

Abstract
Variable clinical responses, tumor heterogeneity, and drug resistance reduce long-term survival outcomes for metastatic melanoma patients. To guide and accelerate drug development, we characterized tumor responses for five melanoma patient derived xenograft models treated with Vemurafenib. Three BRAFV600E models showed acquired drug resistance, one BRAFV600E model had a complete and durable response, and a BRAFV600V model was expectedly unresponsive. In progressing tumors, a variety of resistance mechanisms to BRAF inhibition were uncovered, including mutant BRAF alternative splicing, NRAS mutation, COT (MAP3K8) overexpression, and increased mutant BRAF gene amplification and copy number. The resistance mechanisms among the patient derived xenograft models were similar to the resistance pathways identified in clinical specimens from patients progressing on BRAF inhibitor therapy. In addition, there was both inter- and intra-patient heterogeneity in resistance mechanisms, accompanied by heterogeneous pERK expression immunostaining profiles. MEK monotherapy of Vemurafenib-resistant tumors caused toxicity and acquired drug resistance. However, tumors were eradicated when Vemurafenib was combined the MEK inhibitor. The diversity of drug responses among the xenograft models; the distinct mechanisms of resistance; and the ability to overcome resistance by the addition of a MEK inhibitor provide a scheduling rationale for clinical trials of next-generation drug combinations.
Keywords: Melanoma, patient derived xenografts (PDX), BRAF, MEK, Vemurafenib, drug resistance
Introduction
Metastatic melanoma is a highly aggressive malignancy that remains a problem despite improved methods for genotyping tumors and approved targeted therapies [1]. Agents targeting BRAFV600E, such as Vemurafenib and dabrafenib, have improved therapeutic options [2-4]. Unfortunately, initial responses are short-lived [5], and work remains to combat the clonal evolution of resistance [6]. Further complicating personalized medicine for malignant melanoma is an ever-expanding range of acquired Vemurafenib resistance mechanisms [7-10]. The fact that multiple resistance mechanisms may be found within different tumors from the same patient [9] and the reported heterogeneity within a single tumor [10-13] adds to the complexity. Thus, two major challenges in oncology are to understand, and to counter, the evolution of drug resistance in tumors under chemotherapeutic selection pressure.
Patient derived xenograft (PDX) models have emerged as one of several promising strategies to investigate these challenges [14]. A prior report using metastatic melanoma PDX models uncovered novel therapeutic opportunities by using intermittent Vemurafenib dosing to forestall drug resistance [15]. Additional studies including the refinement and validation of such PDX model systems are crucial to realize the promise of personalized treatment paradigms. Along these lines, we examined five metastatic melanoma PDX models in which a single tumor from each patient was used to generate multiple tumorgrafts. The characterization of each tumor illuminated the complexities arising from tumor heterogeneity, such as efficacy of targeted therapy, durability of response, and development of acquired drug resistance. We believe the PDX models described here, provide a critical experimental tool to guide future drug development efforts. Importantly, we confirmed that these melanoma PDX models recapitulate the spectrum of Vemurafenib responses that have been previously reported in metastatic melanoma patients.
Materials and methods
Tissue processing, tumor engraftment and treatment of PDX models
All aspects of this study were reviewed and approved by the IRB at the Van Andel Research Institute (VARI). Tumor tissue in excess of that needed for diagnosis was collected after receiving written informed consent prior to study enrollment. Characteristics of five different metastatic melanoma patients are summarized in Table 1. The animal procedures used for PDX models were approved by the VARI IACUC. Upon receipt of a portion of a tumor, the tissue was subdivided by blunt dissection into fragments used for histological study (after formalin fixation), for genomic and proteomic analyses (after snap freezing), for implantation into 6- to 8- week-old athymic nu/nu (nude mice), and for cryopreservation for future grafting. All details regarding the PDX models were previously described [16]. A staggered enrollment protocol was used in which the initiation of treatment for individual mice was based on tumor size and growth kinetics (log phase).
Table 1.
Patient characteristics and comparative mutational status between the patient tumor and first generation PDX model
| Patient | Sex | Age | Tumor Site | BRAF | NRAS | KIT | MET | PIK3CA | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| T0 | F0 | T0 | F0 | T0 | F0 | T0 | F0 | T0 | F0 | ||||
| 1 | Male | 53 | Lymph node | V600E | V600E | WT | WT | WT | WT | WT | WT | WT | WT |
| 2 | Male | 45 | Lymph node | V600E | V600E | WT | WT | WT | WT | WT | WT | WT | WT |
| 3 | Male | 43 | Lymph node | V600E | V600E | WT | WT | WT | WT | WT | WT | WT | WT |
| 4 | Male | 43 | Metastasis, abdominal wall | NA | V600E | WT | WT | WT | WT | WT | WT | WT | WT |
| 5 | Male | 80 | Lymph node | V600V | V600V | WT | WT | WT | WT | WT | WT | WT | WT |
In-vivo drug studies
Vemurafenib
Vemurafenib tablets (240 mg) were manually ground by mortar and pestle and suspended in carboxymethylcellulose (CMC) followed by dilution with dimethyl sulfoxide (DMSO) to form a final milky white suspension in 5% DMSO/1% CMC. Vemurafenib stock (8.3 mg/ml) was administered twice a day, 5 days per week (Monday through Friday) by oral gavage for a final dose of 50 mg/kg body weight.
PD0325901
To prepare the MEK inhibitor PD0325901 (Selleck Chemicals, Houston, TX) stock solution, 100 mg of drug was first diluted in DMSO at a final concentration of 100 mg/mL then diluted to a concentration of 6.3 mg/mL using 1% CMC. The PD0325901 stock solution was administered once or twice a day, 5 days per week (Monday through Friday) by oral gavage for a final dose of 25 mg/kg body weight.
Vehicle control
The control solution was 5% DMSO/1% CMC. Control mice received 150 µl of vehicle on same schedule as mice receiving Vemurafenib treatment or 100 µl of vehicle at same schedule as mice receiving PD0325901 treatment.
Histopathology and immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were routinely processed using hematoxylin and eosin (H&E) staining. Immunohistochemical (IHC) staining was performed using a Discovery XT (Ventana Medical System, Tucson, AZ). Primary antibodies were used to detect pERK (Thr202/Tyr204), pAKT (Ser473), and activated/cleaved caspase 3 (clone 20G11, 736E11, and CS9661, respectively; Cell Signaling Technology, Danvers, MA) and Ki-67 (ab833; Abcam Inc., Cambridge, MA). Ultramap anti-rabbit or goat alkaline phosphatase secondary antibodies were added with a Chromogen Red or Brown reaction product following the manufacturer’s instructions. Slides were viewed on an Olympus BX51 microscope with Nikon Image Software. Scanning magnification images were captured using ScanScope XT, (Aperio; Vista, CA).
Detection of DNA mutations and copy number
Targeted sequencing of tumors was performed using the Ion AmpliSeq Cancer Hotspot Panel v2 and an Ion Torrent PGM instrument following the manufacturer’s instructions. Variant calls were made using the Torrent Variant Caller (version 3.6.63335) with the “Somatic - High Stringency” configuration. Raw variants within the NRAS gene were then filtered such that coverage was > 15 and variant frequencies were > 25%. Copy number variations of the BRAF gene for tumor samples were performed using the TaqMan Copy Number Assay following the manufacturer’s instructions. Normal adult skin-derived human melanocytes were obtained from Zenbio (Research Triangle Park, NC) and grown according to the manufacturer’s recommandation.
Detection and confirmation of alternative BRAF splicing
Analysis of BRAF splice variants was performed in vehicle-treated and Vemurafenib-resistant tumors as previously described [17]. As a control, BRAF V600E A375 melanoma cells were purchased from the American Tissue Culture Collection, and were examined after being grown in RPMI supplemented with 10% FBS, 1% penicillin, and 1% streptomycin.
Relative BRAF mRNA levels
BRAF mRNA was measured from the parental tumor, normal melanocytes, and PDX-derived tumor samples by quantitative RT-qPCR as previously described [15].
Results
Vemurafenib responses show distinct resistance mechanisms in three BRAFV600E models
In the patient 1-derived BRAFV600E model exposed to vehicle alone (Figure 1A; green line) tumors rapidly increased in size within 14 days (n=4). Consistent with clinical trial results [18], the growth of tumors from patient 1 was initially inhibited by Vemurafenib, but later acquired resistance and subsequently began growing (red line, n=11 started on therapy). Upon serial transplantation of the Vemurafenib-resistant tumors, there was immediate and rapid growth in the presence of Vemurafenib (Supplementary Figure 1A). In a repeated experiment using patient 1 tissue, xenograft tumors in 11 mice (vehicle-treated tumors, green line, n=4; Vemurafenib-treated tumors, red line, n=7) recapitulated the acquired resistance (Supplementary Figure 1B). Unlike a previous report in which re-engrafted tumor fragments from PDX models temporarily regained drug sensitivity [15], re-engrafted tumor fragments from the patient 1 PDX model displayed immediate drug resistance.
Figure 1.

BRAFV600E PDX tumor responses to Vemurafenib treatment reveal multiple resistance mechanisms. Mice were treated with either vehicle control or Vemurafenib (50 mg/kg orally, twice daily, 5 d on, 2 d off) for up to 150 days. The structure of Vemurafenib is displayed inside panel A. All vehicle treated tumors from patients 1-3 (green lines - ≥ n=4) rapidly increased in size. Chronic exposure to Vemurafenib (red line, except in panel B) produced differing responses among these PDX models. Y-axis values represent the mean tumor volume ± SEM (except for panel B) for each treatment group. A. Patient 1 tumors showed a significant, uniform response to Vemurafenib (n=11 started on treatment) with tumor growth inhibited for 50 d, after which acquired resistance emerged in all mice. Sampling of whole-tumor sections revealed progressively increased pERK expression by IHC staining (lower left panel) during treatment (day 50) and after resistance (day 70), with inter- and intra-tumor heterogeneity. Alternative splicing of the BRAF V600E gene was found in all four resistant tumors, but not in control vehicle-treated tumors or in A375 cells. Note that the dominant isoform is the 1.1 kb isoform (upper right, single asterisk), not the the 1.7 kb isoform (two asterisks). B. Patient 2 tumor growth is plotted for each individual mouse due to at the heterogeneous response to treatment with Vemurafenib (n=12 started on treatment). Resistant tumors with NRASQ61K mutations (frequency 33-50%) were present in three of four tumors examined; one resistant tumor lacked this point mutation, but had an alternatively spliced BRAFV600E isoform of 1.1 kb. C. Patient 3 tumors showed a significant, uniform response to Vemurafenib (n=13 started on treatment) with tumor growth inhibited for 20 d, after which resistance emerged in all mice. No alternative BRAF splicing or NRAS mutations were observed. Three of the resistant tumors had elevated COT expression (lower right panel) relative to vehicle-treated tumors and to tumors isolated from patient 1. D. Patient 4 tumors displaying a reduction in tumor volume in response to chronic Vemurafenib treatment (n=12 started on treatment) did not develop resistance over the course of the study (80 days). Y-axis values represent the mean tumor volume ± SEM for each treatment group. The fidelity of the PDX model system is confirmed by the patient 5 BRAFV600V tumors which did not respond by growth inhibition to either vehicle (n=5) or Vemurafenib (n=8). Y-axis values represent the mean tumor volume ± SEM for each treatment group.
Because ERK activity has been linked to response [19] and resistance [10] to Vemurafenib, we profiled pERK activation. Despite heterogeneous pERK activation profiles within individual tumors and between tumors, overall expression patterns for pERK were stronger in Vemurafenib-treated tumors on day 50 and in Vemurafenib-resistant tumors on day 70 than in vehicle-control tumors (Figure 1A; IHC-stained sections). The presence of enhanced ERK activation at the advancing margins of metastatic melanoma lesions was noted by another group [20]. To compare pERK expression and cell proliferation status, we used IHC staining for the cell cycle-related antigen, Ki-67 (Supplementary Figure 1C). Overall, the number of Ki-67 positive nuclei was consistent with the proliferative state of the PDX model. Thus, vehicle-treated tumors (day 10) had widespread Ki-67 expression, as did rapidly growing resistant tumors (day 70), while Vemurafenib-treated tumors prior to the onset of resistance (day 50) had less Ki-67 expression.
BRAFV600E mutation analysis derived from patient 1 samples confirmed that the WT vs. mutant allelic frequencies were similar (65-85%; two tumors analyzed/group, data not shown). Thus, Vemurafenib resistance was not accompanied by the emergence of metastatic melanoma clones having a substantially increased frequency of WT (e.g., BRAF V600V) alleles in resistant tumors. Additionally, targeted BRAF resequencing of DNA isolated from control and resistant tumors, found that no single mutation was apparent as being differentially present between the two groups (data not shown). Because previous reports have implicated BRAF V600E alternative splicing as a mechanism of Vemurafenib resistance [10,17], PCR was used to examine BRAF mRNA size. No evidence of alternative splicing was found in either vehicle-treated tumors or A375 cells; only the full length (2.3-kb) BRAF form was detected (Figure 1A). However, all four acquired-resistance tumors harvested on day 70 revealed two alternatively spliced variants, a predominant 1.1-kb isoform and a 1.7-kb isoform. The 1.7-kb variant was shown by Sanger sequencing to be missing exons 8-11, 14, and 15; this is not the same as the 1.7-kb variant described previously in which exons 3-8 were deleted [17]. Sanger sequencing of the 1.1-kb variant identified the previously reported deletion of exons 2-10 [17], which eliminates the Ras binding domain while preserving the kinase domain. This BRAF V600E variant has been identified in Vemurafenib-resistant metastatic melanoma clinical samples [21] and in a clinical setting in which a dabrafenib/trametinib combination therapy was used [22].
The BRAFV600E PDX model derived from patient 2 revealed a rapid increase in the volume of vehicle-treated tumors (Figure 1B, green line, n=5), but a heterogeneous response among Vemurafenib-treated tumors (n=6 started on therapy). While all treated tumors initially decreased in size, four tumors developed drug resistance and two tumors showed sustained growth suppression for up to 150 days (Figure 1B). Examination of the resistant tumors from patient 2 revealed the emergence of an activating NRAS Q61K mutation in three of the four tumors, but not in any of the vehicle-treated tumors (Figure 1B). This resistance mechanism has been previously observed in both metastatic melanoma cell lines [23-25] and in patient tumors [9]. Interestingly, the remaining resistant tumor lacked the activating NRAS mutation but expressed the 1.1-kb variant BRAF V600E, which lacks exons 2-10. To further confirm the differences among this group of resistant tumors, pERK and pAKT were examined by IHC staining. While pERK profiles were similar among all four progressing tumors, the pAKT level was markedly lower in the tumor without NRASQ61K (Supplementary Figure 2D).
The BRAFV600E PDX model derived from patient 3 responded to both vehicle and Vemurafenib similar to tumors from patient 1; both vehicle- and Vemurafenib-treated tumors showed synchronous changes in tumor volume (Figure 1C, vehicle-treated tumors, green line, n=6; Vemurafenib-treated tumors, red line, n=13). While no alternatively spliced BRAFV600E isoforms or NRASQ61K were detected in either vehicle-treated or resistant tumors, the expression of MAP3K8, the gene encoding COT, was elevated in all three of the resistant tumors examined (Figure 1C). By comparison, Vemurafenib-treated tumors from patient 1 did not show increases in COT (Figure 1C).
Vehicle-treated tumors derived from patient 4 also grew rapidly (Figure 1D, green line; n=5) but a sustained reduction in tumor size was observed in all Vemurafenib-treated tumors up to 80 days (red line, n=12 started therapy). From a clinical perspective, patient 4 would reflect complete responders, expected to have a prolonged progression-free survival. The patient 5-derived BRAFV600V model confirmed the expected lack of response to Vemurafenib for a non-BRAF-mutated metastatic melanoma (Figure 1D); vehicle-treated (green line, n=5) and Vemurafenib-treated tumors (red line, n=8) had similar growth kinetics.
Mutant BRAF amplification has been reported in patient derived tumors and in-vitro in cell lines as a mechanism of acquired BRAF inhibitor resistance [21]. More recently, Das Thakur et al. [15] implicated increased BRAFV600E expression and copy number in the Vemurafenib resistance of a single metastatic melanoma patient derived PDX model. We found modest increases in BRAF mRNA among the patients, predominantly in the various Vemurafenib-resistant tumors (Figure 2, left axis, solid bars) relative to parental tumors and to human melanocytes that we used as controls. In contrast, while changes in BRAF copy number were seen in individual tumors, there was no clear pattern among vehicle-treated and Vemurafenib-resistant tumors (Figure 2, right axis, open bars). Thus, in our models, BRAF overexpression in Vemurafenib-resistant metastatic melanoma is predominantly driven by increased transcription or mRNA stability.
Figure 2.

BRAF gene expression and copy number in BRAFV600E PDX models. BRAF gene expression (left axis black columns) and BRAF copy number (right axis, open columns) for PDX tumors versus parental tumors and normal melanocytes as controls. No consistent trends were identified.
Tumors with acquired Vemurafenib resistance are MEK inhibitor-sensitive
To determine whether the Vemurafenib-resistant tumors had become addicted to MEK-mediated signaling, we selected the MEK inhibitor PD0325901 as a second agent to interrupt the RAF-MEK-ERK signaling cascade. Fragments of resistant tumors from patient 1 were transplanted into new recipient mice followed by treatment with Vemurafenib (Figure 3A). Three resistant tumors were allowed to reach approximately 1,000 mm3 (red line) before addition of PD0325901 to the treatment regimen. The rapid growth of the tumors immediately ceased and the tumors regressed (Figure 3A). After 14 days of treatment with PD0325901 plus Vemurafenib, the tumors contained conspicuous paucicellular fibrovascular tracks through the tissue, resulting in islands of small melanoma cells with high nuclear:cytoplasmic ratios (Figure 3B, upper panel), accompanied by low proliferation marker expression, and apoptotic cells (assessed by IHC for Ki-67 and activated caspase 3 positivity, respectively; Figure 3B, lower panels). Because tumors were not examined before day 14 (start of PD0325901 doses), the relative contribution of apoptosis to the rapid loss of tumor volume is uncertain. To verify that the addition of PD0325901 was inhibiting the MAPK signaling cascade, the three Vemurafenib-resistant tumors examined after PD0325901 treatment were found to have decreased pERK expression and Ki-67 expression relative to the majority of Vemurafenib-resistant tumors (compare Figure 3C, upper and lower panels versus, respectively, Figure 1A and Supplementary Figure 1C). Thus, in this PDX mode, tumors that acquired Vemurafenib resistance remain drug-resistant upon re-engraftment, and further, reactivation of MAPK signaling can be targeted using PD0325901, which gives rapid size reduction of these resistant tumors, along with lower pERK and Ki-67 expression.
Figure 3.
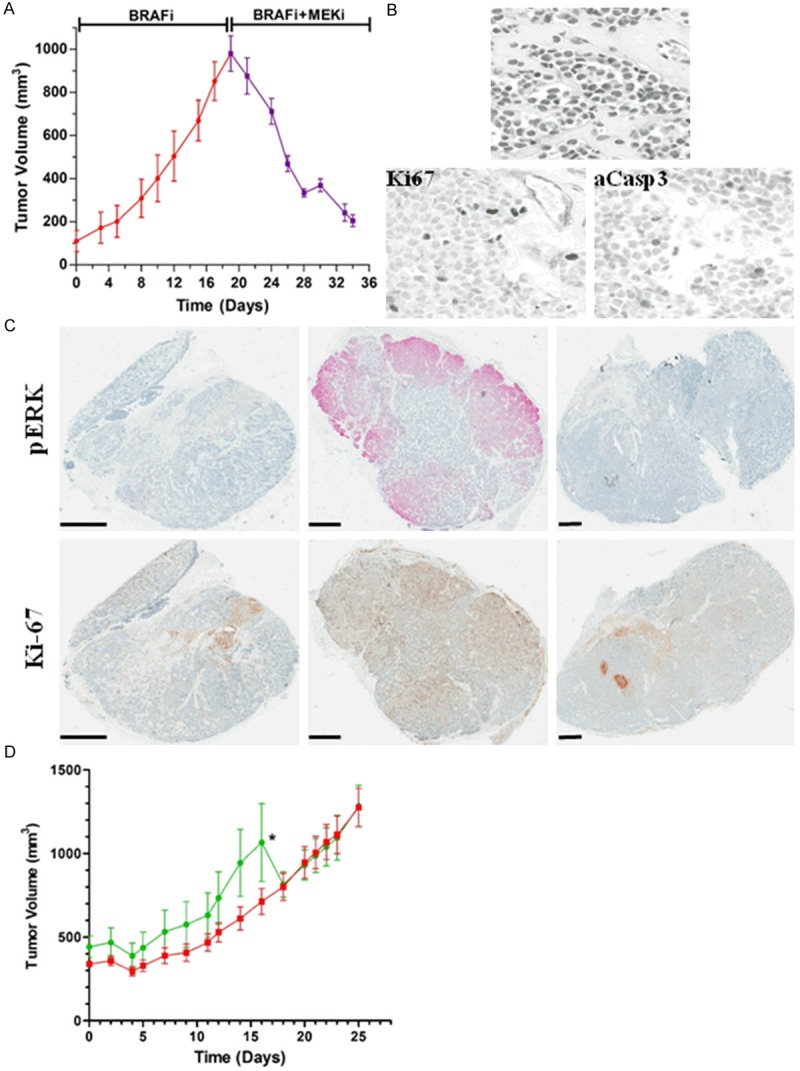
MEK inhibition using PD0325901 inhibits Vemurafenib-resistant tumor growth in patient 1 but not patient 3 tumors. A. Vemurafenib-resistant grew to about 1000 mm3 during Vemurafenib treatment (red line) at which point PD0325901 (25 mg/kg orally, twice daily, 5 days on, 2 days off) triggersed a rapid reduction in tumor size over 14 days of co-treatment (purple line, n=3). B. High power magnification of Vemurafenib-resistant tumors exposed to PD0325901 plus Vemurafenib for 14 d (top image) reveals paucicellular fibrovascular tracks surrounding islands of viable melanoma cells with condensed nuclear chromatin and no nucleoli (H&E stain). Occasional nuclei are IHC-positive for the proliferation-associated marker Ki-67 (bottom left image); scattered apoptotic cells expressing activated caspase 3 were IHC positive (bottom right image). C. Stained whole mounts using (IHC followed by hematoxylin staining) show that both pERK and Ki-67 levels are reduced after Vemurafenib plus PD0325901 treatment. Scale bars indicate 1 mm. D. Vemurafenib-resistant tumors from patient 3 having elevated COT levels were transferred into new recipient mice and were exposed either to Vemurafenib alone (green line, n=5), or Vemurafenib plus PD0325901 (red line, n=6). The asterisk indicates the removal of mice from the study due to large tumor volumes.
Given the complexities of targeting signal transduction pathways, we explored the biological consequences of the elevated COT (MAP3K8) levels observed in Vemurafenib-resistant tumors from patient 3 (Figure 1C). Resistant tumor fragments were re-engrafted into different mice (Figure 3D) and treated with either Vemurafenib (green line) or Vemurafenib plus PD0325901 (red line). Both of these groups grew rapidly. These results are consistent with an earlier report that COT-expressing cell lines are refractory to MEK inhibition [26]. Thus, the different responses of Vemurafenib-resistant tumors from patient 1 (Figure 3A) and patient 3 (Figure 3D) to Vemurafenib plus PD0325901 likely reflect the higher COT expression in patient 3 tumors.
Targeting BRAF and MEK sustains tumor suppression in Vemurafenib-resistant tumors
To determine whether the shrinking of resistant tumors could be sustained using a single daily dose of PD0325901, 12 different tumors derived from patient 1 were examined. Being mindful of previous clinical trials using PD0325901 [27,28], treatment protocols were modified from a twice-daily dosing to single daily dose, and from daily treatment to a 5-day-on-2 day off schedule to avoid toxicity. We observed that tumor size reduction was sustained when PD0325901 was delivered once a day together with Vemurafenib; no resistance was developed during the extended period of treatment (Figure 4A). Several animals were euthanized after 3 weeks of single daily dosing, and an additional four mice were observed for 100 days (30 days after discontinuation of PD0325901). Furthermore, when we discontinued PD0325901 treatment for the four mice at day 70 and followed them for an additional 30 days, there was no tumor regrowth. The tumors remained barely palpable or non-palpable, and when the tumor sites were examined, two sites showed no evidence of tumor. At the third tumor site we observed a fibrocalsific, pigmented nodule that was devoid of melanoma cells (Supplementary Figure 3A). At the fourth site a collection of viable basaloid-appearing melanoma cells remained (Supplementary Figure 3B).
Figure 4.
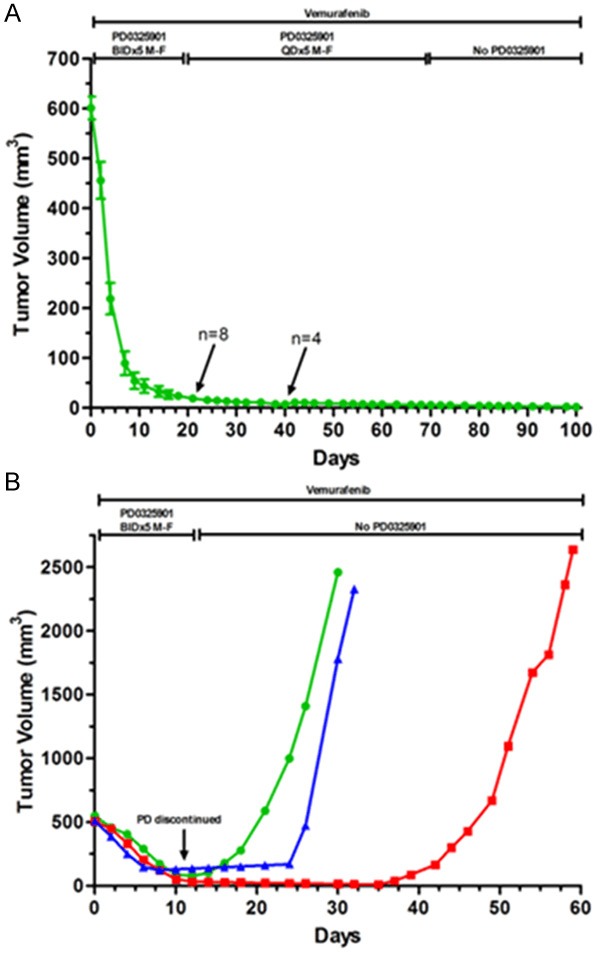
Prolonged tumor size reduction of Vemurafenib-resistant tumors by once-daily dosing of PD0325901. A. Lowering the twice-daily dose of PD0325901 to once per day (25 mg/kg orally, Monday -Friday) sufficesd to maintain tumor suppression in Vemurafenib-resistant tumors; all tumor cells were eradicated in 3 of 4 tumors which had PD0325901 removed on day 70 and which were examined on day 100. B. When Vemurafenib-resistant tumors (500 mm3) were treated with PD0325901 (25 mg/kg orally, twice a day) their size decreased, and remained so until PD0325901 was removed, at which time there was rapid re-growth of the tumors.
To verify that viable metastatic melanoma cells remain after a more-abbreviated combination treatment of Vemurafenib plus PD0325901, three different tumors were taken off PD0325901 but maintained on Vemurafenib; these tumors displayed rapid regrowth with slightly different times to the onset of logarithmic growth (Figure 4B). Thus, by targeting MEK, tumors were unable to grow in vivo, and with prolonged dual treatment, near-complete tumor eradication of Vemurafenib-resistant tumors was accomplished.
Discussion
Almost every patient receiving targeted therapy develops drug resistance. In this study, we initially characterized the growth responses of five different metastatic melanoma patient derived xenograft models to Vemurafenib for up to 150 days. Our results matched clinical observations, and provided mechanistic insight into acquired Vemurafenib resistance on a patient by-patient and lesion-by lesion basis, including evidence of NRAS mutation, mutant BRAF alternative splicing, increased COT expression, and increased BRAF mRNA levels. The relative frequency of the various drug resistance mechanisms seen in our PDX tumors reflected clinical experience using BRAF inhibitors [10]. Thus, our metastatic melanoma PDX models are now confirmed as clinically relevant in vivo systems that can extend knowledge of potential genetic pathways by which tumors escape from targeted therapy.
Melanoma has been recognized for decades as more than just one disease [29]. The basis for multiple drug resistance mechanisms reflects this tumor heterogeneity, which in turn likely reflects the high mutational burden of metastatic melanoma lesions. Human genomes can now be characterized in less than a month [30] portending greater clinical potential for identification of druggable driver mutations. However, enthusiasm for deep genome sequencing must be tempered by the realization that drug resistance is virtually a universal and lethal event for single agent targeted therapy [6]. In this study, we discovered that drug resistant tumors, originating from a single parental metastatic melanoma lesion, could develop distinct resistance mechanisms. In patient 2 tumors, even though there was simultaneous emergence of drug resistance, three tumors contained activated-NRAS mutant cells, while the fourth tumor acquired a BRAFV600E splice variant. The identical BRAF splice variant was not only present in resistant tumors from patient 1, but also was identified among metastatic melanoma patients who were drug resistant in clinical studies [10,17]. One resistant tumor from patient 1 also contained increased number of mutant BRAF copies, although whether an individual melanoma cell has both BRAFV600E alternative splicing and increased copy number remains to be determined. The challenges presented by intratumoral heterogeneity are not unique to melanoma. Distinct cellular sub-populations with differential tumorigenicity have been isolated from 2 breast cancer PDX tumors [31].
Having established the clinical relevancy of our models regarding Vemurafenib, we next assessed the impact of inhibiting MEK signaling. As previously reported [10,19], we found increased pERK in patients developing Vemurafenib resistance. This led us to ask if Vemurafenib-resistant tumors were addicted to this reactivated MAPK signaling pathway, and hence would be sensitive to MEK inhibition. Because broad inhibition of MAPK signaling is required to trigger the killing of metastatic melanoma cells [19], we sought to confirm and extend in vitro studies combining Vemurafenib with PD0325901. This strategy was successfully used to halt disease progression involving BRAFV600E alternative splicing (patient 1), but has not been verified with resistant tumors having NRAS mutations. Vemurafenib resistance in patient 3 samples associated with increased COT expression, that has been reported to activate ERK by both MEK-dependent and MEK-independent mechanisms [26]. Thus, the clinical success of adding a MEK inhibitor to a BRAF inhibitor-resistant tumor will depend on the context of the resistance mechanism.
A previous report in which BRAF V600E amplification was identified as a resistance mechanism indicated that a Vemurafenib drug holiday might be of value [15]; this was subsequently confirmed in three metastatic melanoma pa-tients [9,32]. Such completely opposite ap-proaches - to withdraw or maintain BRAF inhibition upon progression - likely reflects the underlying resistance mechanisms. Based on our results, we propose the metastatic melanoma PDX models will be a useful tool to optimize the schedule of multi-drug combinations that will counter evolution on a patient by-patient and lesion-by-lesion approach.
Combining our results with those of others highlights the complexities of drug resistance and clonal evolution in metastatic melanoma patients receiving BRAF inhibitor therapy, and also demonstrates the utility of PDX models for identifying solutions for these challenges. Taken together, these studies indicate the translational value and relevance of PDX models for these two agents in the clinic and future studies that target BRAF and/or MEK. Further studies are indicated to define the phenotype and signaling pathways operative in residual tumor cells and guide future drug development and therapeutic strategies. These PDX models will be useful for testing triple therapeutic combinations, which would be challenging in the clinic, but likely essential to combat the evolutionary dynamics of cancer drug resistance.
Acknowledgements
We thank Bree Berghuis, Lisa Turner of the Pathology and Biorepository Core and Stephanie Scott for technical assistance. RNA arrays were performed and generated by staff members at the Clinical Reference Laboratory (Lenexa, Kansas). This work was supported by Michigan State University College of Human Medicine and by Van Andel Research Institute as well as by individual supporters of Van Andel Research Institute through the Purple Community and Annual Giving programs.
Disclosure of conflict of interest
The authors declare no conflict of interest.
Supporting Information
References
- 1.Tronnier M, Mitteldorf C. Treating advanced melanoma: current insights and opportunities. Cancer Manag Res. 2014;6:349–356. doi: 10.2147/CMAR.S49494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. Improved survival with Vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WHJ, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan RJ, Flaherty KT. Major therapeutic developments and current challenges in advanced melanoma. Br J Dermatol. 2014;170:36–44. doi: 10.1111/bjd.12698. [DOI] [PubMed] [Google Scholar]
- 6.Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–851. doi: 10.1056/NEJMra1204892. [DOI] [PubMed] [Google Scholar]
- 7.Ravnan MC, Matalka MS. Vemurafenib in patients with BRAF V600E mutation-positive advanced melanoma. Clin Ther. 2012;34:1474–1486. doi: 10.1016/j.clinthera.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Kim KB, Weber JS, Hersey P, Long GV, Lawrence D, Ott PA, Amaravadi RK, Lewis KD, Puzanov I, Lo RS, Koehler A, Kockx M, Spleiss O, Schell-Steven A, Gilbert HN, Cockey L, Bollag G, Lee RJ, Joe AK, Sosman JA, Ribas A. Pharmacodynamic effects and mechanisms of resistance to Vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013;31:1767–1774. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 9.Romano E, Pradervand S, Paillusson A, Weber J, Harshman K, Muehlethaler K, Speiser D, Peters S, Rimoldi D, Michielin O. Identification of multiple mechanisms of resistance to Vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin Cancer Res. 2013;19:5749–5757. doi: 10.1158/1078-0432.CCR-13-0661. [DOI] [PubMed] [Google Scholar]
- 10.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, Glaspy JA, Sosman JA, van Baren N, Long GV, Ribas A, Lo RS. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilmott JS, Tembe V, Howle JR, Sharma R, Thompson JF, Rizos H, Lo RS, Kefford RF, Scolyer RA, Long GV. Intratumoral molecular heterogeneity in a BRAF-mutant, BRAF inhibitor-resistant melanoma: a case illustrating the challenges for personalized medicine. Mol Cancer Ther. 2012;11:2704–2708. doi: 10.1158/1535-7163.MCT-12-0530. [DOI] [PubMed] [Google Scholar]
- 12.Yancovitz M, Litterman A, Yoon J, Ng E, Shapiro RL, Berman RS, Pavlick AC, Darvishian F, Christos P, Mazumdar M, Osman I, Polsky D. Intra- and inter-tumor heterogeneity of BRAF(V600E))mutations in primary and metastatic melanoma. PLoS One. 2012;7:e29336. doi: 10.1371/journal.pone.0029336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boursault L, Haddad V, Vergier B, Cappellen D, Verdon S, Bellocq JP, Jouary T, Merlio JP. Tumor homogeneity between primary and metastatic sites for BRAF status in metastatic melanoma determined by immunohistochemical and molecular testing. PLoS One. 2013;8:e70826. doi: 10.1371/journal.pone.0070826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merlino G, Flaherty K, Acquavella N, Day CP, Aplin A, Holmen S, Topalian S, Van Dyke T, Herlyn M. Meeting report: The future of preclinical mouse models in melanoma treatment is now. Pigment Cell Melanoma Res. 2013;26:E8–E14. doi: 10.1111/pcmr.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling Vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monsma DJ, Monks NR, Cherba DM, Dylewski D, Eugster E, Jahn H, Srikanth S, Scott SB, Richardson PJ, Everts RE, Ishkin A, Nikolsky Y, Resau JH, Sigler R, Nickoloff BJ, Webb CP. Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. J Transl Med. 2012;10:125. doi: 10.1186/1479-5876-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, Misteli T, Chapman PB, Sosman JA, Graeber TG, Ribas A, Lo RS, Rosen N, Solit DB. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaherty KT, Yasothan U, Kirkpatrick P. Vemurafenib. Nat Rev Drug Discov. 2011;10:811–812. doi: 10.1038/nrd3579. [DOI] [PubMed] [Google Scholar]
- 19.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D'Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leong SP, Mihm MCJ, Murphy GF, Hoon DS, Kashani-Sabet M, Agarwala SS, Zager JS, Hauschild A, Sondak VK, Guild V, Kirkwood JM. Progression of cutaneous melanoma: implications for treatment. Clin Exp Metastasis. 2012;29:775–796. doi: 10.1007/s10585-012-9521-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV, Nelson SF, Ribas A, Lo RS. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, Johannessen CM, McKenna A, Cibulskis K, Kryukov G, Hodis E, Lawrence DP, Fisher S, Getz G, Gabriel SB, Carter SL, Flaherty KT, Wargo JA, Garraway LA. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan FM, Kugel CH, Dadpey N, Shao Y, Abel EV, Aplin AE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem. 2012;287:41797–41807. doi: 10.1074/jbc.M112.390906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–920. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- 26.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB, DePrimo SE, Bentivegna S, Wilner KD, Tan W, Ricart AD. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–1937. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 28.Boasberg PD, Redfern CH, Daniels GA, Bodkin D, Garrett CR, Ricart AD. Pilot study of PD-0325901 in previously treated patients with advanced melanoma, breast cancer, and colon cancer. Cancer Chemother Pharmacol. 2011;68:547–552. doi: 10.1007/s00280-011-1620-1. [DOI] [PubMed] [Google Scholar]
- 29.Pimiento JM, Larkin EM, Smalley KS, Wiersma GL, Monks NR, Fedorenko IV, Peterson CA, Nickoloff BJ. Melanoma genotypes and phenotypes get personal. Lab Invest. 2013;93:858–867. doi: 10.1038/labinvest.2013.84. [DOI] [PubMed] [Google Scholar]
- 30.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, Barrette T, Everett J, Siddiqui J, Kunju LP, Navone N, Araujo JC, Troncoso P, Logothetis CJ, Innis JW, Smith DC, Lao CD, Kim SY, Roberts JS, Gruber SB, Pienta KJ, Talpaz M, Chinnaiyan AM. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skrbo N, Hjortland GO, Kristian A, Holm R, Nord S, Prasmickaite L, Engebraaten O, Maelandsmo GM, Sorlie T, Andersen K. Differential in vivo tumorigenicity of distinct subpopulations from a luminal-like breast cancer xenograft. PLoS One. 2014;9:e113278. doi: 10.1371/journal.pone.0113278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seghers AC, Wilgenhof S, Lebbe C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012;22:466–472. doi: 10.1097/CMR.0b013e3283541541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.