay.
Enhanced anti-tumor effect of SAHA and GDC-0449 in mouse tumor xenografts
To assess potential clinical efficacy of combined HDAC and HH pathway inhibition, the effects of SAHA and GDC-0449 were evaluated in SqCC/Y1 mouse tumor xenografts (Figure 6). As single agents, SAHA had modest growth delay while GDC-0449 had no impact on tumor growth in comparison to controls (Figure 7A). When SAHA and GDC-0449 combined, there was a more pronounced tumor growth delay than either agent alone. This in turn, resulted in prolonged survival of mice that received SAHA and GDC-0449 (Figure 7B).
Figure 7.
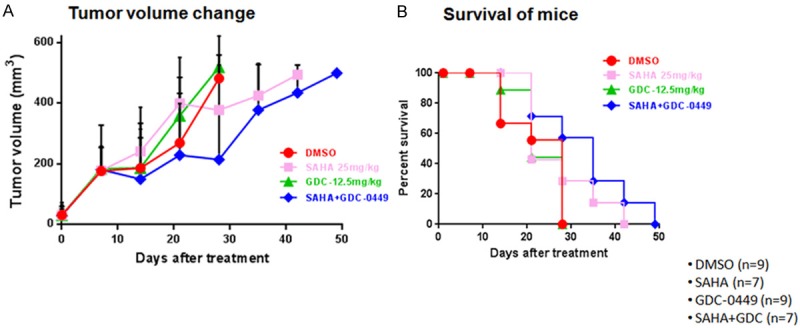
In vivo effect of SAHA and GDC-0449 on the SqCC/Y1 cell line. A. Tumor growth delay with SAHA, GDC-0449, and SAHA + GDC-0449. B. Mouse survival associated with single and combined treatment of SAHA and GDC-0449.
Discussion
Advanced aerodigestive tract malignancies are classically resistant to chemotherapy and radiation therapy, and we have presented translational data to support combined HDAC and HH pathway inhibition as a strategy to improve therapeutic responses for these cancers. We have found that combined targeting of HDACs with SAHA and Smo by GDC-0449 not only synergistically suppressed cellular proliferation in multiple aerodigestive cancer cell lines, but also enhanced suppression of HR. The purpose of this translational study is to provide rationale to evaluate the effect of combined HDAC and HH pathway inhibition in patients with advanced solid tumors.
A key finding in our study was that SAHA and GDC-0449 cooperatively suppressed cell cycle progression associated with the induction of the cyclin-dependant kinase inhibitor p21waf. Induction of p21waf in a p53-independent fashion by HDAC inhibitors in cancer cells is a well-established phenomenon [36], but our data suggests that the HH pathway represents a mechanism of resistance to HDAC inhibitor induced p21waf expression. Our findings are consistent with previous data in pancreatic cancer showing that SAHA and the Smo antagonist SANT-1 synergistically induce p21waf expression in the pancreatic cancer cell lines Panc-1 and BxPC-3. It is notable that the phenomenon occurs in the cell lines SqCC/Y1, Panc-1, and BxPC-3, where p53 expression is absent [37,38]. Dysregulation of p53 expression is one of the most common findings in cancer [39], and we have shown that targeting HDACs and Smo can induce p21waf expression an cell cycle arrest in a cooperative manner independent of p53 signaling.
Our findings are also in line with emerging data showing epigenetic modulation by HDACs is a fundamental regulator of the HH pathway [40-42]. The mechanistic basis for this interaction in medulloblastoma, as reported by Canettieri et al. [20], was shown to be mediated by HDAC1-induced activation of Gli-1 and Gli-2 transcriptional activity. Additionally, it was shown that the E3 ubiquitin ligase complex negatively regulates the HH pathway by promoting degradation of HDAC1 [20]. Thus, our strategy to chemically inhibit HDACs with SAHA was hypothesized to have a similar suppressive effect on HH signaling. Interestingly, we have shown that SAHA induced expression of Gli-1 and Gli-2, but that GDC-0449 could abrogate this up-regulation, providing insight into HH pathway up-regulation as a mechanism of resistance to HDAC inhibition. Moreover, we have shown a cooperative suppression of the Ptc-1 and Smo expression by SAHA and GDC-0449 showing that the combination of HDAC and Smo inhibition may overcome resistance to HH pathway activity. Further evaluation of the mechanistic basis for the interaction between HDACs and HH signaling may reveal more fundamental mechanisms of therapeutic resistance.
With respect to radiation sensitization in a combined modality strategy, in addition to accumulating cells in G1 by reducing cell population in radioresistant S/G2 phase, SAHA and GDC-0449 cooperatively regulated Rad51 and HR. While SAHA has been established to have a radiation sensitization effect through the suppression of Rad51 expression and HR [3,35], the role of HH signaling in HR and DNA repair is not well understood. We present novel evidence that inhibition of the HH pathway with GDC-0449 down-regulates Rad51 in a dose-dependent fashion. Moreover, we have shown that SAHA and GDC-0449 cooperatively down-regulate Rad51, suggesting non-redundant regulation of Rad51 by HDACs and HH signaling. Since Rad51 is essential for recruitment of repair proteins to sites of DNA DSBs during HR [43], its cooperative down-regulation by SAHA and GDC-0449 provides a potential basis for the pronounced enhancement of DNA DSBs as evidenced by the induction of γ-H2AX and 53BP1 foci formation when combined with IR.
There are a number of potential future directions of investigation identified in this study that warrant mention. The potential regulation of Rad51 signaling by the HH pathway is novel, and our ongoing work will aim to clarify the basis for this interaction. Additionally, while there is a pronounced suppression of colony formation and radiation sensitization by the combination of SAHA, GDC-0449, and IR, understanding the underlying mechanism of cell death may yield further insights into the therapeutic applications of this strategy. Another direction for future study would be further evaluation p21waf transcriptional regulation by HDACs and the HH pathway. As the cooperative induction of p21waf has now been observed in response to combined inhibition of HDACs and the HH pathway in multiple tumor cell lines, this mechanism of cell cycle arrest may represent an opportunity to improve tumor responses in many solid cancer histologies.
Clinical data already exists for combining radiation therapy with SAHA for whole brain radiotherapy, where the recommended dose of SAHA was 300 mg daily in phase I clinical trial [6]. A phase I clinical trial will be needed to address the safety profile of combined HDAC and Smo inhibition in humans, with a separate phase I trial to assess toxicity with the addition of concurrent radiation therapy. We anticipate that aerodigestive tract tumors will benefit from this combination, and we foresee locally recurrent HNC re-irradiation as an appropriate setting to test this concept since there are few good treatment options for these patients. Given the activity of this combination in multiple tumor cell lines from a variety of primary tumor histologies, we speculate that combined HDAC and Smo inhibition has the potential to expand the therapeutic application for agents such as SAHA and GDC-0449 to new disease sites.
In summary, we present pre-clinical data showing that combined targeting of HDACs and HH pathway with SAHA and GDC-0449 respectively, cooperatively suppress tumor grown and suppress HR in aerodigestive cancers.
Acknowledgements
We express our gratitude to Dr. Michael Story, Ph.D., Dr. Asaithamby Aroumougame, Ph.D., and Dr. Guillermo Palchik, Ph.D. for their input and discussion in the production of this manuscript. S.G.C. is a resident in the Department of Radiation Oncology at UT Southwestern Medical Center. The contributions of J.S.Y., S.G.C., and H.P. were supported by a start-up grant from the Department of Radiation Oncology at the University of Texas at Southwestern Medical Center. The contributions of T.K.P. and N.H. were supported by the National Institutes of Health grants R01 CA129537, R01 CA154320 and GM109768. J.S.Y. would like to acknowledge the Anchorage and Valley Radiation Centers for use of book and travel funds to finance the publication fee for this manuscript.
Disclosure of conflict of interest
The authors have no conflicts of interest to disclose.
Abbreviations
- HDAC
histone deacetylases
- HH
hedgehog
- Smo
smoothened
- Ptc-1
patched-1
- RT-PCR
real time-polymerase chain reaction
- SAHA
suberoylanilide hydroxamic acid
- Gy
Gray or joules/kilogram
- Gli
glioma associated oncogene
- HR
homologous recombination
- GFP
green fluorescent protein
- DSB
DNA double strand break
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, Rassool FV. HDAC inhibitors: roles of DNA damage and repair. Adv Cancer Res. 2012;116:87–129. doi: 10.1016/B978-0-12-394387-3.00003-3. [DOI] [PubMed] [Google Scholar]
- 4.Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. 2006;5:1967–1974. doi: 10.1158/1535-7163.MCT-06-0022. [DOI] [PubMed] [Google Scholar]
- 5.Mueller S, Yang X, Sottero TL, Gragg A, Prasad G, Polley MY, Weiss WA, Matthay KK, Davidoff AM, DuBois SG, Haas-Kogan DA. Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: efficacy and underlying mechanisms. Cancer Lett. 2011;306:223–229. doi: 10.1016/j.canlet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi W, Lawrence YR, Choy H, Werner-Wasik M, Andrews DW, Evans JJ, Judy KD, Farrell CJ, Moshel Y, Berger AC, Bar-Ad V, Dicker AP. Vorinostat as a radiosensitizer for brain metastasis: a phase I clinical trial. J Neurooncol. 2014;118:313–9. doi: 10.1007/s11060-014-1433-2. [DOI] [PubMed] [Google Scholar]
- 7.Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, Abrahamsen TW, Flatmark K. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459–464. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 8.Reguart N, Rosell R, Cardenal F, Cardona AF, Isla D, Palmero R, Moran T, Rolfo C, Pallares MC, Insa A, Carcereny E, Majem M, De Castro J, Queralt C, Molina MA, Taron M. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer. 2014;84:161–167. doi: 10.1016/j.lungcan.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 9.Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoffski P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26:483–488. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 10.Blumenschein GR Jr, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C, Frankel SR. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26:81–87. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 11.Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 12.Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805:181–208. doi: 10.1016/j.bbcan.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 15.Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang ZJ, Dutton JW, Wang F, Wechsler-Reya RJ. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23:157–170. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 17.Chung CH, Dignam JJ, Hammond ME, Klimowicz AC, Petrillo SK, Magliocco A, Jordan R, Trotti A, Spencer S, Cooper JS, Le QT, Ang KK. Glioma-associated oncogene family zinc finger 1 expression and metastasis in patients with head and neck squamous cell carcinoma treated with radiation therapy (RTOG 9003) J. Clin. Oncol. 2011;29:1326–1334. doi: 10.1200/JCO.2010.32.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey HM, Lum BL, Darbonne WC, Marsters JC Jr, de Sauvage FJ, Low JA. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 19.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, Pelloni M, De Simone G, Pedone EM, Gallinari P, Giorgi A, Steinkuhler C, Vitagliano L, Pedone C, Schinin ME, Screpanti I, Gulino A. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–142. doi: 10.1038/ncb2013. [DOI] [PubMed] [Google Scholar]
- 21.Chun SG, Zhou W, Yee NS. Combined targeting of histone deacetylases and hedgehog signaling enhances cytoxicity in pancreatic cancer. Cancer Biol Ther. 2009;8:1328–1339. doi: 10.4161/cbt.8.14.8633. [DOI] [PubMed] [Google Scholar]
- 22.Freeman JW, Wang Y, Giles FJ. Epigenetic modulation and attacking the hedgehog pathway: potentially synergistic therapeutic targets for pancreatic cancer. Cancer Biol Ther. 2009;8:1340–1342. doi: 10.4161/cbt.8.14.8756. [DOI] [PubMed] [Google Scholar]
- 23.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 24.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colleaux L, D’Auriol L, Galibert F, Dujon B. Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci U S A. 1988;85:6022–6026. doi: 10.1073/pnas.85.16.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartlerode A, Odate S, Shim I, Brown J, Scully R. Cell cycle-dependent induction of homologous recombination by a tightly regulated I-SceI fusion protein. PLoS One. 2011;6:e16501. doi: 10.1371/journal.pone.0016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, Gupta A, Wellinger RJ, Zhang J, Powell SN, Roti Roti JL, Lieberman HB, Pandita TK. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006;26:1850–1864. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta A, Hunt CR, Hegde ML, Chakraborty S, Udayakumar D, Horikoshi N, Singh M, Ramnarain DB, Hittelman WN, Namjoshi S, Asaithamby A, Hazra TK, Ludwig T, Pandita RK, Tyler JK, Pandita TK. MOF Phosphorylation by ATM Regulates 53BP1-Mediated Double-Strand Break Repair Pathway Choice. Cell Rep. 2014;8:177–189. doi: 10.1016/j.celrep.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CS, Pandita TK. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene. 2003;22:131–146. doi: 10.1038/sj.onc.1206063. [DOI] [PubMed] [Google Scholar]
- 30.Pandita TK. Effect of temperature variation on sister chromatid exchange frequency in cultured human lymphocytes. Hum Genet. 1983;63:189–190. doi: 10.1007/BF00291543. [DOI] [PubMed] [Google Scholar]
- 31.Kim SB, Pandita RK, Eskiocak U, Ly P, Kaisani A, Kumar R, Cornelius C, Wright WE, Pandita TK, Shay JW. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc Natl Acad Sci U S A. 2012;109:E2949–2955. doi: 10.1073/pnas.1207718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Briscoe J, Vincent JP. Hedgehog threads to spread. Nat Cell Biol. 2013;15:1265–1267. doi: 10.1038/ncb2878. [DOI] [PubMed] [Google Scholar]
- 33.Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004;32:3683–3688. doi: 10.1093/nar/gkh703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Wong P, Radany EH, Stark JM, Laulier C, Wong JY. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol Cancer Res. 2012;10:1052–1064. doi: 10.1158/1541-7786.MCR-11-0587. [DOI] [PubMed] [Google Scholar]
- 36.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 37.Reiss M, Brash DE, Munoz-Antonia T, Simon JA, Ziegler A, Vellucci VF, Zhou ZL. Status of the p53 tumor suppressor gene in human squamous carcinoma cell lines. Oncol Res. 1992;4:349–357. [PubMed] [Google Scholar]
- 38.Moore PS, Sipos B, Orlandini S, Sorio C, Real FX, Lemoine NR, Gress T, Bassi C, Kloppel G, Kalthoff H, Ungefroren H, Lohr M, Scarpa A. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001;439:798–802. doi: 10.1007/s004280100474. [DOI] [PubMed] [Google Scholar]
- 39.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 40.Coni S, Antonucci L, D’Amico D, Di Magno L, Infante P, De Smaele E, Giannini G, Di Marcotullio L, Screpanti I, Gulino A, Canettieri G. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS One. 2013;8:e65718. doi: 10.1371/journal.pone.0065718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Smaele E, Di Marcotullio L, Moretti M, Pelloni M, Occhione MA, Infante P, Cucchi D, Greco A, Pietrosanti L, Todorovic J, Coni S, Canettieri G, Ferretti E, Bei R, Maroder M, Screpanti I, Gulino A. Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia. 2011;13:374–385. doi: 10.1593/neo.101630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gulino A, Di Marcotullio L, Canettieri G, De Smaele E, Screpanti I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam Horm. 2012;88:211–227. doi: 10.1016/B978-0-12-394622-5.00009-2. [DOI] [PubMed] [Google Scholar]
- 43.Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]