

Abstract
Cervical cancer is a common and devastating female cancer worldwide. The etiology of cervical cancer has been largely attributed to human papillomavirus (HPV) infection and activation of the P13K/AKT/mTOR (mammalian target of rapamycin) pathway. However, the limited HPV-directed therapy, as well as therapeutic approach targeting P13K/AKT/mTOR pathway, has not yet been established or effective. A deeper understanding of cervical carcinogenesis and finding of novel candidate molecules for cervical cancer therapeutics is largely warranted. The unconventional prefoldin RPB5 interactor (URI or URI1), a known transcription factor involving the TOR signaling pathway, has recently been implicated a role in multiple tumorigenesis. We recently reported significant upregulation of URI in precancerous cervical intra-epithelial neoplasia (CIN) and invasive cervical cancer, suggesting its role in cervical carcinogenesis. However, the effect and underlying mechanism of URI in cervical cancer development have never been elucidated. Here, we aimed to investigate the in vitro effect of URI on cervical cancer using two cervical cancer cell lines CaSki and C33A, which are HPV-positive and HPV-negative respectively. We have shown that forced over-expression of URI in C33A and CaSki cells markedly promoted cell growth, while down-regulation of URI mediated by siRNA inhibited cell proliferation. We have found that URI over-expression enhanced resistance of cervical cancer cells to cisplatin. In contrast, knockdown of URI promoted apoptosis by influencing cell response to cisplatin, supporting URI as an oncogenic protein for cervical cancer cells. We have also shown that URI promoted the migration and invasive capacity of cervical cancer cells by up-regulation of Vimentin, a mesenchymal cell migration marker relating to the epithelial-mesenchymal transition (EMT) program. Our data support an important function of URI in the biological behavior of cervical cancer cells and provide novel mechanistic insights into the role of URI in cervical cancer progression and possibly, metastasis.
Keywords: URI, C33A and CaSki cell lines, cisplatin, cervical cancer invasive capacity, vimentin, epithelial-mesenchymal transition
Introduction
Cervical cancer is the third most commonly diagnosed female cancer [1]. It is also the fourth leading cause of cancer death in females worldwide, with more than 85% of the cases and deaths occur in developing countries [1]. Therapeutic resistance is a common phenomenon in cervical cancer, especially in patients with advanced, recurrent, and metastatic disease. These are also the main reasons causing cervical cancer death [2]. The etiology of cervical cancer has been largely attributed to infection of human papillomavirus (HPV). However, HPV has been associated with multiple cancer types, while not all females with HPV infection develop cervical cancer. There are also patients with cervical cancer that are HPV-negative. In addition, the limited HPV-directed therapy to date on cervical cancer has not been very effective [2]. These findings suggest that there exist additional biomarkers or molecular targets that impact cervical carcinogenesis.
One of the targets involving cervical cancer tumorigenesis could be the oncogenic protein URI, the unconventional prefoldin RPB5 interactor. URI, also called URI1 or RMP, was originally identified as a transcriptional repressor that interacts with RPB5, a subunit of all three RNA polymerases [3,4]. Interestingly, URI has been shown to participate in the TOR (Target of Rapamycin) signaling pathway, suggesting its potential function during cancer development and therapy [5]. Indeed, we have previously shown that URI regulates cell apoptosis and is required for the proliferation of hepatocellular carcinoma (HCC) [6]. URI over-expression was detected both in ovarian cancer cell lines and in ovarian carcinomas and has been associated with its therapeutic resistance [7]. URI was also shown to be essential for androgen receptor signaling, a pathway involving prostate cancer progression [8]. In addition to solid tumors, a recent study has indicated that URI is involved in tumorigenesis of multiple myeloma and is associated with its chemotherapeutic resistance, possibly by activation of the IL-6/STAT3 pathway [9]. Together, the above findings demonstrated that URI participate in multiple oncogenesis. Intriguingly, our recent study has shown that URI is up-regulated in pre-cancerous cervical intra-epithelial neoplasias (CINs) and cervical cancer, suggesting that URI may play a role in cervical carcinogenesis [10]. However, the effect and potential mechanism of URI regulation of cervical cancer cell survival, growth and invasion has never been elucidated.
In this study, we examined the in vitro function of URI on cervical cancer development and its role in therapeutic response to cisplatin using two cervical cancer cell lines. These cell lines represent HPV-positive (CaSki) and HPV-ne-gative (C33A) cells respectively [11]. We detected URI expression in both cell lines without significant difference. By manipulating URI expression using siRNA and gene transfection approaches, we have shown that URI promotes proliferation, migration, and the invasion capacity of cervical cancer cells and may enhance their resistance to cisplatin. The underlying mechanism of URI and cell invasion and its potential as a therapeutic target for cervical cancer were also analyzed.
Materials and methods
Cell culture
The human cervical cancer cell lines C33A (ATCC #HTB-31) and CaSki (ATCC #CRL-1550) were purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institute of Biological Sciences Chinese Academy of Sciences (Shanghai, China). C33A cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Corning, USA) and CaSki cells were grown in RPMI-1640 (Corning, USA). All cells were supplemented with 10% fetal bovine serum (Gibco, New Zealand) and 1% penicillin/streptomycin (Invitrogen) and cultured at 37°C in a humidified incubator containing 5% CO2.
Cell transfection
To knockdown URI expression, three small interfering RNA (siRNA) sequences targeting URI: siRNA-A- rArGrArArGrGrUrArGrArUrArArUrGrArCrUrArUrArArUGC; siRNA-B - rGrGrArArGrArArArGrCrUrArCrArUrGrArArUrUrArArUTG; siRNA-C - rGrArArCrUrArGrArGrArGrArCrArGrGrArArGrArArUrUGC and scramble control sequence: Scrambled-rCrGrUrUrArArUrCrGrCrGrUrArUrArArUrArCrGrCrGrUAT were purchased from Origene Technologies, Inc. Transfection of siRNAs and the scrambled control duplex in C33A and CaSki cells was performed using siTran 1.0 reagent (Origene) according to the manufacturer’s instructions. To over-express URI, the URI expression plasmid pCMV6-URI and its control plasmid, pCMV6-entry (Origene) were transfected into cells in Opti-MEM (Invitrogen, Carlsbad, CA) using the Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). Untransfected cells were used as blank control.
Total RNA extraction, cDNA synthesis, RT-PCR and qRT-PCR
After 24 h transfection, total RNAs were extracted from C33A and CaSki cells using Trizol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized by reverse transcription using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) following manufacturer provided protocol. RT-PCR was performed to examine expression of selected genes using standard protocol. PCR products were subjected to electrophoresis in 1.2% agarose gel and stained with ethidium bromide. All experiments were performed in triplicate and the data shown are representative. qRT-PCR was performed to quantify gene expression using the Bio-Rad CFXTM96 Detection System and SYBR Green real-time PCR master mix (Bio-Rad, Hercules, CA). The mean threshold cycle number (CT values) of target genes was normalized to endogenous GAPDH and calculated using the 2-ΔΔCt method. The relative mRNA levels of treated samples were compared to that of control samples, which were arbitrarily set to 1 [12,13]. The specific primer sequences of selected genes GAPDH, GAPDH1, URI, E-cadherin, Snail, and Vimentin are shown in Table 1.
Table 1.
Specific primer sequences
| Gene | Forward (5’-3’) | Reverse (5’-3’) | Amplicon |
|---|---|---|---|
| GAPDH1 | CATGAGAAGTATGACAACAGCCT | AGTCCTTCCACGATACCAAAGT | 113 bp |
| URI | TTTGCAGAAAATGAGCGATG | GCAATTCGGTGTTTTGCTTT | 93 bp |
| GAPDH | TCTCTGCTCCTCCTGTTCGA | GCGCCCAATACGACCAAATC | 122 bp |
| E-cadherin | GGCCTTAGAGGTGGGTGACT | GGCTGTGCCTTCCTACAGAC | 132 bp |
| Snail | GCCCCACAGGACTTTGATGA | CAAAAACCCACGCAGACAGG | 153 bp |
| Vimentin | GGACCAGCTAACCAACGACA | AAGGTCAAGACGTGCCAGAG | 178 bp |
Western blot analysis
After 48 h transfection, cells were washed with cold phosphate-buffered saline (PBS) collected and lysed in RIPA buffer (Beyotime Biotechnology, CA, China) containing protease inhibitor cocktail (Kangchen, Shanghai, China). Cells were placed on ice for 30 min, and then centrifuged at 14000 rpm for 10 min to remove cellular debris. The supernatant was collected and the protein concentration was determined by BCA-assay (Eppendorf, Hamburg, Germany). 50 μg of total protein were subjected to SDS-PAGE and subsequently transferred onto Immobilon-P membranes (Millipore, Billerica, USA), which were then blocked with 5% nonfat milk for 1 h under constant shaking. These membranes were then treated with either mouse anti-human URI antibody or rabbit anti-human β-actin antibody (Santa Cruz Biotechnology, CA, USA) at 4°C overnight. After washing with TBST containing 0.1% Tween 20 three times, the membranes were incubated with horseradish peroxidase conjugated goat anti-mouse IgG antibody and goat anti-rabbit IgG antibody (Fcmacs Biotechnology, CA, China) at room temperature for 1 h followed by detection using an enhanced chemiluminescence system (Minichemi, China). Anti-actin was used to ensure equal loading and scanned images of the X-ray films were subjected to densitometry analysis. Western blot assay was performed three times and data from representing one set of experiment was shown.
Cell viability assay
Cell viability was determined using a cell counting kit-8 (CCK-8) at four time points (0, 1, 2, and 3 days respectively) in accordance with the manufacturer’s protocol (Vazyme Biotech, Nanjing, China). After 48 h transfection, cells were seeded at 5000 per well in 96-well plates. 1/10 volume of CCK-8 was then added to each well and incubated for an additional 2 h at 37°C. The optical density (OD) was measured at 450 nm wavelength with a microplate reader (Bio-Rad Model 680, Richmond, CA, USA). Cells from each group were added to 6 wells and experiment was performed in triplicate. CCK-8 assay was used to test the effects of cisplatin intervention on growth and proliferation of cervical cancer cells at different concentrations. The inhibitory concentrations of 50% proliferation (IC50) of cisplatin were calculated by GraphPad Prism software version 5.0. The experiment was performed in triplicate.
Wound healing assay
For wound-healing migration assay, C33A and CaSki cells (5×105) were transfected with or without pCMV6-entry/pCMV6-URI and seeded on 6-well plates. After 48 hours of transfection, the monolayer cells (~90% confluence) were scratched a straight line with a sterile 100-ml-pipette tip. The floating cells were removed with PBS and cultured again in DMEM and RPMI-1640 medium containing 1% FBS. The migrated distance of C33A and CaSki cells was monitored and imaged at four different time points (0 h, 6 h, 12 h and 24 h) under a microscope (100×). The experiment was conducted independently in triplicate.
Transwell cell migration assay
The migration ability of cervical cancer cells was assessed by calculating the cells that passed through a polycarbonate membrane (8-mm pore size) using a 24-well transwell chamber (Corning Costar, New York, USA). After 48 hours of transfection, C33A and CaSki cells (1×105 cells) were suspended in 150 µl serum-free DMEM and RPMI-1640 medium and cultured in the upper transwell chamber for 24 h at 37°C, the lower chamber was filled with 600 µl of DMEM and RPMI-1640 medium supplemented with 10% FBS. The non-migrated cells attached to the upper surface of the membrane were removed with a sterile cotton swab, followed by washing with PBS. The migration cells on the bottom surface of membrane were fixed with 4% paraformaldehyde solution for 30 minutes, washed with PBS and stained with 0.5% crystal violet for 25 min at room temperature. The numbers of cells were calculated under a microscope in nine random fields (200×). The experiment was conducted three times.
Apoptosis assay
Apoptosis assay was performed using the FITC Annexin V apoptosis detection kit (BD PharmingenTM, CA, USA) according to manufacturer suggested protocol. In brief, at 48 h after transfection, the cells were collected and washed twice with cold PBS, resuspended in 500 µl 1x Annexin V binding buffer at a concentration of 1×106 cells/ml. 100 µl of the cell suspension (1×105 cells) was then transferred to a 5 ml culture tube and add 5 µl each of Annexin V-FITC and propidium iodide (PI) with gentle vortex. The cells were kept in dark for 15 min at RT (25°C) before adding 400 µl of 1x binging buffer to each tube and subjected to flow cytometry analysis (BD AccuriTM C6 system) within 1 h. Meanwhile, after transfection for 24 hours, C33A and CaSki cells were also treated with different concentrations of cisplatin for additional 48 h and subjected to similar apoptosis assay. All these experiments were repeated three times.
Statistical analysis
All data were presented as the mean ± SEM. Differences in different groups were analyzed by one-way ANOVA using the GraphPad Prism software version 5.0. P<0.05 was considered statistically significant.
Results
URI expression and transfection in cervical cancer cells
We have examined URI expression in cervical cancer cell lines, CaSki and C33A, which are HPV positive and negative respectively [11]. The results showed that URI mRNA transcript is expressed both in C33A and in CaSki cancer cells by RT-PCR and qRT-PCR analysis of RNAs derived from these cells (Figure 1A, 1B, untransfected cells). We also detected URI protein in both cell lines as confirmed by western blot analysis (Figure 1C, untransfected cells). To explore the in vitro function of URI, we have performed transient transfection in above cell lines using URI expression plasmid (pCMV6-URI), the vector only (pCMV6-entry), and no transfection control. Both URI mRNA transcript and protein showed significant upregulation in cells transfected with pCMV6-URI compared to that with pCMV6-entry or untransfected cells (Figure 1A-C, left two panels). We have also performed URI siRNA gene knock-down experiment in above two cell lines. Three candidate URI siRNA sequences (siRNA-A, -B, and -C) and a scrambled control sequence were used for the transfection. All three siRNA sequences lead to reduced URI mRNA and protein expression compared to the untransfected and scrambled controls, with siRNA-A sequence showed the strongest interfering effect on URI (Figure 1A-C, right two panels). Together, the results demonstrated URI expression in C33A and CaSki cervical cancer cell lines. Transfection of pCMV6-URI resulted in increased URI expression while URI siRNAs successfully knocked down URI expression in C33A and CaSki cells.
Figure 1.

URI expression in cervical cancer cells. A. The expression level of URI mRNA in transfected cervical cells was examined by RT-PCR. GAPDH served as a loading control. B. Relative mRNA transcripts of URI were examined using qRT-PCR. C. Western blot analysis to confirm the siRNA mediated knockdown of URI and the pCMV6-URI mediated overexpression of URI in C33A and CaSki cells. β-actin was used as internal control. left two panels of A-C: URI over-expression; right two panels of A-C: URI siRNA transfection. Data was expressed as mean ± SEM of three independent experiments, **p<0.01.
URI promotes proliferation of cervical cancer cells
We have performed above transient transfection studies to enhance or knock-down URI expression in C33A and CaSki cells followed by cell proliferation analysis. CCK-8 assay was used to determine the effect of URI on cell proliferation. Our results showed that over-expressing URI by transient transfection of pCMV6-URI enhanced cell proliferation significantly compared to the vector and untransfected controls (Figure 2A, 2B). Meanwhile, knockdown of URI by siRNA-A in C33A and CaSki cells after transfection for two days and for prolonged cell culture leads to markedly decreased cell proliferation compared to that of scrambled or untransfected controls (Figure 2C, 2D). These results together suggest that URI promotes proliferation of cervical cancer cells.
Figure 2.
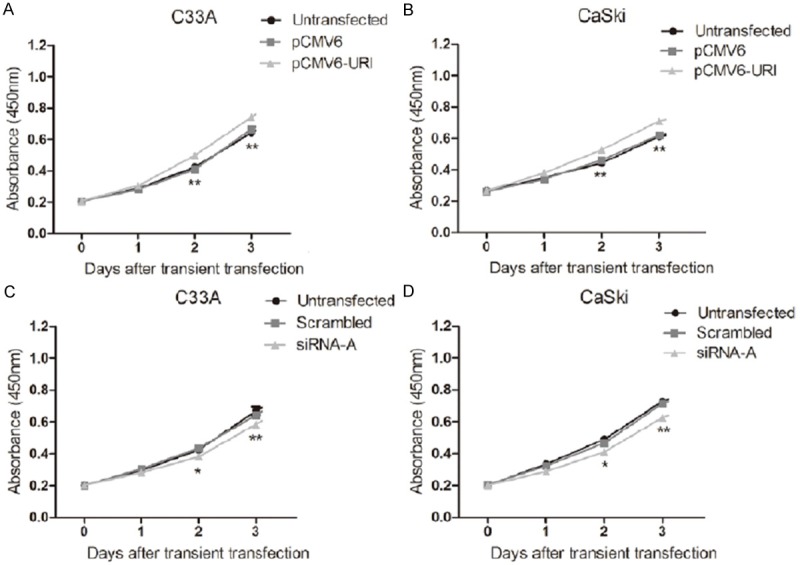
URI effects on cell proliferation of cervical cancer cells. A, B. Cells were transfected with pCMV6-URI, pCMV6-entry, and untransfected control. The viability was assessed after 48 h transfection using a CCK-8 assay at four time points (0, 1, 2, and 3 days respectively). Up-regulation of URI increased cell viability of C33A and CaSki cells. C, D. Cells were transfected with URI siRNA-A, Knockdown of URI significantly inhibit cell proliferation in C33A and CaSki cells. Analyses were performed in triplicate and the results are expressed as mean ± SEM, *p<0.05, **p<0.01.
Effect of URI on cervical cancer cell migration and invasion
To determine if URI plays a role in cervical cancer metastasis, we examined the influence of URI on migration and invasion of C33A and CaSki cancer cells by wound healing and transwell assay. The wound healing cell migration assay has shown that over-expressing URI in C33A and CaSki cells by transfection of pCMV6-URI enhanced their healing process (closing of the scratched wound) compared to untransfected cells or cells transfected with the pCMV6-entry empty vector (Figure 3A, 3B). The CaSki cells showed slight faster capacity of cell migration than that of the C33A cells. On the contrary, URI siRNA-A transfected C33A and CaSki cells delayed the healing process compared to untransfected cells or cells transfected with scrambled control sequence, while the healing ability may differ between C33A and CaSki cells (Figure 3C, 3D). For transwell assay, we detected significantly increased number of C33A and CaSki cells that passed through the polycarbonate membrane in the cells transfected with pCMV6-URI compared to control groups (Figure 4A), whereas siRNA-A knock-down of URI in C33A and CaSki cells significantly decreased the number of cells passing through the well (Figure 4B). These results provided evidence that URI enhances the migration and invasion capacity of cervical cancer cells.
Figure 3.
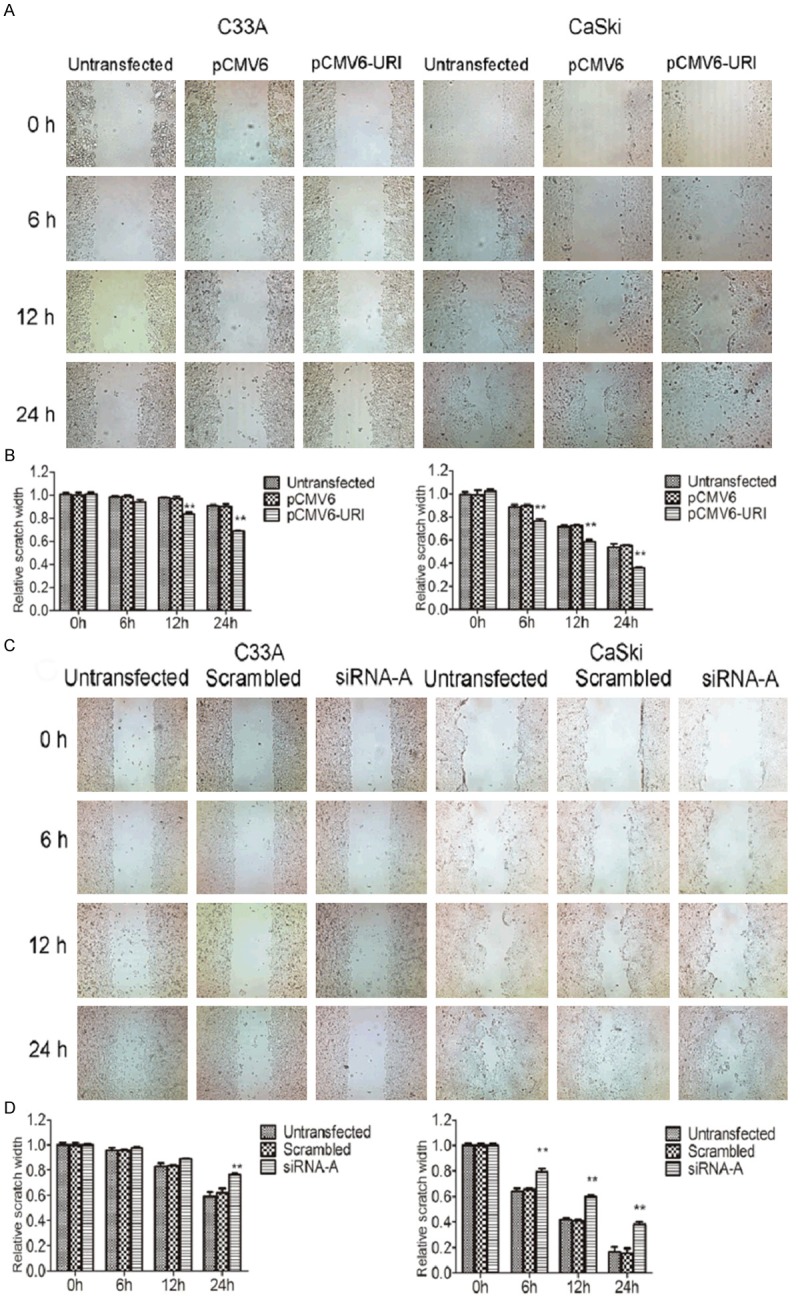
Effect of URI on migration of cervical cancer cells by wound healing assay. A. Scratch wounds were made on confluent monolayer cultures after 48 h of transfection. Images of wound group were taken at 0, 6, 12 and 24 h after wound (upper panel). Overexpression of URI presented a faster wound recovery compared to the control groups at 12 h and 24 h after scratching the line. B. Bar graph represented the relative migration distance. C. Knockdown of URI1 exhibited a slower wound recovery compared to the control groups at 24 h after the scratching. D. Bar graph represented the relative migration distance of cells. The results presented represent mean of three independent experiments. Data was expressed as mean ± SEM of three separated experiments, **P<0.01.
Figure 4.

Effect of URI on migration/invasion of cervical cancer cells by transwell assay. At 48 h after transfection, cells were reseeded in the upper Transwell chamber for 24 h and then stained with crystal violet. A. Up-regulation of URI significantly increased the migrated ability of C33A and CaSki cells. The number of pCMV6-URI cells on the filter surface was larger than untransfected and pCMV6 cells. B. Down-regulation of URI inhibits cell migration of C33A and CaSki cells. All the experiments were performed in triplicate. URI siRNA group contained significantly less migrated cells than the control groups. The mean value of invaded cells was shown in right panel. Data was mean ± SEM, *p<0.05, **P<0.01.
URI and EMT-related mark gene expression in cervical cancer cells
To elucidate the potential mechanism of URI affecting cervical cancer cell migration and invasion, we selectively examined expression of genes E-Cadherin, Snail, and Vimentin in C33A and CaSki cells. These genes are known to be related to the epithelial-mensenchymal transition (EMT) that may be involved in cancer migration, invasion or metastasis of cervical cancer cells [14]. We examined the correlation of these genes with URI by manipulating URI expression in C33A and CaSki cells. The results showed that over-expression or depletion of URI does not significantly change the expression levels of E-Cadherin (Figure 5A) and Snail (Figure 5B) in both C33A and CaSki cells. However, compared with control groups, we detected significantly increased Vimentin mRNA levels in cells over-expressing URI, whereas knocking down of URI in cervical cancer cells leads to decreased Vimentin expression (Figure 5C), suggesting a role of URI in regulation of Vimentin, and therefore, a role in cervical cancer invasion or metastasis as previously described [15].
Figure 5.
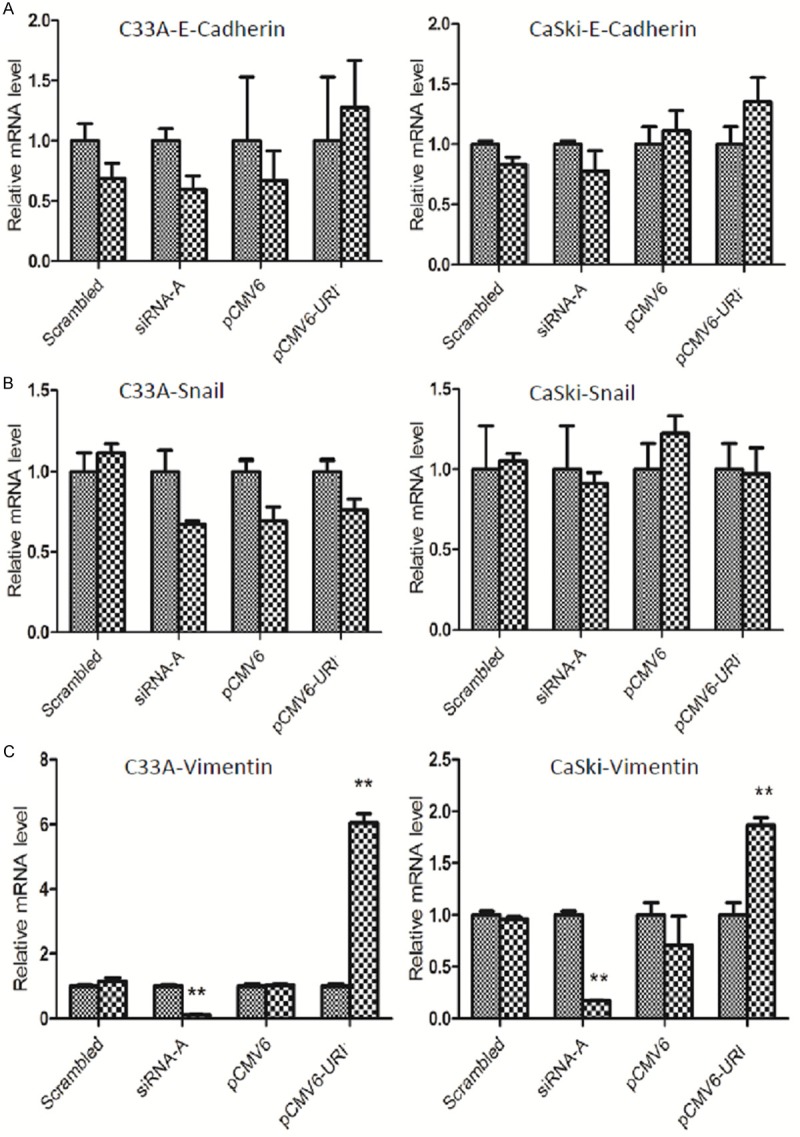
EMT related marker gene expression in URI-treated cervical cancer cells. E-cadherin, Snail and Vimentin mRNA expression by qRT-PCR in pCMV6-URI and siRNA-A transfected cells. E-Cadherin (A) and Snail (B) mRNA expression has no significant change between groups with altered URI expression in both C33A cells and CaSki cells. Vimentin (C) mRNA level was significantly increased in C33A and CaSki cells transfected with pCMV6-URI while URI knockdown in CaSki and C33A cells significantly down-regulated Vimentin expression compared to cells without URI treatment (set as 1 in graph), **P<0.01.
URI1 regulation of cisplatin response to cervical cancer cells
Cisplatin has been widely used for the treatment of various cancers. Cisplatin resistance is the main reason causing treatment failure and cancer death, including cervical cancer [16]. To examine whether URI plays a role in cisplatin resistance in cervical cancer, we measured the cell viability of C33A and CaSki cells after transfection with URI/URI siRNA and exposed to different concentration of cisplatin. Due to their different response to cisplatin, after transfection for 24 hours, C33A cells were treated with cisplatin ranging from 0 to 100 μM, while CaSki cells ranging from 0 to 50 μM, for 48 hours. The results showed that forced over-expression of URI in both C33A and CaSki cells dose-dependently increased their resistance to cisplatin (Figure 6A, 6B). The mean IC50 of C33A and CaSki cells transfected with pCMV6-URI was significantly higher than untransfected and pCMV6 vector transfected cells (Figure 6C). Meanwhile, down-regulation of URI by URI siRNA-A transfection in both C33A and CaSki cells enhanced their sensitivity to cisplatin, also in a dose-dependent manner (Figure 6D, 6E). The mean IC50 of URI siRNA-A transfected C33A and CaSki cells was significantly lower than that of untransfected cells and scrambled control (Figure 6F). Together, these results demonstrated that URI enhances cisplatin resistance to cervical cancer cells.
Figure 6.
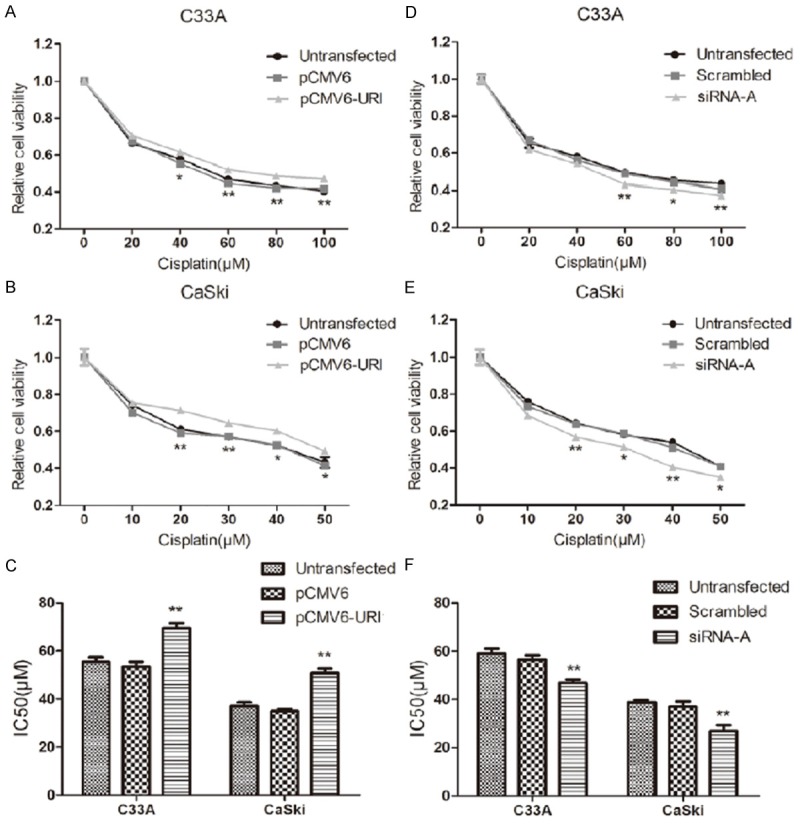
Effect of cisplatin on C33A and CaSki cell growth. At 24 hours after pCMV6-URI and URI siRNA-A transfection, C33A and Caski cells were exposed to different doses of cisplatin for 48 h. Cell viability was determined by CCK-8 assay at 450 nm. A and B. C33A and CaSki cells transfected with pCMV6-URI showed a cisplatin resistance. C. Graphic representation of the mean IC50 values of cisplatin in C33A and CaSki cells over-expressing URI. D and E. Knockdown of URI enhanced response of C33A and CaSki cells to cisplatin. F. Graphic representation of the mean IC50 values of cisplatin in C33A and CaSki cells knockdown of URI. Data was mean ± SEM, *p<0.05, **P<0.01.
Impact of URI expression on cervical cancer cell apoptosis
As cisplatin-induced DNA damage has been implicated a role in activation of both intrinsic and extrinsic apoptotic pathways [17,18], we next proceed to evaluate whether URI expression is associated with cisplatin-induced apoptosis in cervical cancer cells. After transfection for 24 hours with URI siRNA-A, C33A and CaSki cells were exposed to cisplatin treatment for 48 h and subjected to Annexin-V-FITC/propidium iodide labeling followed by flow cytometry analysis. The results showed that the total cell apoptosis rates of the URI siRNA transfected groups treated with cisplatin were significantly higher compared with corresponding negative control groups in both C33A and CaSki cells (Figure 7). We also performed similar apoptotic assay in C33A and CaSki cells by transiently over-expressing URI as described above. However, the proportion of apoptotic cells did not show significant difference between URI over-expression groups and that of control groups (data not shown). These data suggested that URI knockdown may promote apoptosis by enhancing cervical cancer cell response to cisplatin.
Figure 7.
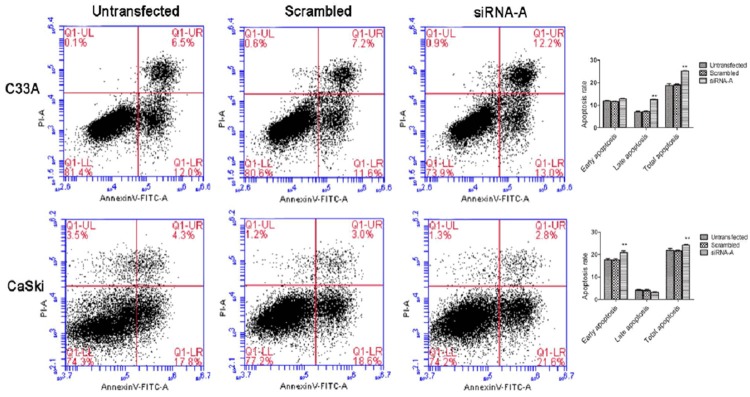
The impact of URI Knockdown on cell apoptosis induced by cisplatin. At 24 h after URI siRNA-A transfection, cells were exposed to cisplatin treatment for 48 h, and apoptosis assay was performed using the annexin V-FITC apoptosis detection kit. Cells in the right lower and right upper quadrants were considered as early and late apoptotic cells respectively. Cells down-regulation of URI have a higher apoptosis rate compared with control groups, **P<0.01.
Discussion
URI and HPV in cervical cancer
HPV infection has been demonstrated as the primary cause for cervical cancer [19]. Oncoproteins from high-risk HPV subtypes, HPV-16 and HPV-18, are known to play major roles in cervical cancer tumorigenesis, possibly through inactivation of RB1 and P53 signaling pathway [11,20-23]. Recently, we reported detection of upregulated URI expression in precancerous CIN and invasive cervical cancer compared to normal epithelial cells of the cervix [10]. Given the growing evidence of URI participating in multiple tumorigeneses [6-10], this finding suggests a role of URI in cervical cancer development. To explore whether URI associates with HPV and affect cervical cancer oncogenesis, we examined URI expression in two established HPV+(CaSki) and HPV-(C33A) cervical cancer cell lines. C33A (ATCC# HTB-31) is a known cervical cancer cell line that does not contain any HPV copies, while CaSki (ATCC #CRL-1550) contains hundreds of integrated HPV16 copies along with some HPV18-related sequences [11]. We detected similar levels of URI mRNA transcript and protein in C33A and CaSki cell lines as demonstrated by (q)RT-PCR and western blot analyses (Figure 1). We have performed URI in vitro gain- and loss-of-function studies in C33A and CaSki cells to determine the correlation between URI expression and the cancer cell biological features, including cell growth, proliferation, migration/invasion and apoptosis. While URI over-expression or depletion indeed influence the oncogenic behavior of C33A and CaSki cancer cells, no difference was noticed between these two cell lines upon URI treatment (Figures 2, 3, 4, 5, 6 and 7), suggesting that URI may have an impact on cervical cancer tumorigenesis independent of HPV or work indirectly with HPV.
Effect of URI on cervical cancer cell oncogenic property
URI was previously shown to be amplified and overexpressed in ovarian cancer cell lines and human ovarian carcinomas. URI also mediates resistance to cisplatin in ovarian cancer cells, and thus, considered an oncogene [7]. As to cervical cancer, we have recently detected increased URI expression in precancerous CIN and invasive cancer [10]. In our current study, we have shown that over-expression of URI in C33A and CaSki cervical cancer cells promote cell proliferation, while knockdown of URI led to inhibition of proliferation. In addition, we have found that up-regulated URI expression enhanced resistance of cervical cancer cells to cisplatin, while down-regulation of URI promoted apoptosis by enhancing cell response to cisplatin. These results correspond well with previous findings in ovarian cancer and support URI as an oncogenic protein in cervical cancer pathogenesis. We have further investigated URI’s function during cervical cancer oncogenesis by examining the migration ability of C33A and CaSki cells using wound healing and transwell cell migration assay. We have shown that cells with forced URI over-expression by transfection with pCMV6-URI migrated faster and were more invasive than that of vector and untransfected controls. Meanwhile, cells subjected to URI knockdown by siRNA-A transfection showed slower migration rate and were less aggressive. These results demonstrated that URI expression is associated with migration and invasion of cervical cancer cells, and possibly, plays a role in cervical cancer metastasis.
Potential mechanism of URI in cervical cancer invasion/metastasis
To explore the potential mechanism, we investigated the correlation of URI with epithelial-mesenchymal transition (EMT) program. EMT denotes a process by which epithelial cells lose epithelial characteristic features but acquire migratory and invasive properties to become mesenchymal cells [24]. Notably, EMT program has been implicated as an important molecular mechanism for cervical cancer metastasis [14]. Among the multiple molecules or factors, Snail has been recognized as a central transcription factor that controls the EMT program in cervical cancer by repressing E-cadherin gene expression [14,25,26]. It has also been shown that mesenchymal cells, characterized by up-regulation of N-cadherin and Vimentin, may acquire increased migratory and invasive potentials and promote metastasis during tumor progression [14]. Interestingly, we did not detect any expression changes of genes Snail and E-cadherin upon modulation of URI expression in C33A and CaSki cells. However, we detected significantly upregulated Vimentin mRNA transcript in cells over-expressing URI, while decreased Vimentin expression was observed in cells cells depleted of URI, suggesting a role of URI in regulation of Vimentin. Vimentin is a major member of the III intermediate filament (IF) protein family [27]. As a known marker of mesenchymal transition, Vimentin has been shown to be over-expressed in many cancers and has been associated with tumor metastasis [28-31]. Vimentin was previously detected only in invasive cervical carcinomas and lymph node metastases, but not in CIN III lesions, suggesting a clear association between Vimentin expression and metastatic progression of of cervical cancer [29]. Here, we surmise that URI enhances the migration and invasion of cervical cancer cells, possibly through modulation of Vimentin expression.
In summary
We have shown that modulation of URI expression in cervical cancer cells affects their oncogenic behavior, including cell proliferation, migration and invasion, as well as resistance to cisplatin. The correlation of URI with Vimentin expression in cervical cancer cells suggests a potential mechanism of URI in cervical cancer tumorigenesis or metastasis and potentially, as a therapeutic target. We notice that much remains to be determined regarding the role of URI in cervical cancer pathogenesis. For instance, HPV infection has been an established etiology for cervical cancer. Due to extensive work, we limited current research to only one of each HPV+ or HPV- cervical cancer cell lines and with transient in vitro studies. Further investigation using multiple HPV-related cervical cancer cell lines is warranted. URI may cause tumorigenesis through other mechanism such as DNA damage that has recently been described [32,33]. Also, the correlation of URI with activated P13K/AKT/mTOR pathway, a pathway that frequently occurs in metastatic and recurrent cervical carcinomas, remains elusive. However, to best of our knowledge, this study serves as a pioneer research and our findings of URI in relation to oncogenic properties of C33A and CaSki cells provide novel insights and possible novel therapeutic strategies toward cervical cancer.
Acknowledgements
We are grateful to Dr. Genbao Shao for his help in cell transfection studies. We also appreciate the assistance from Dr. Hai Qian for the Transwell assay. This work was supported by the 2014 Rush University Medical Center Pilot Project (Q.Z., J.G. Y.L.), the Innovation Program of Jiangsu Province (No. 480, 2013, Q.Z.), and the 2013 innovative training project for college students in Jiangsu Province (awarded to Y.C., J.D., J.G., and Q.Z.).
Disclosure of conflict of interest
All authors have no conflict of interest.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA CancerJ Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Duenas-Gonzalez A, Serrano-Olvera A, Cetina L, Coronel J. New molecular targets against cervical cancer. Int J Womens Health. 2014;6:1023–1031. doi: 10.2147/IJWH.S49471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorjsuren D, Lin Y, Wei W, Yamashita T, Nomura T, Hayashi N, Murakami S. RMP, a novel RNA polymerase II subunit 5-interacting protein, counteracts transactivation by hepatitis B virus X protein. Mol Cell Biol. 1998;18:7546–7555. doi: 10.1128/mcb.18.12.7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mita P, Savas JN, Ha S, Djouder N, Yates JR 3rd, Logan SK. Analysis of URI Nuclear Interaction with RPB5 and Components of the R2TP/Prefoldin-Like Complex. PLoS One. 2013;8:e63879. doi: 10.1371/journal.pone.0063879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gstaiger M, Luke B, Hess D, Oakeley EJ, Wirbelauer C, Blondel M, Vigneron M, Peter M, Krek W. Control of nutrient-sensitive transcription programs by the unconventional prefoldin URI. Science. 2003;302:1208–1212. doi: 10.1126/science.1088401. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Gu J, Zheng Q, Li M, Lian X, Miao J, Jiang J, Wei W. RPB5-mediating protein is required for the proliferation of hepatocellular carcinoma cells. J Biol Chem. 2011;286:11865–11874. doi: 10.1074/jbc.M110.136929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theurillat JP, Metzler SC, Henzi N, Djouder N, Helbling M, Zimmermann AK, Jacob F, Soltermann A, Caduff R, Heinzelmann-Schwarz V, Moch H, Krek W. URI is an oncogene amplified in ovarian cancer cells and is required for their survival. Cancer Cell. 2011;19:317–332. doi: 10.1016/j.ccr.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 8.Mita P, Savas JN, Djouder N, Yates JR 3rd, Ha S, Ruoff R, Schafler ED, Nwachukwu JC, Tanese N, Cowan NJ, Zavadil J, Garabedian MJ, Logan SK. Regulation of androgen receptor-mediated transcription by RPB5 binding protein URI/RMP. Mol Cell Biol. 2011;31:3639–3652. doi: 10.1128/MCB.05429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan JL, Zhang J, Dong LW, Fu WJ, Du J, Shi HG, Jiang H, Ye F, Xi H, Zhang CY, Hou J, Wang HY. URI regulates tumorigenicity and chemotherapeutic resistance of multiple myeloma by modulating IL-6 transcription. Cell Death Dis. 2014;5:e1126. doi: 10.1038/cddis.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu J, Li X, Liang Y, Qiao L, Ran D, Lu Y, Li X, Wei W, Zheng Q. Upregulation of URI/RMP gene expression in cervical cancer by high-throughput tissue microarray analysis. Int J Clin Exp Pathol. 2013;6:669–677. [PMC free article] [PubMed] [Google Scholar]
- 11.Wrede D, Tidy JA, Crook T, Lane D, Vousden KH. Expression of RB and p53 proteins in HPV-positive and HPV-negative cervical carcinoma cell lines. Mol Carcinog. 1991;4:171–175. doi: 10.1002/mc.2940040302. [DOI] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MY, Shen MR. Epithelial-mesenchymal transition in cervical carcinoma. Am J Transl Res. 2012;4:1–13. [PMC free article] [PubMed] [Google Scholar]
- 15.Gilles C, Polette M, Piette J, Delvigne AC, Thompson EW, Foidart JW, Birembaut P. Vimentin expression in cervical carcinomas: association with invasive and migratory potential. J Pathol. 1996;180:175–180. doi: 10.1002/(SICI)1096-9896(199610)180:2<175::AID-PATH630>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 16.Moore DH. Chemotherapy for recurrent cervical carcinoma. Curr Opin Oncol. 2006;18:516–519. doi: 10.1097/01.cco.0000239893.21161.51. [DOI] [PubMed] [Google Scholar]
- 17.Sato S, Kigawa J, Minagawa Y, Okada M, Shimada M, Takahashi M, Kamazawa S, Terakawa N. Chemosensitivity and p53-dependent apoptosis in epithelial ovarian carcinoma. Cancer. 1999;86:1307–1313. [PubMed] [Google Scholar]
- 18.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin-Drubin ME, Meyers J, Munger K. Cancer associated human papillomaviruses. Curr Opin Virol. 2012;2:459–466. doi: 10.1016/j.coviro.2012.05.004. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004;109:157–162. doi: 10.1002/ijc.11691. [DOI] [PubMed] [Google Scholar]
- 21.Dehn D, Torkko KC, Shroyer KR. Human papillomavirus testing and molecular markers of cervical dysplasia and carcinoma. Cancer. 2007;111:1–14. doi: 10.1002/cncr.22425. [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin-Drubin ME, Münger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143:195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrede D, Tidy JA, Crook T, Lane D, Vousden KH. Expression of RB and p53 proteins in HPV-positive and HPV-negative cervical carcinoma cell lines. Mol Carcinog. 1991;4:171–175. doi: 10.1002/mc.2940040302. [DOI] [PubMed] [Google Scholar]
- 24.Thiery JP, Acloque H. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Battle E, Sancho E, Franci C, Domingguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 26.Cano A, perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchyal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 27.Green KJ, Bohringer M, Gocken T, Jones JC. Intermediate filament associated proteins. Adv Protein Chem. 2005;70:143–202. doi: 10.1016/S0065-3233(05)70006-1. [DOI] [PubMed] [Google Scholar]
- 28.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sleeman JP, Thiery JP. SnapShot: The epithelial-mesenchymal transition. Cell. 2011;145:162.e1. doi: 10.1016/j.cell.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 30.McInroy L, Määttä A. Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem Biophys Res Commun. 2007;360:109–114. doi: 10.1016/j.bbrc.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 31.Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB, Ivaska J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30:1436–1448. doi: 10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- 32.Tummala KS, Goes AL, Yilmaz M, Grana O, Bakiri L, Ruppen I, Ximenez-Embun P, Sheshappanavar V, Rodriguez-justo M, Piasano DG, Wagner EF, Djouder N. Inhibition of de NovoNAD(+) synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell. 2014;26:826–839. doi: 10.1016/j.ccell.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, Xu Y, Zhou W, Zhong L, Wen Z, Yu H, Chen S, Shen J, Chen H, She Q, Jiang J, Miao J, Wei W. The viral oncoprotein HBx of Hepatitis B virus promotes the growth of hepatocellular carcinoma through cooperating with the cellular oncoprotein RMP. Int J Biol Sci. 2014;10:1181–1192. doi: 10.7150/ijbs.10275. [DOI] [PMC free article] [PubMed] [Google Scholar]