

Abstract
Introduction
The median survival of patients with glioblastoma multiforme (astrocytoma grade 4) remains less than 18 months despite radical surgery, radiotherapy and systemic chemotherapy. Surgical implantation of chemotherapy eluting wafers into the resection cavity has been shown to improve length of survival but the current licensed therapy has several drawbacks. This paper investigates in vivo efficacy of a novel drug eluting paste in glioblastoma.
Methods
Poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG) self-sintering paste was loaded with the chemotherapeutic agent etoposide and delivered surgically into partially resected tumours in a flank murine glioblastoma xenograft model.
Results
Surgical delivery of the paste was successful and practical, with no toxicity or surgical morbidity to the animals. The paste was retained in the tumour cavity, and preliminary results suggest a useful antitumour and antiangiogenic effect, particularly at higher doses. Bioluminescent imaging was not affected significantly by the presence of the paste in the tumour.
Conclusions
Chemotherapy loaded PLGA/PEG paste seems to be a promising technology capable of delivering active drugs into partially resected tumours. The preliminary results of this study suggest efficacy with no toxicity and will lead to larger scale efficacy studies in orthotopic glioblastoma models.
Keywords: Glioblastoma, Drug delivery systems, Neurosurgery
Glioblastoma multiforme (GBM) (astrocytoma grade 4) is an aggressive, rapidly growing malignant brain tumour that remains incurable. Current optimal therapy consists of neurosurgical resection to as radical a degree as the tumour and neuroanatomy allows, followed by radical radiotherapy (60Gy) with adjuvant chemotherapy. Despite this multimodal approach, median survival remains poor at 14 months or less.1 It is clear that progress in management of high grade brain tumours has lagged behind that of other cancers with survival times only increasing minimally in recent years.
Complete surgical removal of these tumours is almost never possible owing to their infiltrative nature. Even when all tumour visible to the naked eye or on conventional preoperative magnetic resonance imaging is resected, remnant tumour cells remain in what appears macroscopically or radiologically to be normal brain.2 These infiltrative cells are resistant to radiotherapy, the dose of which is limited by neurotoxicity to normal brain.
Improvements in survival have been achieved with the chemotherapeutic agent temozolomide, which has been shown to be especially beneficial in individuals with a methylated promoter sequence for the methylguanine methyltransferase gene.3 Other forms of systemic chemotherapy, including molecularly targeted therapy, have not been shown to have significant survival benefits in humans, partly because of the difficulties of delivering adequate concentrations of agents across the blood–brain barrier (BBB) to achieve a dose lethal to tumour cells in the brain. Indeed, no therapy based on the molecular biology of GBM has yet shown efficacy in a phase III trial.4
One surgical approach that has demonstrated positive impact on survival has been the use of carmustine impregnated wafers (Gliadel®; Arbor Pharmaceuticals, Atlanta, GA, US) to deliver intracavity chemotherapy.5 Implanted by the operating neurosurgeon after maximal tumour resection has occurred (at least 90% to comply with National Institute for Health and Care Excellence [NICE] guidelines for product usage in the National Health Service [NHS]),6,7 these biodegradable polymer wafers release the chemotherapeutic alkylating agent carmustine, which spreads by diffusion into the surrounding brain parenchyma and has antineoplastic effects against the remnant infiltrating tumour cells that cannot be removed surgically without causing catastrophic morbidity.
Gliadel® has been shown in a randomised multicentre phase III trial to have a small but statistically significant survival benefit in GBM of 2–3 months.7 Despite this evidence, and its subsequent licensing and approval for NHS funding, uptake has been limited by concerns regarding possible increases in operative complication rates, particularly wound healing and infection. Some surgical series have reported a significant increase in wound infections following craniotomy,8,9 leading to increased hospital stays for patients who already have limited life expectancy.
The drug release kinetics for Gliadel® show a high burst of release occurring in the first few days after implantation, with this surge of cytotoxic agent potentially responsible for the impaired wound healing observed.10 Conformity of the rigid wafer to the irregular resection cavity wall can also be poor as wafers can displace with gravity. In addition, Gliadel® uses a monotherapeutic approach that may swiftly be countered by the rise of resistant tumour clones in the GBM.
A novel temperature sensitive and biodegradable drug carrying paste based on poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG) has been developed in Nottingham.11,12 PLGA is non-toxic with US Food and Drug Administration approval for certain clinical applications such as in resorbable sutures. The PLGA/PEG particles in the Nottingham formulation form a malleable paste at room temperature when mixed with water or saline. The particles then fuse together at body temperature, causing the paste to solidify gently, with no exothermic reaction.13 In vitro studies have demonstrated release of active chemotherapeutic agents from the formulation over three weeks at body temperature.14 The controlled release of multiple drugs concurrently is possible (Gould TW et al, unpublished data).
The effect of therapy on blood vessel development in a tumour is a key component of an anticancer drug. Reduction in blood vessel growth in the tumour will reduce the supply of nutrients to a tumour, slowing growth kinetics by depriving the tumour of vital substrates including oxygen.15 Evidence is also accumulating that antiangiogenic therapy may depend on the destruction of the perivascular tissue niche that is particularly favourable for the growth of GBM cells,16 particularly stem-like cells, which may confer resistance to oncological therapy.17
This study examined the effects of etoposide loaded PLGA/PEG paste in a murine flank/human GBM xenograft. The surgical relevance of this model was enhanced by undertaking partial resection of the tumour to mimic the clinical situation where even radical surgery leaves remnant tumour behind, concealed in apparently normal brain. The characteristics were studied of the paste in situ in the animal and its effects on the imaging techniques used to monitor tumour growth. The effects of drug release on the surrounding tumour were also investigated with particular focus on markers of blood vessels and angiogenesis. Furthermore, this study presents preliminary data suggesting a useful effect in controlling tumour growth although statistical examination of survival benefit will be required in future larger therapy experiments.
Methods
Particle production was as described previously14 at a ratio of 93.5%:6.5% PLGA:PEG. Particles were sieved to achieve a consistent sizing of 100–200µm. They were mixed with the etoposide drug solution at a ratio of 1:0.6 particles:solution (weight/volume). Drug concentration was at either 80mg/kg, 160mg/kg or 320mg/kg as per the animals’ body weight. Etoposide was purchased from Sigma (Gillingham, Dorset, UK) and resuspended in dimethyl sulfoxide.
This study was approved by the University of Nottingham local ethical review committee and the UK Home Office (licence number PPL 40/3559) after consideration of the justification of animal research and good animal welfare. Fifteen 4–6-week-old male MF1 nude mice (three mice with equivalent sized tumours per arm) were maintained under standard conditions as detailed in the Animals (Scientific Procedures) Act 1986, and studies were conducted and reported in compliance with the 2010 NC3Rs ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.18
U87 or U373 human GBM cells tagged with a bioluminescent marker were injected subcutaneously into the animals’ left flank and the tumour was grown for 15 days while monitored using the IVIS® Spectrum bioluminescent imaging system (PerkinElmer, Cambridge, UK). Mice with satisfactory tumour take and growth rates underwent partial tumour resection. The previous flank incision was reopened and a biopsy punch and fine suction tip used to resect the tumour back to near the tumour–tissue interface, resecting approximately 75% of visible tumour, thereby mimicking the surgical technique used in human patients undergoing comparable surgery for GBM.
Etoposide loaded PLGA/PEG matrices (80mg/kg, 160mg/kg or 320mg/kg) (experimental arms) or blank PLGA/PEG matrices (control arm) were moulded into the resection cavity. Mice were monitored by two and three-dimensional bioluminescent imaging of established tumours prior to implantation and weekly until termination to assess tumour growth. The tumour was positioned uppermost to gain maximum imaging sensitivity. Animals were weighed daily by an experienced technician, any adverse effects noted and sacrificed using cervical dislocation to ameliorate suffering once their clinical condition deteriorated. Mice were sacrificed at a maximum of 28 days following implantation of PLGA/PEG.
Anti-Ki-67 (monoclonal mouse antihuman Ki-67 antigen, clone MIB-1; Dako, Ely, UK) and anti-VEGF (monoclonal mouse antihuman vascular endothelial growth factor, clone VG1; Dako) were used at 1:50 concentration, and anti-endoglin (ab49679; Abcam, Cambridge, UK) was applied at 1:100. All primary antibodies were applied for one hour at room temperature. A secondary antibody (100µl horseradish peroxidase conjugated rabbit antimouse; Dako) was applied for 30 minutes at room temperature. 2µl of 3,3'-diaminobenzidine chromogen in 98µl of REAL™ substrate buffer (Dako) was applied before haematoxylin counterstaining and mounting.
Results
PLGA/PEG does not interfere significantly with bioluminescent imaging
In order to examine the difference between paste and tissue, a tumour was excised from a mouse rejected from the main study owing to a low bioluminescence of less than 2 × 106 photons per second total flux as measured through the intact skin. This large tumour was dissected immediately after imaging while still bioluminescent and was imaged to obtain a base level photon reading. It was then coated with a blank PLGA/PEG matrix to assess the level of light blockade (Figs 1A and 1B). When the tumour was excised, the unimpeded flux measurement ex vivo rose to 9 × 107 photons per second. Coating with PLGA/PEG reduced flux to 3.6 × 107 photons per second (Fig 1C). The PLGA/PEG coating therefore causes a low level of signal blockade (a 2.7:1 reduction). Skin alone impedes light signal by a 5:1 reduction. The effect of surgical resection was easily visible using bioluminescent imaging, with the cavity clearly visualised, allowing standardisation for the amount of resection undertaken (Fig 1D).
Figure 1.

Effects of poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG) on bioluminescent monitoring of tumours Top: Xenograft of U87 human glioblastoma cell line grown in murine flank, excised, with bioluminescence measured before (A) and after (B) application of layer of PLGA/PEG, demonstrating reduction with coating of the biomaterial Bottom: Comparison of flux levels between coated and non-coated tumours demonstrating some reduction but to a lesser degree than with normal tissues (C), and bioluminescent imaging of mouse demonstrating tumour with resected core (arrow) (D)
PLGA/PEG effects remain localised in vivo
The tumour implanted with PLGA/PEG was excised 28 days following implantation, and examined macroscopically and microscopically. The paste remained entirely in the resection cavity with no spillage into the surrounding flank tissues of the animal (Fig 2A). There was good adherence to the cavity wall with coverage of the entire cavity, providing intimate contact between the maximum extent of the tumour and the drug delivery source. The histology findings on Figure 2B illustrate PLGA/PEG forming a close approximation to the tumour (even allowing for a degree of tissue retraction following fixation). In an ex vivo ovine brain, the paste remained attached even when spread thinly as a 2mm thick lining to a brain resection cavity (Fig 2C). The PLGA/PEG retained its characteristic microparticulate structure after implantation (Fig 2D) with particles of approximately 100µm.
Figure 2.
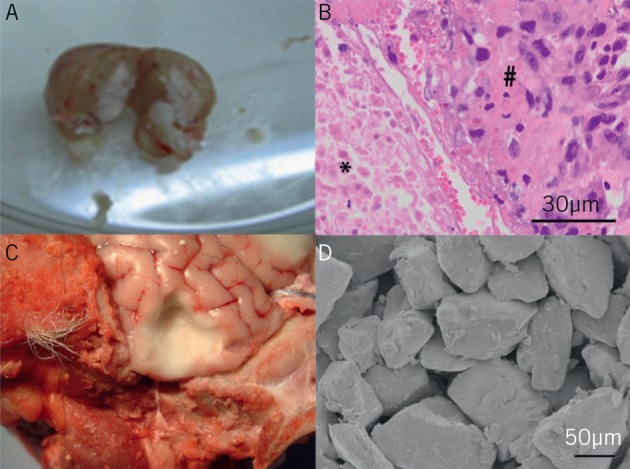
Poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG) application in vivo A: Excised U373 human glioblastoma murine flank xenograft 28 days following implantation, dissected in half, demonstrating central pale core of PLGA/PEG retained in surrounding tumour
B: Histology of tumour on left demonstrating PLGA/PEG matrix (*) closely opposed to tumour (#)
C: Ex vivo ovine model with cavity resection in brain, lined with pale PLGA/PEG sintered into place, and retaining shape and apposition to cavity walls
D: Scanning electron micrography of PLGA/PEG matrix microparticles
Animal weights for all groups, including the higher doses of etoposide delivery, continued to increase throughout the study period, indicating preservation of the animals’ general health. Systemic delivery of etoposide is associated with typical cytotoxic adverse events but in these experiments, no such events were observed, suggesting no significant systemic effect from the localised delivery of the chemotherapy (Fig 3).
Figure 3.
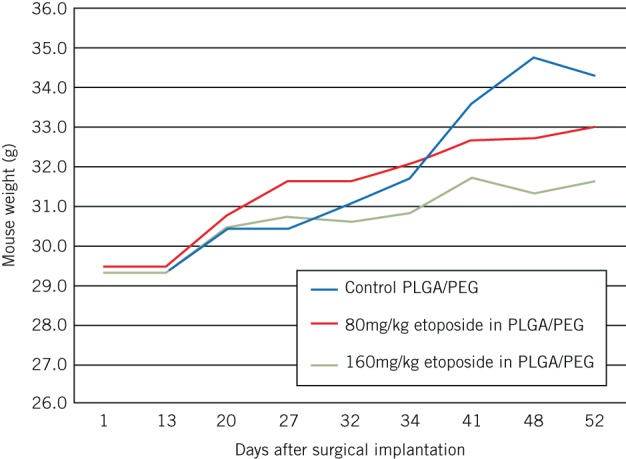
Animal weights after surgical implantation of blank poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG), PLGA/PEG with 80mg/kg etoposide and PLGA/PEG with 160mg/kg etoposide
Localised drug release has antiangiogenic effects
Histological examination of tumours resected after 28 days demonstrated tumour necrosis in the immediate vicinity, as reported previously.14 The tumour tissue was also stained using the Ki-67 (MIB-1) marker for cell proliferation and markedly decreased levels of proliferating cells were found after exposure to the locally released etoposide compared with animals implanted with blank PLGA/PEG paste (Fig 4). Similarly, levels of vascular endothelial growth factor staining were reduced significantly following 28 days of exposure to the etoposide loaded PLGA/PEG but not with blank PLGA/PEG. Immunohistochemistry was also performed to examine the number of vessels staining positive for the neoangiogenic marker endoglin. In a similar fashion, levels of new vessels in the adjacent tumour were reduced after usage of etoposide loaded but not blank PLGA/PEG (Fig 4).
Figure 4.
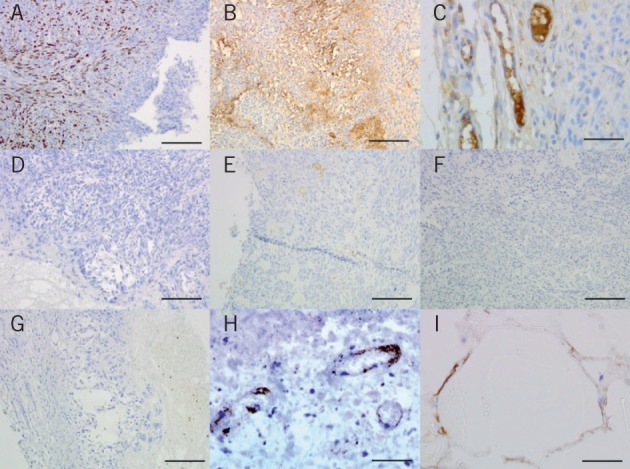
Immunohistochemistry in murine flank xenografts A&D: Staining for Ki-67 proliferation marker in a U87 murine flank xenograft partially resected and treated with blank poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG) (A) or PLGA/PEG loaded with 160mg/kg etoposide (D), showing reduction in tumour cell proliferation in response to active etoposide release
B&E: Staining for vascular endothelial growth factor (VEGF) in a U87 murine flank xenograft partially resected and treated with blank PLGA/PEG (B) or PLGA/PEG loaded with 160mg/kg etoposide (E), showing reduction in VEGF positivity
C&F: Staining for the angiogenic vessel marker endoglin, showing new vessel formation in a U87 murine flank xenograft treated with blank PLGA/PEG (C) but not for PLGA/PEG loaded with 160mg/kg etoposide (F)
G: Low power view of an excised U87 murine flank xenograft treated with PLGA/PEG loaded with 160mg/kg etoposide showing the inner zone of necrosis, more viable tumour centrally and the outer capsule of normal mouse tissue
H: Immunostaining for endoglin in a U87 murine flank xenograft treated with PLGA/PEG loaded with 160mg/kg etoposide showing damaged blood vessels in highly necrotic tissue
I: Immunostaining for VEGF with tumour cells growing around blank PLGA/PEG particles showing the non-toxic nature of the carrier biomaterial
Intracavity etoposide release slows tumour growth
Serial examination of the bioluminescence levels in tumours demonstrated substantial reduction in growth in tumours treated with the higher dose (160mg/kg) of etoposide, which persisted until 21 days following implantation (Fig 5). After this time, tumour regrowth began to occur at the 28-day timepoint, consistent with our in vitro data, which suggests drug release ceases at the 21-day timepoint. Tumour regrowth at this stage was rapid as the tumour ‘rebounded’ and the remnant tumour cells could now grow unopposed. In the clinical situation, patients would normally commence radiotherapy and temozolomide three weeks after surgery, which should prevent a similar phenomenon. At the highest etoposide dosage level, substantial reduction in tumour bioluminescence occurred, a correlate of reduced tumour activity and burden.
Figure 5.

In vivo effects of drug eluting poly(lactic-co-glycolic acid)/poly(ethylene glycol) (PLGA/PEG)
A: Tumour growth following partial tumour resection and implantation of blank PLGA/PEG, PLGA/PEG with 80mg/kg etoposide and PLGA/PEG with 160mg/kg etoposide. The higher dose PLGA/PEG with etoposide demonstrated reduction in tumour growth in the animals until day 21, when in vitro studies predicted drug release would stop. By the day 28 measurement, tumour growth had resumed at a rapid rate.
B: Sample bioluminescent image of two mice with partial resection of a U373 flank xenograft, mouse on left with blank PLGA/PEG and mouse on right with PLGA/PEG loaded with 320mg/kg etoposide, showing considerable reduction in bioluminescence and corresponding reduction in tumour load
Discussion
GBM remains an essentially incurable malignant brain tumour for which surgery can reduce disease volume and prolong survival. However, disease recurrence is almost inevitable as infiltrative malignant cell populations remain in the surrounding brain parenchyma. Adjuvant oncological treatment with radiotherapy and temozolomide improves survival but still cannot eradicate the invasive cancer cells. The choice and efficacy of chemotherapy agents is limited by their ability to cross the BBB. Escalating systemic doses of drug to achieve effective levels in the brain itself results in unacceptable toxicity (eg bone marrow suppression).
One approach to achieve high concentrations of chemotherapy in the brain but low toxic doses systemically is to surgically implant chemotherapy depot material to release the drug over a period of time. This directly bypasses the BBB, enhancing the local dose while minimising exposure of other organs such as the bone marrow and liver. The only NICE approved application of this technique is Gliadel® (carmustine) wafers.7 Although they do prolong survival by a short but significant amount, their efficacy is limited by technical factors. They have poor resection cavity conformity, delivery is limited to one agent and drug release is rapid with all drug eluted off the wafers by one week.10 Most patients in the UK wait three weeks between surgery and commencement of adjuvant therapy. Surgically implanted PLGA/PEG with its gradual drug release over 3–4 weeks has the potential to fill this ‘therapy gap’ as a possibly more effective delivery system for intracavity chemotherapy.
It has been demonstrated previously that PLGA/PEG has physical characteristics that make it extremely attractive surgically.14 It is applied as a malleable paste at room temperature, allowing close tissue approximation to the infiltrated brain parenchyma, and sinters gently (in a non-exothermic fashion) at body temperature, causing retention at the site of application. It can deliver a combination of chemotherapeutic agents over a prolonged time course, and the released agents retain cytotoxicity in vitro and in vivo. It is not affected by clinically relevant doses of radiation, it is non-toxic, and identifiable on computed tomography and magnetic resonance imaging in relation to the brain.
In the current study, we demonstrate that PLGA/PEG does not interfere with the bioluminescent imaging system used to assess tumour growth. There is a possibility that the paste could interfere marginally with a weaker signal if it is positioned directly between the camera and the signal source but at a much lower level than that observed in pre-existing tissue such as skin, muscle and bone. If used as a packing agent in a debulked tumour, the paste is likely to reduce light signal from the tissue compartment below it at a low level and have minimal effect on the rest of the remaining tumour other than alteration of the direction of light refraction. PLGA/PEG retains its position in the tumour in a living animal and does not spread outside of the target region.
Tumour necrosis has been demonstrated previously in response to the release of the cytotoxic agent etoposide from PLGA/PEG paste in vivo.14 The present study extended these findings to show reduction in other critical markers of tumour growth such as cell proliferation, release of proangiogenic factors and the development of new blood vessels. This suggests that the released drug is having meaningful antineoplastic effects, and interfering with tumour growth beyond the zone of obvious cell death and necrosis.
Disruption of the development of new microvasculature19 has been associated with disruption of the supportive tumour microenvironment (the perivascular niche) that allows GBM to grow and invade so rapidly.20 In particular, this disruption of the blood vessel network may deny tumour stem-like cells a supportive refuge in which they can resist the effects of adjuvant oncological therapy.21–23 The current ‘therapy gap’ can therefore be used as a time for surgically implanted therapy to target the residual invasive cell population and potentially act as a sensitiser for radio or chemotherapies.
Even with this limited examination of a relatively low dose of single agent etoposide, antitumour effects can be demonstrated in the animals. With the caveat of low sample numbers, there appears to be a drug response in the high dose etoposide loaded PLGA/PEG group compared with the control PLGA/PEG group and the 80mg/kg etoposide loaded PLGA/PEG group over 21 days following implantation. By day 28, the tumours in this responsive group appear to be escaping drug control, as may be expected from the in vitro drug release kinetics, whereby drug release tailed off at the 21-day mark.14 No substantial side effects were demonstrated in any group due to systemic toxicity.
Efforts will now focus on higher doses of etoposide and combining this with multiple agents such as temozolomide or a poly(ADP-ribose) polymerase inhibitor. PLGA/PEG has the potential to deliver a wide variety or combinations of agents and may even be suitable for delivering viruses or genetic material. This trial was terminated at 28 days following surgery, and future studies will focus on animal survival and be powered to examine this statistically.
Clearly, the current animal model is limited by implantation of the tumour in the animal flank rather than in the brain. We are currently engaged in developing an orthotopic partial resection brain tumour model, which will allow toxicity and efficacy testing in a setting equivalent to the intended clinical usage in humans. Although no toxicity was seen in the current model, we would anticipate that systemic exposure to the drug would be reduced by its direct placement in the central nervous system and beyond the BBB. This may allow higher doses of the drug to be used although direct neurotoxicity will have to be monitored. Tumour response is also dependent on the surrounding microenvironment and this will have to be evaluated carefully in an orthotopic setting as a prelude to any future clinical human trial.
Conclusions
Surgical intracavity drug delivery using the PLGA/PEG system is a safe and effective way of releasing chemotherapeutic agents (in this case, etoposide) in a living animal. It has the potential to be a useful adjunct to current therapy for GBM, seemingly extending survival for this highly aggressive tumour.
References
- 1.Stupp R, Mason WP, van den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–996. [DOI] [PubMed] [Google Scholar]
- 2.Price SJ, Jena R, Burnet NG et al. Improved delineation of glioma margins and regions of infiltration with the use of diffusion tensor imaging: an image-guided biopsy study. Am J Neuroradiol 2006; 27: 1,969–1,974. [PMC free article] [PubMed] [Google Scholar]
- 3.Hegi ME, Diserens AC, Gorlia T et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352: 997–1,003. [DOI] [PubMed] [Google Scholar]
- 4.Patel M, Vogelbaum MA, Barnett GH et al. Molecular targeted therapy in recurrent glioblastoma: current challenges and future directions. Expert Opin Investig Drugs 2012; 21: 1,247–1,266. [DOI] [PubMed] [Google Scholar]
- 5.Brem H, Piantadosi S, Burger PC et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet 1995; 345: 1,008–1,012. [DOI] [PubMed] [Google Scholar]
- 6.National Institute for Health and Clinical Excellence. Improving Outcomes for People with Brain and Other CNS Tumours. London: NICE; 2006. [Google Scholar]
- 7.Westphal M, Hilt DC, Bortey E et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol 2003; 5: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGovern PC, Lautenbach E, Brennan PJ et al. Risk factors for postcraniotomy surgical site infection after 1,3-bis (2-chloroethyl)-1-nitrosourea (Gliadel) wafer placement. Clin Infect Dis 2003; 36: 759–765. [DOI] [PubMed] [Google Scholar]
- 9.Perry J, Chambers A, Spithoff K, Laperriere N. Gliadel wafers in the treatment of malignant glioma: a systematic review. Curr Oncol 2007; 14: 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet 2002; 41: 403–419. [DOI] [PubMed] [Google Scholar]
- 11.Rahman CV, Kuhn G, White LJ et al. PLGA/PEG-hydrogel composite scaffolds with controllable mechanical properties. J Biomed Mater Res B Appl Biomater 2013; 101: 648–655. [DOI] [PubMed] [Google Scholar]
- 12.Rahman CV, Ben-David D, Dhillon A et al. Controlled release of BMP-2 from a sintered polymer scaffold enhances bone repair in a mouse calvarial defect model. J Tissue Eng Regen Med 2014; 8: 59–66. [DOI] [PubMed] [Google Scholar]
- 13.Dhillon A, Schneider P, Kuhn G et al. Analysis of sintered polymer scaffolds using concomitant synchrotron computed tomography and in situ mechanical testing. J Mater Sci Mater Med 2011; 22: 2,599–2,605. [DOI] [PubMed] [Google Scholar]
- 14.Rahman CV, Smith SJ, Morgan PS et al. Adjuvant chemotherapy for brain tumors delivered via a novel intra-cavity moldable polymer matrix. PloS One 2013; 8: e77435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahman R, Smith S, Rahman C, Grundy R. Antiangiogenic therapy and mechanisms of tumor resistance in malignant glioma. J Oncol 2010; 251231. [DOI] [PMC free article] [PubMed]
- 16.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell 2011; 8: 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seidel S, Garvalov BK, Wirta V et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010; 133: 983–995. [DOI] [PubMed] [Google Scholar]
- 18.Kilkenny C, Browne WJ, Cuthill IC et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SJ, Tilly H, Ward JH et al. CD105 (Endoglin) exerts prognostic effects via its role in the microvascular niche of paediatric high grade glioma. Acta Neuropathol 2012; 124: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calabrese C, Poppleton H, Kocak M et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11: 69–82. [DOI] [PubMed] [Google Scholar]
- 21.Heddleston JM, Li Z, McLendon RE et al. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009; 8: 3,274–3,284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan N, Pires IM, Bencokova Z et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res 2010; 70: 8,045–8,054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Straussman R, Morikawa T, Shee K et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487: 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]