

Abstract
The histone lysine demethylase KDM4 subfamily, comprised of four members (A, B, C, and D), play critical roles in controlling transcription, chromatin architecture and cellular differentiation. We previously demonstrated that KDM4C is significantly amplified and overexpressed in aggressive basal-like breast cancers and functions as a transforming oncogene. However, information regarding the genomic and transcriptomic alterations of the KDM4 subfamily in different subtypes of breast cancer remains largely incomplete. Here, we conducted a meta-analysis of KDM4A, B, C and D in breast cancer and identified associations among recurrent copy number alterations, gene expression and breast cancer subtypes. We demonstrated that KDM4A and D are also significantly overexpressed in basal-like breast cancer, whereas KDM4B overexpression is more dominant in estrogen-receptor-positive, luminal breast cancer. Next, we investigated the therapeutic potential of a novel histone demethylase inhibitor, NCDM-32B, in breast cancer. The treatment of basal breast cancer cell lines with NCDM-32B resulted in the decrease of cell viability and anchorage independent growth in soft agar. Furthermore, we found that NCDM-32B impaired several critical pathways that drive cellular proliferation and transformation in breast cancer. Our findings demonstrate genetic amplification and overexpression of the KDM4 demethylases in different subtypes of breast cancer. Furthermore, histone methylation is reversible and KDM4 demethylases are druggable targets. Thus, KDM4 inhibitors may serve as a novel therapeutic approach for a subset of aggressive breast cancer.
Keywords: KDM4, GASC1, histone demethylase, gene amplification, breast cancer
Introduction
Breast cancer is a heterogeneous disease, consisting of tumors with varying pathologic and molecular characteristics. The primary biological subtypes of breast cancer include Luminal A, Luminal B, human epidermal growth factor receptor 2 (HER2/ERBB2)-enriched, basal-like, and normal-like [1]. Both Luminal A and Luminal B breast cancers are estrogen receptor (ER) positive, but Luminal B cancers have poorer outcomes [2]. Basal-like breast cancer accounts for some of the most aggressive types of breast cancer, marked by high rates of relapse, visceral metastases, and poor prognoses [1,3]. Novel therapeutic regimens are likely to result from a growing understanding of both genetic and epigenetic abnormalities that accumulate in different types of breast cancer, as well as the identification of new subtype-specific therapeutic targets.
Histone lysine demethylases (KDMs) regulate histone methylation dynamics and play critical roles in modulating chromatin architecture, gene transcription, and cellular differentiation [4-6]. The fundamental unit of chromatin is a nucleosome that is composed of two copies of each core histone: H2A, H2B, H3, and H4. The core histones are predominantly globular except for the N-terminal tails, which are targets for posttranslational modifications [7]. Lysine (K) methylation, controlled by histone lysine methyltransferases and KDMs, is one of the most common modifications on histone tails. Methylation of lysines can result in different transcriptional and biological outcomes depending on the site and degree of methylation [mono-, di-, or trimethylation (me1/me2/me3)]. The KDM4 demethylases A, B, C and D, were the first identified demethylases to act on trimethylated lysines [8,9]. KDM4A, B and C are uniquely defined by N-terminal Jumonji N (JmjN) and JmjC domains, followed by C-terminal plant homeodomain (PHD) and Tudor domains [6]. KDM4A, B and C catalyze the demethylation of H3K9me3/me2 with a preference for the trimethyl state, a histone mark associated with gene repression and found in heterochromatin [10-12]. KDM4A, B and C also catalyze the demethylation of H3K36me3, a mark linked to transcriptional elongation, albeit at a lower rate [12]. In comparison, KDM4D lacks the C-terminal region, including the PHD and Tudor domains, and mainly catalyzes the demethylation of H3K9me3/me2 [13].
Dysregulation of the KDM4 demethylases has been documented in a variety of cancers, including lymphoma, medulloblastoma, and breast, prostate, colorectal, lung, gastric, esophageal, renal cancers [14-20]. The KDM4C gene, originally termed GASC1 (gene amplified in squamous cell carcinoma 1) was identified and cloned from the 9p24 amplified region of esophageal cancer cell lines [21]. Previously we demonstrated that KDM4C is significantly amplified and overexpressed in aggressive basal-like breast cancers [18]. KDM4C serves as a transforming oncogene: stable KDM4C overexpression in human non-tumorigenic mammary epithelial MCF10A cells induces transformative phenotypes, whereas KDM4C knockdown in breast cancer cells inhibits proliferation in vitro and in vivo [18,20]. Furthermore, KDM4A and B are co-activators of the estrogen receptor (ER) and stimulate the transcriptional potential of the ER in breast cancer [22-24]. Recent evidence has suggested that alteration of the KDM4 demethylases is associated with breast cancer. However, our current knowledge of the specificity of KDM4 demethylases in different types of breast cancer is still incomplete. Fur-thermore, targeting epigenetic proteins such as KDMs is currently a highly active frontier of anti-cancer drug development. Here, we conducted a meta-analysis of KDM4A, B, C and D in breast cancer and identified associations among recurrent copy number alterations, gene expression and breast cancer subtypes. We tested a novel inhibitor of the KDM4 demethylases, small molecule NCDM-32B, for its ability to attenuate breast cancer cell growth. We investigated the downstream pathways that are altered by NCDM-32B in basal breast cancer. Our studies demonstrate different patterns of DNA copy number, mRNA, and protein expression levels of the four KDM4 subfamily members across the subtypes of breast cancer. Furthermore, KDM4 inhibitors may serve as a novel therapeutic approach for a subset of aggressive breast cancer.
Materials and methods
Cell culture
The cultures for the SUM series of breast cancer cell lines and an immortalized, non-transformed human mammary epithelial MCF10A cell line have been described in detail previously [25,26]. The Colo824 cell line was obtained from DSMZ (Braunschweig, Germany), and HCC70, HCC1937, HCC1428, HCC1954, MDB-MA-468, T47D, and ZR-75-1 cell lines were obtained from ATCC (Manassas, VA, USA).
The Cancer Genome Atlas (TCGA) data for breast cancer
The DNA copy number, mutation, and RNA sequencing datasets of 976 breast cancer samples used in this research were obtained from the cBio Cancer Genomics Portal [13,27]. The copy number of each KDM was generated from the copy number analysis algorithms GISTIC (Genomic Identification of Significant Targets in Cancer) and categorized as copy number level per gene: “-2” is a deep loss (possibly a homozygous deletion), “-1” is a heterozygous deletion, “0” is diploid, “1” indicates a low-level gain, and “2” is a high-level amplification. For mRNA expression data, the relative expression of an individual gene and the gene expression distribution in a reference population were analyzed. The reference population was either all tumors that are diploid for the gene in question, or, when available, normal adjacent tissue. The returned value indicates the number of standard deviations away from the mean of expression in the reference population (Z-score). Somatic mutation data were obtained from exome sequencing [13,27]. The breast cancer subtype information was from a previous publication and the cBio Cancer Genomics Portal [3,13,27].
Examination of cell growth
Cell growth was assessed by using a Coulter counter or the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. For the MTT assay, cells were seeded in 24-well plates at a density of 6-8 × 103 cells per well and allowed to attach overnight. At designated time points, thiazolyl blue tetrazolium bromide (Molecular Probes, Carlsbad, CA, USA) was added to each well (final concentration 0.5 mg/ml) and incubated for 4 h at 37°C. After removing the growth medium, dimethyl sulfoxide was added. Absorbance of the solution was read at a test wavelength of 540 nm against a reference wavelength of 570 nm. Soft agar assays were done as previously described [25]. Briefly, dishes were coated with a 1:1 mix of the appropriate 2 × medium for the cell line being studied and 1% Bactoagar. Cells were plated in 12-well plate, fed thrice per week for 3 to 4 weeks, stained with 500 μg/mL p-iodonitrotetrazolium violet (Sigma, St. Louis, MO) overnight, and counted with an automated mammalian cell colony counter (Oxford Optronix GELCOUNT, Oxford, United Kingdom).
Immunoblotting and antibodies
Whole cell lysates were prepared by scraping cells from the dishes into cold RIPA lysis buffer. Nuclear protein extracts from breast cancer cells and the MCF10A cells were prepared with an NE-PER Nuclear Protein Extraction Kit (Thermo Scientific, Rockford, IL, USA). Histone proteins from cells were isolated with the EpiQuik Total Histone Extraction Kit (Epigentek, Farmingdale, NY, USA). Antibodies used in the study included anti-KDM4D (Abcam ab93694, Cambridge, MA, USA), anti-KDM4B and C (Bethyl Laboratories A301-478A, Montgomery, TX, USA), anti-KDM4A, anti-H3, anti-H3K4me2, anti-H3K4me1, anti-H3K9me1, anti-H3K36me3, anti-H3K36me2 (Cell Signaling, Danvers, MA, USA), anti-H3K4me3, anti- H3K9me3, anti-H3K36me1 (Active Motif, Carlsbad, CA, USA).
mRNA microarray analysis
Total RNA from NCDM-32B treated HCC1954 breast cancer cells and non-treated control cells was extracted using an RNeasy Plus Mini Kit (Qiagen) and then used for subsequent mRNA expression profiling. The quality and quantity of total RNA were assessed by optical density (Nanodrop ND-2000 spectrophotometer) and Agilent Bioanalyzer. Human HT-12 v4 Expression BeadChip (Illumina) arrays were performed by the Genomics Core of the Karmanos Cancer Institute. Data were processed for quality control and normalized across compared arrays by quantile normalization. Pathway and gene ontology analysis was performed using Ingenuity Pathway Analysis software.
Statistical analysis
Statistical analyses were performed using R software (http://www.r-project.org) and Stata [28]. The correlations between copy numbers and mRNA levels of each KDM from 976 se-quenced breast cancer specimens were analyzed using Spearman, Kendall, and Pearson correlation tests. The Spearman and Kendall tests are rank correlations: the Spearman coefficient relates the two variables while conserving the order of data points, and the Kendall coefficient measures the number of ranks that match in the data set. Although the Pearson correlation coefficient is the most widely used, it was deemed the least relevant to our study, as it measures only the strength of linear relationships and ignores all others. We used the “cor” function in R statistical software for computation, specifying in the code which type of test we wanted (Spearman, Kendall, or Pearson). The difference in mRNA expression level for each KDM between the basal-like and the other cancer subtypes was calculated using Student’s t-test.
Results
Genetic alterations of KDM4 histone demethylases across breast cancer subtypes
To systematically investigate genetic alterations of the KDM4 demethylases in breast cancer, we first analyzed the genome sequencing data of 976 primary breast cancer samples from The Cancer Genome Atlas (TCGA) database via cBioPortal [13,27]. In cBioPortal, copy numbers are computed using the GISTIC algorithm, which identifies the putative copy number level as high-level amplification, low-level gain, diploid, heterozygous deletion, or homozygous deletion [13,27]. For mRNA expression analysis, the Z-score is used to determine whether a gene is upregulated or downregulated relative to normal adjacent tissue or relative to all other tumor samples that are diploid for the gene. As shown in Figure S1, we found that KDM4C had the highest frequency (13.4%) of genetic alteration, including high-level amplification, homozygous deletion, mutation, upregulation, and downregulation, whereas KDM4B had the lowest frequency (6.1%) of genetic alteration among the 976 TCGA breast cancer specimens. For KDM4C, 2.5% of samples had high-level amplification and 0.7% had homozygous deletion, whereas 1.9% of samples had high-level amplification or homozygous deletion of KDM4A, B and D (Figure 1A).
Figure 1.
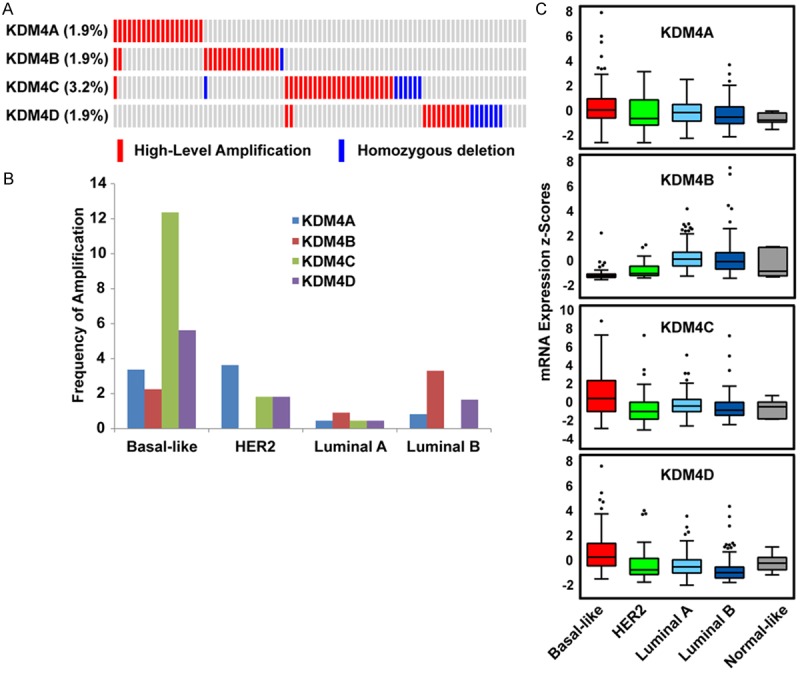
KDM4A-D copy number and expression levels in different subtypes breast cancer. A. High-level amplification and homozygous deletion of KDM4 subfamily members in TCGA breast cancer dataset (n=976). Data are displayed with the Oncoprint tool from cBioPortal. B. Frequencies of high-level amplification of KDM4A, B, C, and D in different subtypes of breast cancer. C. Expression levels of KDM4A, B, C, and D across five subtypes of breast cancer based on TCGA database. The differences in KDM4A, B, C, and D mRNA levels among breast cancer subtypes are statistically significant (P < 0.001).
Correlations between gene expression and copy number have been used widely to prioritize driver oncogenes in human cancer, because mRNA expression can successfully translate the effect of elevated copy number to cancer initiation and progression. Thus, we examined the association between copy number and mRNA expression of KDM4A, B, C and D by using Spearman’s rank correlation in TCGA breast cancer specimens. We found that the correlation between copy number and mRNA expression was strongest for KDM4C (R=0.61), followed by KDM4A (R=0.52); remaining KDM4 members had correlation coefficients less than 0.50 (R=0.44 for KDM4D, and R=0.28 for KDM4B) (Figure S2).
Next, we determined whether the high-level amplification or expression level of each KDM4 member was related to breast cancer subtype. Of the TCGA breast cancer samples, 493 had subtype data available, including 8 normal-like, 220 Luminal A, 121 Luminal B, 55 HER2+, and 89 basal-like breast cancers. Due to the small sample size (n=8) of the normal-like subtype, those samples were excluded from this analysis. KDM4C and KDM4D amplification was found to have the highest frequency (12.4% and 3.6%, respectively) in basal-like breast cancer. The highest frequency of KDM4A amplification (3.6%) was in HER2-subtype samples, whereas KDM4B amplification (3.3%) was highest in Luminal B breast cancer. To determine whether mRNA expression of each KDM4 subfamily member is associated with a specific subtype of breast cancer, we compared the mRNA expression levels of KDM4A, B, C and D across different subtypes of breast cancer specimens. We found that the mRNA expression levels of KDM4A, C, and D were significantly higher in basal-like breast cancer (P=0.005, 6.01E-05, and 3.04E-07, respectively), whereas KDM4B mRNA was highly expressed in luminal breast cancer and less expressed in basal-like breast cancer (Figure 1C). Taken together, these data indicate that amplification and overexpression of KDM4A, C, and D are more prevalent in aggressive, basal-like breast cancers, while KDM4B overexpression is more prevalent in ER+, luminal breast cancers.
Expression of KDM4 demethylases and histone methylation marks in breast cancer cell lines
Breast cancer cell lines retain many of the molecular characteristics of the tumors from which they were derived [29]. To identify suitable cellular models to test the effects of KDM4 inhibitors on breast cancer growth and progression, we examined the relative mRNA and protein expression levels of KDM4A, B, C and D in a panel of breast cancer cell lines. MCF10A, an immortalized but non-tumorigenic breast epithelial cell line, was used as the control. Quantitative RT-PCR (qRT-PCR) assays were used to measure the mRNA expression level of KDM4A, B, C and D in 17 breast cancer cell lines, including nine basal-like, two HER2+, and six Luminal lines. To determine KDM4A, B, C and D protein abundance more precisely relative to their histone demethylase function, nuclear extracts from eight breast cancer cell lines and MCF10A were isolated and probed with antibodies that recognize KDM4A, B, C and D. As shown in Figure 2A and 2B, we found that KDM4A mRNA is overexpressed in several ER+ luminal cell lines (such as HCC1428) and basal-like cell lines (suchas MDA-MB-468). KDM4B was strikingly overexpressed at the mRNA level in luminal cell lines, yet nuclear protein was also abundant in basal-like lines, such as Colo824. Consistent with the data from primary breast cancer specimens and our previous findings, KDM4C is highly overexpressed in a set of basal-like breast cancer cell lines, including HCC1954 and Colo824, which both contain high-level KDM4C gene amplification (Note: Expression analysis indicate that the HCC1954 line belongs to the basal-like subtype even though it contains HER2 amplification) [18]. Strikingly, western blot analysis did not detect KDM4C protein expression in nuclear extracts from four ER+ Luminal breast cancer cell lines. Similar to KDM4A, KDM4D is also overexpressed at the mRNA level in various ER+ luminal and basal-like cell lines. Interestingly, HCC1954 and Colo824 cells also showed KDM4D (11q21) gene amplification. However, KDM4D protein in these nuclear extracts was likely ubiquitously expressed in ER+ and basal breast cancers as well as MCF10A cells (Figure 2B).
Figure 2.
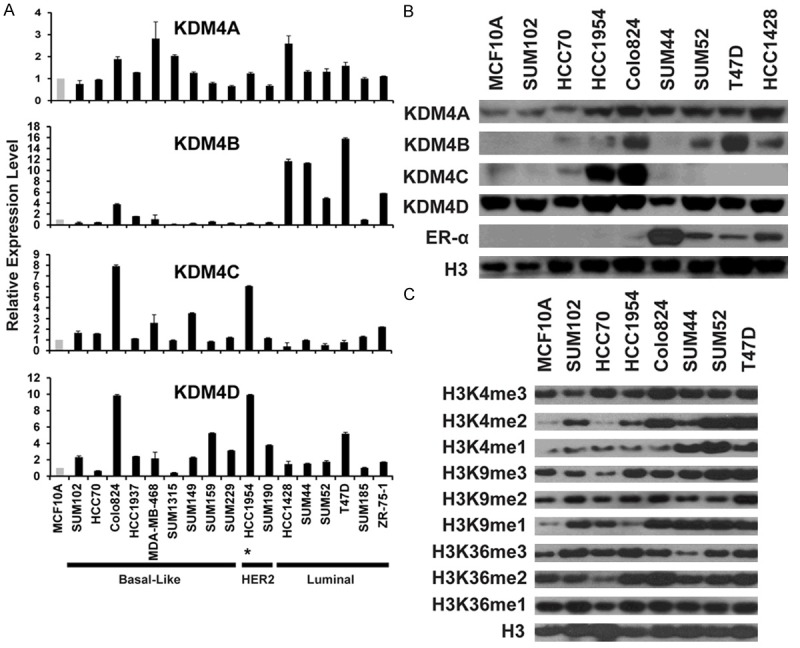
KDM4 and H3 mark expression in breast cancer. A. mRNA expression levels of KDM4 demethylases in a panel of breast cancer cell lines were determined by qRT-PCR. mRNA expression levels in the MCF10A cells, an immortalized but nontumorigenic breast epithelial cell line, were arbitrarily set as 1. Relative expression levels in breast cancer cell lines are shown as fold changes compared with that of MCF10A cells. *The HCC1954 line belongs to the basal-like subtype even though it contains HER2 amplification. B. KDM4A, B, C, D and ER protein levels were analyzed by western blot in eight breast cancer cell lines and MCF10A line. Total H3 was used as the loading control. C. Global H3K4, K9, and K36 methylation levels in seven breast cancer cell lines and MCF10A line. Histone extracts were prepared from cells using the EpiQuik Total Histone Extraction Kit. Global histone methylation levels were detected by western blot assays with specific histone methylation antibodies. Total H3 level was used as the loading control.
KDM4 proteins catalyze the demethylation of H3K9me3/me2 and H3K36me3/me2 marks. Next, we assessed the global methylation (H3K4, H3K9, and H3K36) levels in a panel of breast cancer cell lines, as well as MCF10A cells, by western blot. As shown in Figure 2C, global levels of H3K4me3/me2/me1, H3K9me3/me2/me1, and H3K36me3/me2/me1 vary among the different breast cancer cell lines. Overall, basal breast cancer cell lines showed moderately lower levels of histone lysine methylation, including H3K9me3/me2 and H3K4me2 at global levels. It should be noted that histone methylation is a dynamic process in the nucleus, and methylation patterns are also specific to any gene locus in a particular cell type. However, global levels of histone methylation are one way to measure KDM inhibitor activity, and the breast cancer cell lines with clearly defined histone methylation levels will be useful in establishing a model to further investigate the biological and functional roles of histone-modifying regulators in breast cancer, as well as to develop novel anti-cancer epidrugs.
KDM4 inhibitor NCDM-32B impairs viability and transforming phenotypes of basal breast cancer
Given the significant roles of KDM4 demethylases in the development of various cancers, there is rapidly growing interest in developing highly potent and selective inhibitors [6]. The enzymatic activity of the KDM4 subfamily depends on the JmjC catalytic domain which requires alpha-ketoglutarate as a key cofactor for the demethylation reaction. Based on the crystal structure of the KDM4A JmjC domain, a series of small-molecule inhibitors were designed and synthesized, including the pro-drug NCDM-32B (Figure 3A) [30-32]. In enzyme assays, NCDM-32B showed potent and selective KDM4 inhibition, with IC50 values of 3.0 and 1.0 μM for KDM4A and KDM4C, respectively [30,32]. We tested the effect of NCDM-32B on the behavior of basal breast cancer cell lines HCC1954 and Colo824 because these cell lines have the highest expression of KDM4C. As shown in Figure 3B, in HCC1954 cells, NCDM-32B treatment resulted in the increased global H3K9me3/me2 levels, while H3K4me3/me2 levels were not affected. NCDM-32B did not affect KDM4C protein level (data not shown). To analyze cell proliferation, we treated HCC1954, Colo824, and MCF10A cells with NCDM-32B. We found that NCDM-32B impaired the viability of HCC1954 and Colo824 cells in a dose-dependent manner and had only a slight effect on the control MCF10A cells, even at higher concentrations of 50 or 100 μM (Figure 3C). In addition, because anchorage-independent growth of cancer cells in vitro is a key aspect of the tumor phenotype, particularly with respect to metastatic potential, we performed similar studies to assess this feature [14]. Of note, the Colo824 breast cancer cell line was originally established from a metastatic site in a breast cancer patient and is known to form robust colonies in soft agar [33]. We found that NCDM-32B dramatically inhibited Colo824 anchorage-independent growth in soft agar (Figure 3D). Thus, our studies indicate that NCDM-32B inhibited KDM4 demethylase activity, decreased cell viability and anchorage-independent growth in soft agar of basal breast cancer cells.
Figure 3.
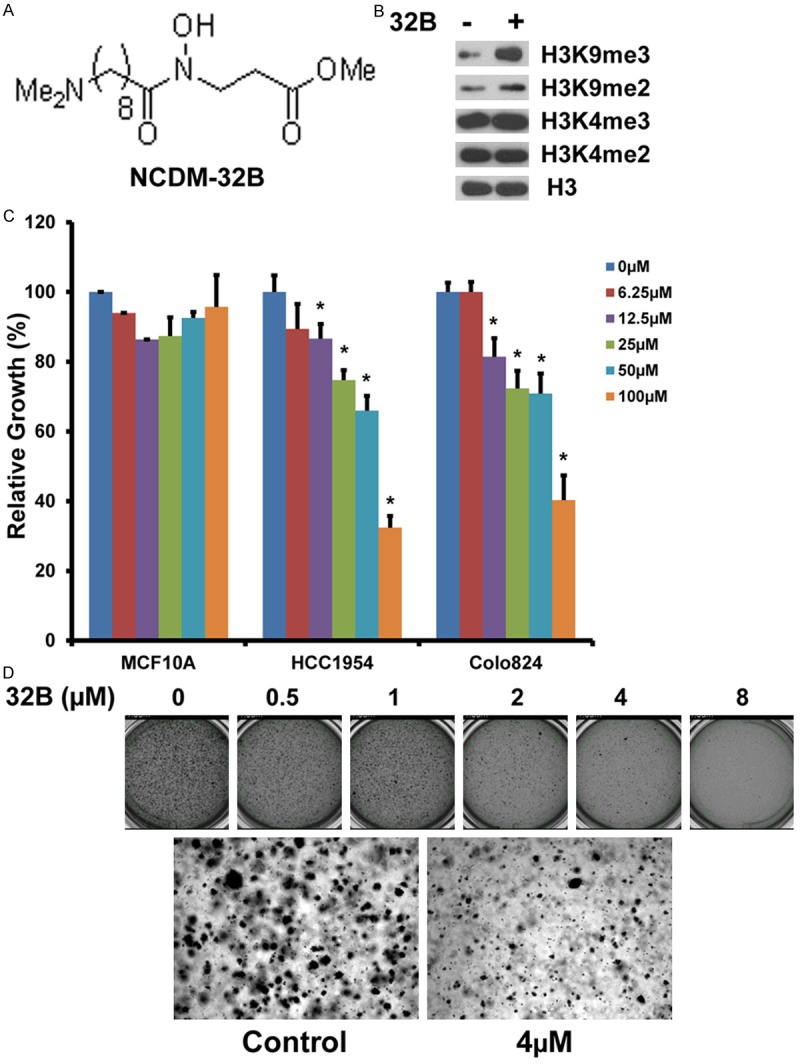
Effects of inhibitor NCDM-32B on breast cancer growth and transformation. A. Chemical structure of the KDM4 inhibitor NCDM-32B. B. Western blot of H3K4me3/me2 and H3K9me3/me2 levels in HCC1954 cells with or without NCDM-32B (40 µM) treatment for 48 hours. C. Growth-inhibitory effect of NCDM-32B on HCC1954 and Colo824 breast cancer cells and non-tumorigenic MCF10A cells. D. The growth of Colo824 breast cancer cells in soft agar was also significantly inhibited by NCDM-32B treatment in a dose-dependent manner. Data are expressed as mean ± SD. *p < 0.05.
KDM4 inhibitor NCDM-32B affects cell growth pathways
To examine changes in gene expression induced by NCDM-32B, we conducted mRNA microarray analysis in HCC1954 cells after NCDM-32B treatment. Cells treated with NCDM-32B were compared with corresponding controls, and a cutoff of log2 change > 0.4 was assigned to select the genes modulated by NCDM-32B. We found that, in HCC1954 cells, 597 genes were upregulated, and 463 genes were downregulated by NCDM-32B treatment. Ingenuity pathway analysis and gene ontology analysis showed that genes altered by NCDM-32B were enriched in pathways that control cellular growth and proliferation, DNA replication, recombination, and repair, as well as in cell cycle progression (Table S1). The top canonical pathways included mismatch repair in eukaryotes, cell cycle control of chromosomal replication, the role of BRCA1 in the DNA damage response, hereditary breast cancer signaling, and interferon signaling (Figure 4A). We also validated our mRNA microarray data in both HCC1954 and Colo824 cells after NCDM-32B treatment (Figure 4B). Notably, we found that several classical oncogenes, including the met proto-oncogene (MET), and key cell-cycle regulator genes, including cell division cycle 26 (Cdc26, a component of the cell cycle anaphase-promoting complex) and cyclin-dependent kinase 6 (CDK6), were inhibited by NCDM-32B. These data suggest that NCDM-32B contributes to the impairment of several critical pathways that drive cell growth and transformation in breast cancer.
Figure 4.
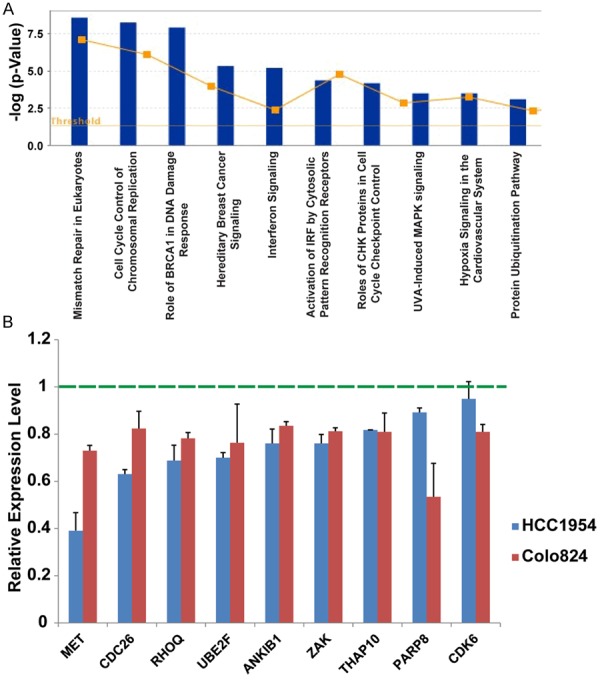
Gene expression changes with NCDM-32B inhibitor treatment in breast cancer. A. Ingenuity Pathway Analysis of the top pathways affected in differentially expressed genes in breast cancer after NCDM-32B treatment. The Y-axis is an inverse indication of P value or significance. B. qRT-PCR-validated mRNA microarray data in HCC1954 and Colo824 cells after NCDM-32B treatment.
Discussion
Utilizing The Cancer Genome Atlas breast cancer database and cell line models, we analyzed KDM4A, B, C and D gene amplification and expression relative to different breast cancer subtypes. Consistent with our previous findings, KDM4C amplification had the highest frequency (12.4%) in basal-like breast cancer compared with other subtypes [18]. Furthermore, we found high expression of KDM4A, C, and D in the basal type and KDM4B in ER+ luminal-type breast cancers. We also tested a novel KDM4 inhibitor, NCDM-32B, and found that it decreased cell viability and anchorage independent growth in soft agar of basal breast cancer cells. NCDM-32B inhibited both the activity of KDM4 demethylases and gene expression of several pathways related to cellular growth and transforming phenotypes.
Within the KDM4 subfamily, KDM4A, B, and C show a high degree of homology in sequence and domain organization [6]. Although KDM4 demethylases all catalyze via the same demethylation reaction, recent evidence indicates that their normal cellular functions are not completely redundant. Possible reasons for these unique functions are: (1) a distinct pattern of intracellular location, (2) cell-type-specific ex-pression, (3) selective recruitment of the different KDM4 demethylases to their target genes, and (4) demethylase specific non-histone proteins. Indeed, recent studies revealed that KDM4A is equally present in the cytoplasm and nucleus, KDM4B is more prevalent in the nucleus, and KDM4C strongly associates with chromatin [34,35]. Furthermore, meta-analyses of next-generation sequencing profiles revealed considerable variation in mRNA expression levels of the KDM4 subfamily members in different normal human tissues [6]. By querying proteomic profiling of 30 normal fetal and adult human tissues in the Human Proteome Map database, we also found a distinct expression pattern of KDM4 family (Figure S3). It was also reported that protein levels of KDM4A and B, but not KDM4C, are highly regulated by ubiquitination and the proteasome since KDM4A and B are direct substrates of the E3 ubiquitins RNF8 and RNF168 [36]. In contrast, inositol pyrophosphates regulate KDM4C-dependent histone demethylation [37]. More recent studies showed that KDM4B and C have distinct and combinatorial functions in mouse embryonic stem cell identity [38]. Most likely, the four KDM4 subfamily members have substantially overlapping but not completely redundant functions, and their functions are cell-type specific and context-dependent.
The basal-like subtype of breast cancer is often aggressive and has a poorer prognosis [1,3]. Previous studies from our lab demonstrated that KDM4C is prevalently amplified and overexpressed in the basal-like subtype [18]. Using a large-scale cancer genomics data set, we validated that KDM4C is amplified at high levels in 12.4% of basal-like tumors. If we incorporate low level-gain (39 of 89 cases, 43.8%), then more than half of basal-like tumors had an increased KDM4C copy number. Furthermore, we revealed that KDM4A, C, and D had high mRNA expression levels in basal-like breast cancer. As shown in Figures 1A and S1, high-level amplification and expression of KDM4A, C, and D tended toward mutual exclusivity. Thus, KDM4A, C, and D might individually contribute to the aggressive phenotypes of basal-like breast cancer.
Considering that epigenetic changes are reversible and histone demethylases are druggable, the KDM4 members are promising therapeutic targets. The NCDM-32B inhibitor was synthesized on the basis of the crystal structure model of the KDM4A Jumonji domain complexed with alpha-ketoglutarate [30-32]. In vitro biochemical assays demonstrated that NCDM-32B inhibits the activity of KDM4 subfamily demethylases with high selectivity for KDM4C (9000-fold) compared to other demethylase members [30,31]. We have shown that NCDM-32B impaired the proliferation and transforming phenotypes of KDM4C-amplified HCC1954 and Colo824 cells in vitro. Furthermore, we observed that NCDM-32B suppressed expression of a classical oncogene, MET, as well as other genes related to cellular growth and proliferation. Elevated MET expression and activation is associated with basal-like breast cancer [39,40]. Future investigations are required to further investigate the mechanism of NCDM-32B inhibition of MET by directly blocking the demethylase function of KDM4 subfamily, particularly KDM4C.
In conclusion, our findings add layers of information to the genomic and transcriptomic profiles of the KDM4 subfamily in different subtypes of breast cancer. First, we corroborated our earlier findings of amplification and overexpression of KDM4C in breast cancer, with greater prevalence in basal-like subtypes. We also presented new data revealing different patterns of DNA, mRNA, and protein levels of the four KDM4 members across the subtypes of breast cancer in breast cancer specimens and cell lines. We provide the first evidence that a novel KDM4 demethylase inhibitor, the small molecule NCDM-32B, decreases cell viability and anchorage-independent growth in soft agar of basal breast cancer cells. These findings lay the foundation for future studies to preclinically validate KDM4 inhibition as a therapeutic strategy for different subtypes of breast cancer.
Acknowledgements
This work was partially supported by grants from the NIH/NCI R21CA175244-01A1 and the Office of Research on Women’s Health (OWRH), the Mary Kay Foundation Cancer Research Grant Program, and the Karmanos Cancer Institute-SRIG to Dr. Z-Q.Y.; by funding from the Cancer Biology Graduate Program, Wayne State University School of Medicine, to A.H.; and by funding from the Natural Science foundation for Colleges and Universities in Jiangsu Province (10KJD180004) to Qin Ye. The Genomics core is supported, in part, by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute at Wayne State University. We thank Lanxin Liu, Jinling Hou and Xiaogang Wang for technical contributions, and Yuanyuan Jiang for critical reading of the manuscript. We thank Dr. Stephen P. Ethier for providing the SUM breast cancer cell lines and for his continuous encouragement.
Disclosure of conflict of interest
None of the authors declare conflicts of interest with this work.
Supporting Information
References
- 1.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 2.Creighton CJ. The molecular profile of luminal B breast cancer. Biologics. 2012;6:289–297. doi: 10.2147/BTT.S29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 5.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labbe RM, Holowatyj A, Yang ZQ. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2014;6:1–15. [PMC free article] [PubMed] [Google Scholar]
- 7.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 9.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 10.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hillringhaus L, Yue WW, Rose NR, Ng SS, Gileadi C, Loenarz C, Bello SH, Bray JE, Schofield CJ, Oppermann U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J Biol Chem. 2011;286:41616–41625. doi: 10.1074/jbc.M111.283689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinatzer U, Gollinger M, Mullauer L, Raderer M, Chott A, Streubel B. Mucosa-associated lymphoid tissue lymphoma: novel translocations including rearrangements of ODZ2, JMJD2C, and CNN3. Clin Cancer Res. 2008;14:6426–6431. doi: 10.1158/1078-0432.CCR-08-0702. [DOI] [PubMed] [Google Scholar]
- 16.Italiano A, Attias R, Aurias A, Perot G, Burel-Vandenbos F, Otto J, Venissac N, Pedeutour F. Molecular cytogenetic characterization of a metastatic lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genet Cytogenet. 2006;167:122–130. doi: 10.1016/j.cancergencyto.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Suikki HE, Kujala PM, Tammela TL, van Weerden WM, Vessella RL, Visakorpi T. Genetic alterations and changes in expression of histone demethylases in prostate cancer. Prostate. 2010;70:889–898. doi: 10.1002/pros.21123. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, Yang ZQ. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28:4491–4500. doi: 10.1038/onc.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G, Wright GW, Lenz G, Ngo VN, Shaffer AL, Xu W, Zhao H, Yang Y, Lamy L, Davis RE, Xiao W, Powell J, Maloney D, Thomas CJ, Moller P, Rosenwald A, Ott G, Muller-Hermelink HK, Savage K, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Weisenburger DD, Chan WC, Gascoyne RD, Levens D, Staudt LM. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell. 2010;18:590–605. doi: 10.1016/j.ccr.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109:E3367–3376. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60:4735–4739. [PubMed] [Google Scholar]
- 22.Berry WL, Shin S, Lightfoot SA, Janknecht R. Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol. 2012;41:1701–1706. doi: 10.3892/ijo.2012.1618. [DOI] [PubMed] [Google Scholar]
- 23.Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, Wakeham A, Miyagishi M, Mak TW, Okada H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6:e17830. doi: 10.1371/journal.pone.0017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaughan L, Stockley J, Coffey K, O’Neill D, Jones DL, Wade M, Wright J, Moore M, Tse S, Rogerson L, Robson CN. KDM4B is a Master Regulator of the Estrogen Receptor Signalling Cascade. Nucleic Acids Res. 2013;41:6892–6904. doi: 10.1093/nar/gkt469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang ZQ, Streicher KL, Ray ME, Abrams J, Ethier SP. Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res. 2006;66:11632–11643. doi: 10.1158/0008-5472.CAN-06-2946. [DOI] [PubMed] [Google Scholar]
- 26.Forozan F, Veldman R, Ammerman CA, Parsa NZ, Kallioniemi A, Kallioniemi OP, Ethier SP. Molecular cytogenetic analysis of 11 new breast cancer cell lines. Br J Cancer. 1999;81:1328–1334. doi: 10.1038/sj.bjc.6695007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao JJ, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun YC, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Kimball S, Liu H, Holowatyj A, Yang ZQ. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget. 2015;6:2466–2482. doi: 10.18632/oncotarget.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prat A, Karginova O, Parker JS, Fan C, He X, Bixby L, Harrell JC, Roman E, Adamo B, Troester M, Perou CM. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res Treat. 2013;142:237–255. doi: 10.1007/s10549-013-2743-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamada S, Suzuki T, Mino K, Koseki K, Oehme F, Flamme I, Ozasa H, Itoh Y, Ogasawara D, Komaarashi H, Kato A, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem. 2010;53:5629–5638. doi: 10.1021/jm1003655. [DOI] [PubMed] [Google Scholar]
- 31.Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg Med Chem Lett. 2009;19:2852–2855. doi: 10.1016/j.bmcl.2009.03.098. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki T, Miyata N. Lysine demethylases inhibitors. J Med Chem. 2011;54:8236–8250. doi: 10.1021/jm201048w. [DOI] [PubMed] [Google Scholar]
- 33.Savelyeva L, Claas A, An H, Weber RG, Lichter P, Schwab M. Retention of polysomy at 9p 23-24 during karyotypic evolution in human breast cancer cell line COLO 824. Genes Chromosomes Cancer. 1999;24:87–93. [PubMed] [Google Scholar]
- 34.Kim TD, Fuchs JR, Schwartz E, Abdelhamid D, Etter J, Berry WL, Li C, Ihnat MA, Li PK, Janknecht R. Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. Am J Transl Res. 2014;6:236–247. [PMC free article] [PubMed] [Google Scholar]
- 35.Kupershmit I, Khoury-Haddad H, Awwad SW, Guttmann-Raviv N, Ayoub N. KDM4C (GASC1) lysine demethylase is associated with mitotic chromatin and regulates chromosome segregation during mitosis. Nucleic Acids Res. 2014;42:6168–6182. doi: 10.1093/nar/gku253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burton A, Azevedo C, Andreassi C, Riccio A, Saiardi A. Inositol pyrophosphates regulate JMJD2C-dependent histone demethylation. Proc Natl Acad Sci U S A. 2013;110:18970–18975. doi: 10.1073/pnas.1309699110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das PP, Shao Z, Beyaz S, Apostolou E, Pinello L, De Los Angeles A, O’Brien K, Atsma JM, Fujiwara Y, Nguyen M, Ljuboja D, Guo G, Woo A, Yuan GC, Onder T, Daley G, Hochedlinger K, Kim J, Orkin SH. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol Cell. 2014;53:32–48. doi: 10.1016/j.molcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho-Yen CM, Green AR, Rakha EA, Brentnall AR, Ellis IO, Kermorgant S, Jones JL. C-Met in Invasive Breast Cancer. Cancer. 2014;120:163–171. doi: 10.1002/cncr.28386. [DOI] [PubMed] [Google Scholar]
- 40.Gastaldi S, Sassi F, Accornero P, Torti D, Galimi F, Migliardi G, Molyneux G, Perera T, Comoglio PM, Boccaccio C, Smalley MJ, Bertotti A, Trusolino L. Met signaling regulates growth, repopulating potential and basal cell-fate commitment of mammary luminal progenitors: implications for basal-like breast cancer. Oncogene. 2013;32:1428–1440. doi: 10.1038/onc.2012.154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.